
COVID-19 and Delayed Cerebral Ischemia—More in Common Than First Meets the Eye

 , , , ,
, , , ,
Abstract
:1. Introduction
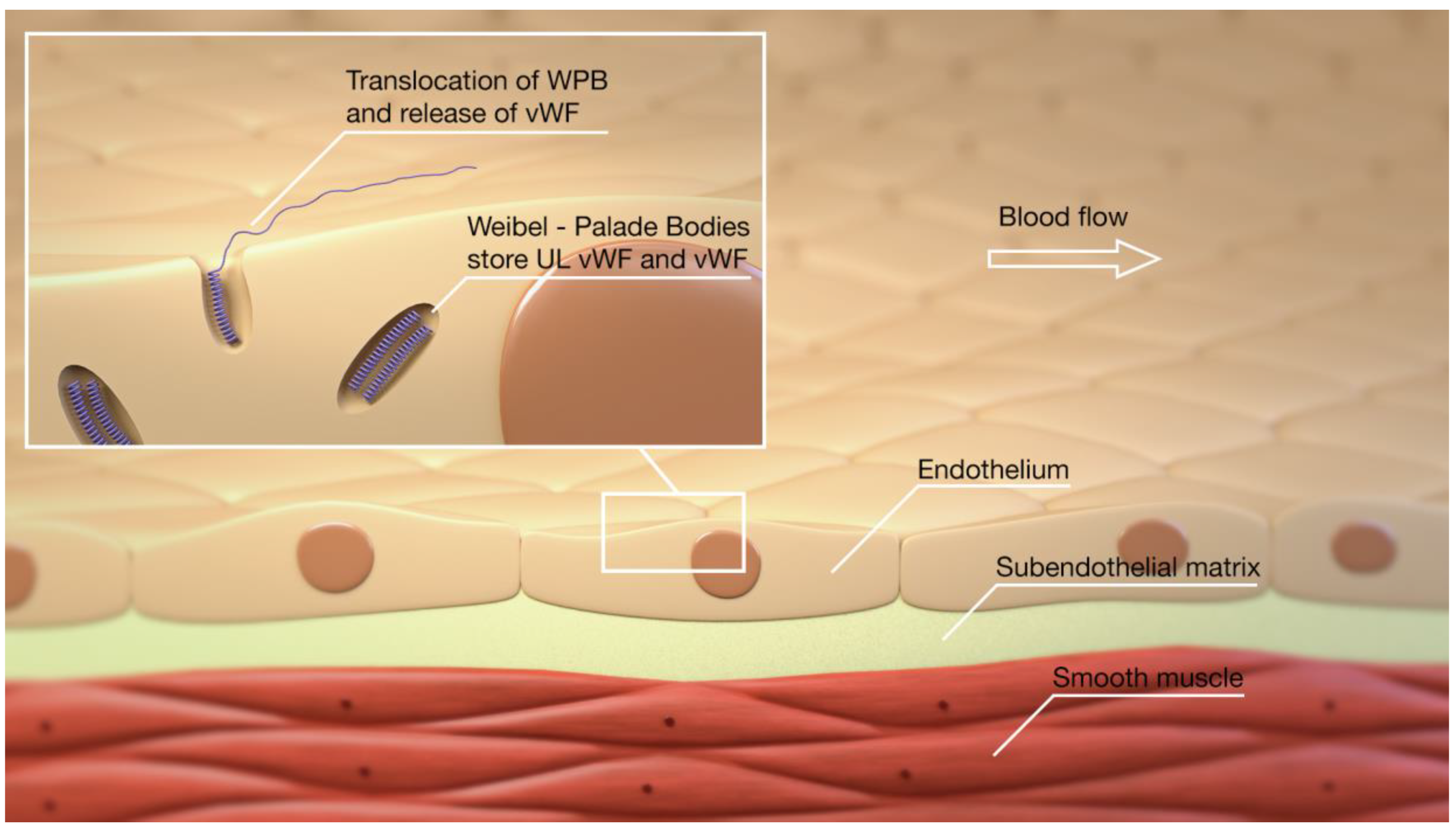
