
Abstract
The novel coronavirus SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2) has led to a global crisis by infecting millions of people across the globe eventually causing multiple deaths. The prominent player of the virus has been known as the spike protein which enters the host system and leads to the infection. The S2 subunit is the most essential in this process of infection as it helps the SARS-CoV-2 to infect the host by binding to the human angiotensin converting enzyme 2 (hACE2), with the help of the receptor binding domain found at the S2 subunit of the virus. Studies also hypothesize that the S glycoproteins present in the virus interacts with different hosts in different ways which might be due to the mutations taking place in the genome of the virus over time. This work aims to decipher the similarities and differences in the sequences of spike proteins from samples of SARS-CoV-2 acquired from different infected individuals in different countries with the help of in silico methods such as multiple sequence alignment and phylogenetic analysis. It also aims to understand the differential infection rates among the infected countries by studying the amino acid composition and interactions of the virus with the host.
Similar content being viewed by others

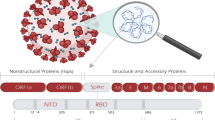

Introduction
SARS-CoV-2 infection lead to a large-scale pandemic distressing several countries around the world. The infection leads to several symptoms such as fever, severe respiratory illness, and pneumonia in the human population (Wrapp et al. 2020). SARS-CoV-2 is a novel coronavirus which was initially reported in the markets of Wuhan, China in November 2019 (Araujo and Naimi 2020). The virus is exceedingly contagious and can be transferred by droplets from the host body. It has been shown to be highly similar to other coronaviruses some of which caused similar diseases such as SARS (severe acute respiratory syndrome) in 2002 and MERS (middle east respiratory syndrome) in 2012 (de Wit et al. 2016). Although, their slow infection rate fortunately did not lead into a pandemic situation. Statistical observations by the world health organisation (WHO) reported that the infection through MERS and SARS took place at the rate of 1000 people in 4 months while, SARS-CoV-2 took 48 days to infect 1000 people. Its rapid rate of infection urged the WHO to affirm it a public health emergency of international concern (PHEIC) (Tarik Jasarevic and Chaib 2020). Prolonged infection with SARS-CoV-2 can cause an increased release of cytokines which may lead to cytokine release syndrome, that is characterized by multiple organ failure and fever (Sun et al. 2020).
SARS-CoV-2 belongs to the family of coronaviridae and sub-family of orthocoronavirinae. The virus is a single stranded positive RNA virus (26 to 32 kilo base pairs) having spike proteins which are crown-like in structure, when viewed under an electron microscope (Periwal et al. 2020). SARS-CoV-2 is very much related to SARS-CoV and is also in close relation to bat coronavirus, as discovered by Zhou et al. (2020). Furthermore, it has been reported that capping loops that cause amplified communication between the viral spike proteins and the human ACE2 cellular receptor in humans are present in human coronavirus but are not present in the bat coronavirus. The virus consists of structural and non-structural proteins. Structural proteins are of four types, spike glycoproteins, envelope proteins, membrane proteins, and nucleocaspid proteins (Forster et al. 2020). Spike proteins emerge from the envelope and aid in host sensitivity and attachment of virus to host (Ortega et al. 2020). Membrane fusion process of infection of the host cell is mediated by the spike glycoproteins present on the surface of the virus. These homotrimeric spike glycoproteins present on the envelope bind to the cellular receptors on the host membrane leading to the viral entry. (Zhang et al. 2020). Spike glycoproteins are made up of two subunits S1 and S2. Each subunit of the trimer is 180 kDa to 200 kDa in size (Ou et al. 2020). The S1 subunit is present within the amine terminal of the S homotrimer. It consists of N-terminal domain (NTD), receptor binding domain (RBD), and receptor binding motif (RBM). Whereas, the S2 subunit is extremely conserved and is present within the C terminal of the sequence. The S2 subunit consists of a fusion peptide (FP), heptad repeat 1 (HR1), heptad repeat 2 (HR2), transmembrane domain (TM) and cytoplasmic domain (CP) (Hillen et al. 2020). Enhanced interactions between the heptad repeat 1 and heptad repeat 2, lead to the stabilization of 6HB structures which cause an enhanced capability of SARS-CoV-2 to contaminate the host (Xia et al. 2020). The spike glycoprotein consists of a furin cleavage site between the two subunits of the S protein. This cleavage site aids in replication of viral protein and differentiates SARS-CoV-2 from all other coronaviruses (Walls et al. 2020). The S proteins present in the virus can be divided by host human proteases at the site of the S2 subunit, this leads to the activation of membrane fusion protein with the help of conformational changes which are irreversible (Walls et al. 2020). SARS-CoV-2 with the help of spike glycoproteins interacts with a receptor called the human angiotensin converting enzyme 2 (hACE2) and infects the human body. The interaction between the viral subunit and enzyme occur via endocytosis with the help of phosphoinositides (Ou et al. 2020). The virus spike glycoprotein belongs to the class I of fusion proteins. The α-helical coiled structure formed is a character of this type of fusion protein. It is also composed of a C terminal region which possess these α-helical formations having a coiled coil structure (Heald-Sargent and Gallagher 2012; Zhang et al. 2020). Open reading frames present the viral genome work as templates for the production of sub-genomic mRNAs and also aid in the termination of transcription. Sub-genomic mRNA is a key player in the replication-transcription complex which causes transcription of the viral genome. There are up to seven open reading frames present in a single coronavirus genome (Xia et al. 2020). The entire structure of the spike glycoprotein consists of 1273 sites, out of which 1 to 667 regions mark the S1 subunit and 668 to 1273 mark the S2 subunit (Fig. 1). Site 336 to 516 consist of the receptor binding domain (RBD) and regions 424 to 494 are responsible for the membrane binding. Similarly, for S2 subunit the region 770 to 788 are made up of fusion proteins, 915 to 949 are the heptad repeat, 1150 to 1185 consist of the heptad repeat 2 and 1190 to 1273 consist of the domains of the transmembrane and cytoplasm.

Schematic representation of the binding of S1 subunit of the SARS-CoV-2 molecule to the ACE2 present in a human cell. The receptor binding domain binds identifies and binds to the ACE2 in the host organism
Studies have reported that the SARS-CoV-2 is highly similar to bat coronavirus, specifically to RaTG13 which reportedly shares a 98% homology to the spike glycoprotein within SARS-CoV-2. A furin recognition site "RRAR" is located within SARS-CoV-2 spike glycoprotein because of an addition inside the S1 or S2 site of division (Wrapp et al. 2020). Moreover, Shang et al. have indicated through a study in SARS-CoV-2 that mutations in spike glycoproteins of novel coronavirus can lead to a change in characteristics of the virus which has been theorised to cause an increase in viral pathogenesis (Shang et al. 2020). It has been noted that the rate of infection varies among countries as per statistics since the outbreak in January 2020 up to March 2021 (Fig. 2) (Othman et al. 2020).

Analysis of the number of SARS-CoV-2 cases in different countries from January 2020 to May 31st 2020
Multiple hypothesis has been developed and tested over the months against the spread of SARS-CoV-2 infections in humans. Miguel B. et al. recommended that the spread of SARS-CoV-2 virus followed a seasonal climate pattern. Based on their in silico studies, the transmission rates were reported to be higher in arid and temperate regions (Araujo and Naimi 2020). Rahila Sardar et al. hypothesised that the mutations in the glycoprotein regions which mediate immune response vary within different geographical regions and may be key in understanding the differences in severity of infection among different countries (Sardar et al. 2020; Fang et al. 2020). S2 subunit plays a crucial role in transmission of infection. The sequence of the surface glycoprotein is reported to be approximately 1273 amino acids in length. It was hypothesized that the possibility of having variation in the spike glycoproteins found in humans from different countries might be high. This might support the hypothesised statement for an augmented pace of infection in the population of certain countries as compared to others. Previous research has shown that the phylogenetic investigation of genomes from diverse geological areas does not have any significant result but showed variable clustering among different countries (Sardar et al. 2020). This suggests that a variation might be possible at an amino acid mutation level which could lead to an increased infection in certain populations around the world. Several other studies have demonstrated the clustering of amino acids in the protein sequences of countries leading to the assumption that a massive exchange was taking place from the epicentre of the disease to other countries via carriers (Begum et al. 2020). The main objective of this study was to understand the mutational changes in the spike glycoproteins between infected populations around the globe. In this study, phylogentic studies of SARS-CoV-2 were carried out along with multiple sequence alignment to understand the variation in spike glycoproteins between infected populations in various countries.
Procedure
Protein sequence retrieval
The surface glycoprotein sequences for SARS-CoV-2 from multiple countries was acquired from the NCBI (National Centre for Biotechnology Information) database for novel coronavirus called NCBI Virus. Surface glycoprotein, S protein and Spike protein; in conjugation with SARS-CoV-2 and the desired country were related as query terms during the search through the database. The sequences were downloaded in their FASTA format and stored in a notepad. All the sequences were made up of 1273 amino acids or sites.
Multiple sequence alignment
All retrieved sequences were aligned using MEGA X (version 10.1.8) using the inbuilt MUSCLE alignment feature. The cluster iterations used UPGMA (un-weighted pair group method with arithmetic man) as a guide, along with 24 as the minimum length of diagonal. A total of 147 sequences were aligned using this software. The aligned data were saved in the form of an excel sheet and the mutation in the sequence was highlighted. The MEGA software is able to align more than 2000 sequences at once in a few minutes. The data were stored with the MSDX suffix and all conserved, singleton, variable and parsimony integrated sites were highlighted. The alignment image was then stored as an image file.
Phylogenetic tree analysis
Using previously aligned protein sequences, a phylogenetic hierarchy was designed to understand the connection between the sequences collected from different geographical locations around the world. MEGA X software (version 10.1.8) was used to prepare the phylogenetic tree. A tree was created by means of maximum likelihood as a statistical base. The analysis had a bootstrap value of 500 replicates. The substitution of amino acids was done using the Jones-Taylor-Thornton (JTT) matrix based Model with uniform rates among different amino acid sites. Missing data and gaps were set to use all sites to ensure an efficient phylogenetic tree. Tree inference options for maximum likelihood heuristics included Nearest-Neighbour-Interchange (NNI) and the initial tree was set to default. Data acquired was stored as a portable document format (PDF) file for further assessment. The amino acid composition was also calculated per sample using the inbuilt tool on MEGA software. On the basis of the phylogenetic tree, pair wise distance between the sequences was calculated using the distance feature in MEGA X software (version 10.1.8).
Outcome & analysis
Sequences collected from NCBI Virus, a public database, were downloaded as a text document. As of 4th May, the sequences were predominantly from China and USA, mainly due to the amount of samples submitted to the database. The sequences retrieved were, ten each from China, India, Hong Kong, Greece, France, Taiwan, Thailand, Australia, USA, and Spain. The other sequences retrieved were Germany (6), Czech Republic (8), Puerto Rico (7), Srilanka (4), Iran (2), Israel (2), South Africa (1), Kazakhstan (4), Malaysia (3), Nepal (1), Pakistan (2), South Korea (4), Italy, (2) and Brazil (2). These sequences were then used to carry out a multiple sequence alignment using MEGA software (version 10.1.8.8). The inbuilt MUSCLE feature was able to sequentially align 147 sequences from various countries around the world. The sequences were composed of total 1273 amino acids. The final alignment displayed, 32 variable sites, 1241 conserved sites. Also, five sites were parsimony informative, which means that these sites consisted of at least two types of amino acids at the site. Also, at least two of those amino acids occurred with a minimum frequency of two. Moreover, the alignment showed 27 singleton sites out of 1273 which illuminates the presence of regions with at least 2 amino acids with 1 repeating several times. The amino acid sequences were > 99% homologues to each other with the exception of single amino acid mutations. Multiple mutations were noted after alignment. The most prominent mutation observed was the substitution of Glycine (G) with Aspartic acid (D) at the 614th position. Based on previous studies, this mutation occurs due to a change in the a triplet code in the RNA sequence when GAU and GAC which code for aspartic acid and GGU and GGC which both code for Glycine undergo a single nucleotide substitution of G to A or vice versa (Fig. 3) (Korber et al. 2020). According to the study, this mutation was visible in many European samples. In our study conducted with multiple countries, we noted that this mutation was more prevalent in Asian countries for instance Taiwan, China, Hong Kong, Malaysia, South Korea and Pakistan. Other countries included, Italy and Brazil. The other substitutions were as shown in Table 1.

MSA of 1247 sequences determined that 63 sequences consisted of the D614G mutation
Another mutation observed was that of a single peptide mutation at the 8th site and the 5th site. It was a substitution of Leucine to Valine in viral samples from Hong Kong and a substitution of Leucine to Phenylalanine in samples from France, respectively. These mutations do not have any major role in functioning of the virus and do not impact transmission in any way known yet. It has been hypothesized that these mutations can be used to identify individuals more susceptible to the viral infection as compared to others (Korber et al. 2020). A mutation in 49th site by the substitution of Histidine and Tyrosine was observed in a sample collected from Taiwan. This mutation occurs in the S1 subunit at the N-terminal Domain but is of not much significance other than aiding in identification of geographical area of the sample collected, as this mutation is unique to Netherlands and Taiwan as of yet. Out of 32 single amino acid substitutions, only 2 were found to be in the binding domain of the viral spike glycoprotein. This mutation was also observed in one of the samples acquired from India as well as Malaysia. The mutation involved the substitution of Arginine to Isoleucine and Proline to Lysine, respectively. Both these mutations are suggested to impact the ability of the domain to attach with the hACE2 of the host. Another peculiar mutation noted was the substitution of a chain of 6 amino acids from site 292 to site 297 in a sample acquired from Malaysia. This mutation showed a substitution of A L D P L S to V M I H F W. Due to lack of data, the reason for this substitution is still unknown and requires further study.
To further assess any homology between the amino acid sequences obtained from different countries, an unrooted phylogenetic tree was depending on Maximum Likelihood among all 147 sequences based on their multiple sequence alignment data obtained earlier. The tree is separated into six clades, as visualised in the condensed tree in Fig. 4. Clade 1 represents a unique mutation in two Sequences, one from China (QJA20044) and India (QJF77846). Clade 2 represents sequences from Hong Kong, Clade 3 represents sequences from Taiwan, Clade 4 represents sequences from France unique due a common mutation, Clade 5 represents sequences from the Czech Republic and Clade 6 represents sequences from Thailand. All of the sequences acquired are unique to each other due to the presence single mutations in their amino acid sequences. These single mutations increase the evolutionary distance between other sequences from other countries. An interesting observation was done by finding out the single sequence similarity from a sample obtained from Nepal (QIB84673), Puerto Rico, Germany and Pakistan. This suggested that the virus was transmitted to other human sources via a common carrier. It was also observed that the single sequence from South Africa (QIZ15537) was highly similar to sequences obtained from samples collected from China. Amino acid composition obtained on the basis of the phylogenetic tree indicate that the sequences are similar in nature with little or no differences apart from single amino acid substitutions. Pair wise distance of amino acids was also calculated using the phylogenetic tree. The analysis by the software was processed via Poisson correction model. This analysis studied all 147 sequences. All regions containing gaps and missing data were removed. There were a total of 1223 sites in the concluding dataset. It was observed that 0.82% was the highest noted distance between the sequences as per the values observed at 0.0082 and the lowest was at 0.

Condensed circular Phylogenetic Tree of predominantly related samples from, Puerto Rico, USA, China, Hong Kong and Australia
Discussion
Upon critical analysis of the data acquired from NCBI Virus database, the protein sequences were aligned to identify multiple or single amino acid mutations which were specifically observed in certain countries along with a mutation which was identified at a global level. The substitution of Glycine to Isoleucine at the 614th position was observed in all countries analysed except for Czech Republic and South Africa. According to previous studies, this mutation was mostly predominant in European countries but has also spread across many other different countries around the world. This mutation has been noted to be associated with enhanced transmission of SARS-CoV-2. Many reasons have been speculated for this to happen. One of which being its structure, as the mutation is present on the surface of the spike glycoprotein. This allows it to make interactions with other subunits of the spike glycoproteins via the interaction of Aspartic acid present in S1 of one spike glycoprotein and Threonine on the S2 subunit of the other spike glycoprotein. This interaction might reduce the interaction between S1 and S2 subunits causing the separation of S1 from bound S2 or it may also cause a change in the way the receptor binding domain binds to human ACE2 in the host (Korber et al. 2020). This mutation can also be associated with immunological changes in the host which can lead to increased susceptibility to infection. This is because of the presence of the mutation in the immunological domain of the spike glycoprotein which leads to high B-Cell response as was earlier seen during the SARS-CoV epidemic in 2002 (Lu et al. 2020).
Initial studies conducted by a group of researchers in Europe discovered that patients with this mutation generally were observed to have a higher load of viral components in their body (Cascella 2021). Due to the lack of studies conducted on this mutation not much could be said for the samples containing these mutations. Furthermore, other than the mutation at the 614th site, multiple single amino acid substitutions were recorded, these were generally specific to a certain country and did not occur in any important region that deals with functionality of protein or aids in receptor binding to enzyme.
Two interesting mutations observed were those that occurred in the receptor binding domain of samples from India and Malaysia. These were found to be isolated mutations. In the case of the sequence from Indian sample, the mutation was present at the 408th site with a substitution of Arginine to Isoleucine, while in the sequence from Malaysian sample the mutation was present at the 491st site with a substitution of Proline to Leucine. Both the mentioned sequences were present in the binding membrane of the receptor binding domain. In previous studies conducted by researchers, the presence of Arginine at the 408th site is preserved in SARS-CoV-2, SARS-CoV and Bat-CoV. This region directly impacts the binding of the viral spike glycoprotein with the ACE2 receptor of the human host. The mutation of the 408th Arginine replaced by Isoleucine has been considered to reduce the ACE2 receptor binding ability of SARS-CoV-2 as it disrupts the glycan-hydrogen bond present at the 408th site coding for Arginine (Jia et al. 2020). Mutation found at the 491st site in the Malaysian sample was the Proline substitution to Leucine which also had the same impact on the binding efficacy of receptor binding membrane to ACE2. Due to lack of samples and further data, this study could not be further tested and therefore calls for further studies.
A sudden increase in the infection in human population in Italy and Australia can also be attributed to the lack of alterations in the RBD of the viral protein. Another reason, for a high number of cases in China, USA, Australia, Thailand, Taiwan and Italy can be attributed to the presence of mutation at the 614th site (Figs. 5 and 6) which has been known to increase the ability of receptor binding domain to interact with the human angiotensin converting enzyme 2 in the host organism. At this position, Aspartic acid is replaced by Glycine. Aspartic acid has an average occurrence of about 5% in all proteins, it is acid in nature, normally used in peptide mapping and proteomic analysis. Its specificity also complements those of trypsin, endoproteinase Lys-C and other proteases. Whereas, Glycine is hydrophobic in nature and reported as a virulent factor in SARS-CoV-2. For example, the average occurrence of Asp, Arg and Lys is about 5, 5, and 6% in all proteins, respectively. Therefore, digestion with Asp-N, generally leads to longer and fewer peptides than tryptic cleavage. Another finding in this study was the identification of possible objectives for the production of fitting vaccines and therapeutics, which could potentially aid in the battle against the SARS-CoV-2 virus (Jia et al. 2020).

Figure represent the WT and Mutated sequence of S2 domain of Spike Glycoprotein of SARS-CoV-2. Aspartic acid is replaced by Glycine at 614 position of S2 domain of Spike Glycoprotein of SARS-CoV-2

Structural 3D representation in ribbon style of WT (614:D(Aspartic acid)) and mutated (614:G(Glycine)) spike glycoprotein of SARS-CoV-2
A major aim of this study was to identify the presence of a mutation within spike proteins of the SARS-CoV-2 in infected populations across the countries which in aim to understand why some countries are affected in a higher rate than others.
With the data available on the National Centre for Biotechnology Information (NCBI) Virus database, we were able to identify a few single amino acid mutations unique to certain populations and one global amino acid mutation, which could give a novel strategy to describe the differential infection rate of SARS-CoV-2 across the globe. A large-scale analysis of these mutations are required, with more samples, to confirm and validate the study.
The mutations identified, especially those in the receptor binding domain can be used as potential targets. Moreover, the phylogenetic analysis helped in showing that most samples were predominantly related to samples collected from, Puerto Rico, USA, China, Hong Kong and Australia. This was mainly because of the number of samples obtained from the database as compared to samples from other countries. To broaden the understanding of geographical source of conduction of SARS-CoV-2, samples from throughout the globe must be collected in larger number and deeper studies into spike glycoprotein must take place. This would aid in the prediction of the spreading of the infection as it would be great strategy to prevent the spread of the infection at a larger number. Due to the sudden onset of diseases such as the recent SARS-CoV-2 and earlier diseases like SARS and MERS, a system where disease transmission can be predicted could prove to be useful in the future.
Conclusions
In silico analysis of surface spike glycoprotein sequences have enabled to identify multiple mutations in different SARS-CoV-2-infected populations. Over the past few months, constant efforts from researches across the globe has contributed to the vaccine development and has been effectively distributing to several countries infected with SARS-CoV-2. Unfortunately, the ability of the virus to attain mutations in its genome has opened up for fast and effective solutions against it. The new mutated strain of SARS-CoV-2 identified in Britain is one of the new strains reported to be having novel mutations helping it become more contagious and infectious. The analysis of spike glycoprotein sequences performed by multiple sequence alignment and phylogenetic tree studies helped in understanding the heterogeneity in S2 subunit of spike glycoprotein of SARS-CoV-2 in different populations. A deeper study into the mutational changes taking place in the regulatory proteins of SARS-CoV-2 would help researchers and clinicians develop better therapeutics to combat the virus. Multiple studies done to identify specific epitopes such as E332-370, E627-651, E440-464 and E694-715, along with MHC-I, MHC-II alleles, B-Cell and IFN-inducing epitopes, could be a great knowledge to be targeted and develop novel effective vaccines (Lizbeth et al. 2020; Rahman et al. 2020) Mutational heterogeneity analysis of more samples along with those of the new variant would advance the development of more specific therapeutics and vaccines.
References
Araujo MB, Naimi B (2020) Spread of SARS-CoV-2 coronavirus likely to be constrained by climate. medRxiv. https://doi.org/10.1101/2020.03.12.20034728
Begum F, Mukherjee D, Thagriki D, Das S, Tripathi PP, Banerjee AK, Ray U (2020) Analyses of spike protein from first deposited sequences of SARS-CoV2 from West Bengal, India. bioRxiv. https://doi.org/10.1101/2020.04.28.066985
Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R (2021) Features, evaluation, and treatment of coronavirus (COVID-19). In: StatPearls [Internet]. StatPearls Publishing, Treasure Island (FL)
de Wit E, van Doremalen N, Falzarano D, Munster VJ (2016) SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14(8):523–534. https://doi.org/10.1038/nrmicro.2016.81
Fang L, Karakiulakis G, Roth M (2020) Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med 8(4):e21. https://doi.org/10.1016/S2213-2600(20)30116-8
Forster P, Forster L, Renfrew C, Forster M (2020) Phylogenetic network analysis of SARS-CoV-2 genomes. Proc Natl Acad Sci 117(17):9241–9243. https://doi.org/10.1073/pnas.2004999117
Heald-Sargent T, Gallagher T (2012) Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 4(4):557–580. https://doi.org/10.3390/v4040557
Hillen HS, Kokic G, Farnung L, Dienemann C, Tegunov D, Cramer P (2020) Structure of replicating SARS-CoV-2 polymerase. Nature. https://doi.org/10.1038/s41586-020-2368-8
Jia Y, Shen G, Zhang Y, Huang K-S, Ho H-Y, Hor W-S, Yang CH, Li C, Wang W-L (2020) Analysis of the mutation dynamics of SARS-CoV-2 reveals the spread history and emergence of RBD mutant with lower ACE2 binding affinity. bioRxiv. https://doi.org/10.1101/2020.04.09.034942
Korber B, Fischer WM, Gnanakaran S, Yoon H et al (2020) Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 Virus. Cell. 182(4):812–827. https://doi.org/10.1016/j.cell.2020.06.043
Lizbeth R-SG, Jazmín G-M, José C-B, Marlet M-A (2020) Immunoinformatics study to search epitopes of spike glycoprotein from SARS-CoV-2 as potential vaccine. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1780944
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu WJ, Wang D, Xu W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, Tan W (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395(10224):565–574. https://doi.org/10.1016/S0140-6736(20)30251-8
Ortega JT, Serrano ML, Pujol FH, Rangel HR (2020) Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: an in silico analysis. EXCLI J 19:410–417. https://doi.org/10.17179/excli2020-1167
Othman H, Bouslama Z, Brandenburg J-T, da Rocha J, Hamdi Y, Ghedira K, Abid NS, Hazelhurst S (2020) In silico study of the spike protein from SARS-CoV-2 interaction with ACE2: similarity with SARS-CoV, hot-spot analysis and effect of the receptor polymorphism. bioRxiv. https://doi.org/10.1101/2020.03.04.976027
Ou X, Liu Y, Lei X, Li P, Mi D, Ren L, Guo L, Guo R, Chen T, Hu J, Xiang Z, Mu Z, Chen X, Hu K, Jin Q, Wang J, Qian Z (2020) Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 11(1):1620. https://doi.org/10.1038/s41467-020-15562-9
Periwal N, Sarma S, Arora P, Sood V (2020) In-silico analysis of SARS-CoV-2 genomes: insights from SARS encoded non-coding RNAs. bioRxiv. https://doi.org/10.1101/2020.03.31.018499
Rahman N, Ali F, Basharat Z, Shehroz M, Khan MK, Jeandet P, Nepovimova E, Kuca K, Khan H (2020) Vaccine design from the ensemble of surface glycoprotein epitopes of SARS-CoV-2: an immunoinformatics approach. Vaccines 8(3):423
Sardar R, Satish D, Birla S, Gupta D (2020) Comparative analyses of SAR-CoV2 genomes from different geographical locations and other coronavirus family genomes reveals unique features potentially consequential to host-virus interaction and pathogenesis. bioRxiv. https://doi.org/10.1101/2020.03.21.001586
Shang J, Wan Y, Liu C, Yount B, Gully K, Yang Y, Auerbach A, Peng G, Baric R, Li F (2020) Structure of mouse coronavirus spike protein complexed with receptor reveals mechanism for viral entry. PLoS Pathog 16(3):e1008392. https://doi.org/10.1371/journal.ppat.1008392
Sun X, Wang T, Cai D, Hu Z, Chen J, Liao H, Zhi L, Wei H, Zhang Z, Qiu Y, Wang J, Wang A (2020) Cytokine storm intervention in the early stages of COVID-19 pneumonia. Cytokine Growth Factor Rev 53:38–42. https://doi.org/10.1016/j.cytogfr.2020.04.002
Tarik Jasarevic CL, Chaib F (2020) Statement on the second meeting of the international health regulations (2005) emergency committee regarding the outbreak of novel coronavirus (2019-nCoV), 30 January 2020 Statement Geneva, Switzerland
Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181(2):281-292.e286. https://doi.org/10.1016/j.cell.2020.02.058
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, Abiona O, Graham BS, McLellan JS (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–1263. https://doi.org/10.1126/science.abb2507
Xia S, Liu M, Wang C, Xu W, Lan Q, Feng S, Qi F, Bao L, Du L, Liu S, Qin C, Sun F, Shi Z, Zhu Y, Jiang S, Lu L (2020) Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res 30(4):343–355. https://doi.org/10.1038/s41422-020-0305-x
Zhang X, Powell K, Li L (2020) Breast cancer stem cells: biomarkers, identification and isolation methods, regulating mechanisms, cellular origin, and beyond. Cancers 12(12):3765
Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao K, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan FX, Wang Y-Y, Xiao G-F, Shi Z-L (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273. https://doi.org/10.1038/s41586-020-2012-7
Acknowledgements
We thank all our lab members for reading the paper and provide their valuable suggestions.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
AM& DK: conceptualised the idea, AM & DK: performed the experiments and analysed the data, AM, NKJ, SKJ, BR and DK: contributed in manuscript preparation, manuscript review and revision.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
All authors have given their consent to publish this article.
Rights and permissions
About this article

Cite this article
Mathur, A., Raj, S., Jha, N.K. et al. Mutational heterogeneity in spike glycoproteins of severe acute respiratory syndrome coronavirus 2. 3 Biotech 11, 236 (2021). https://doi.org/10.1007/s13205-021-02791-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-021-02791-y