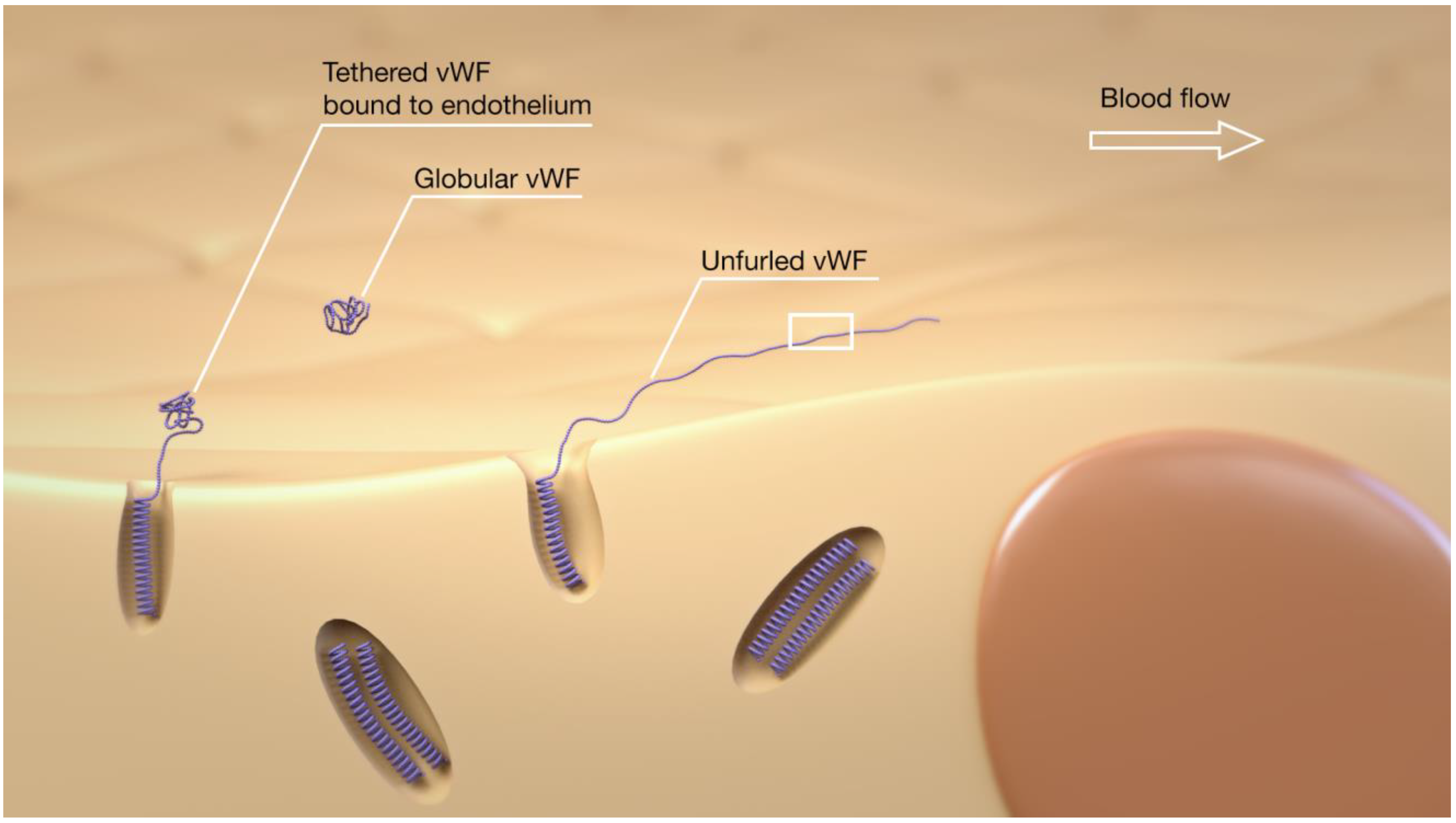
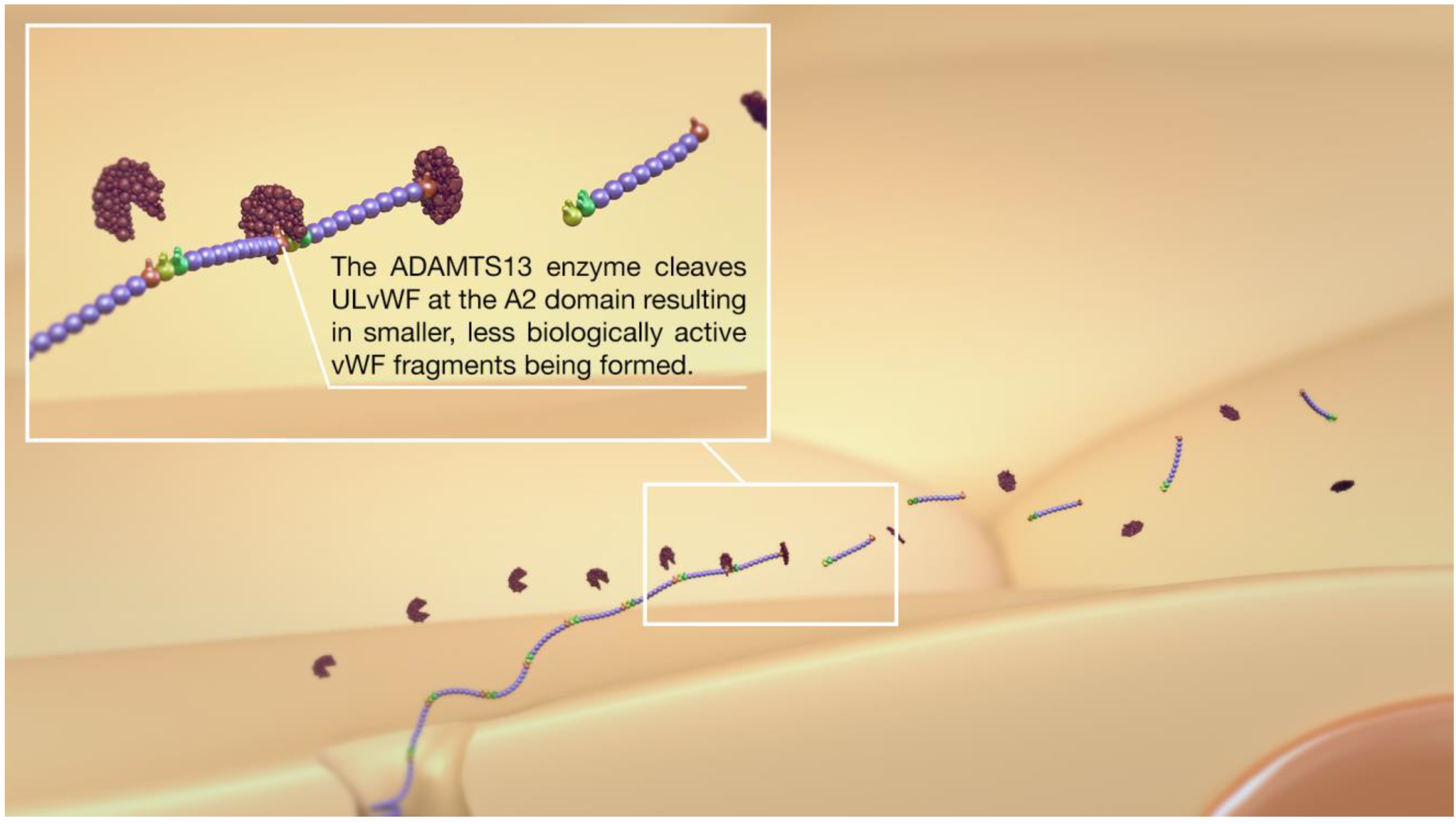
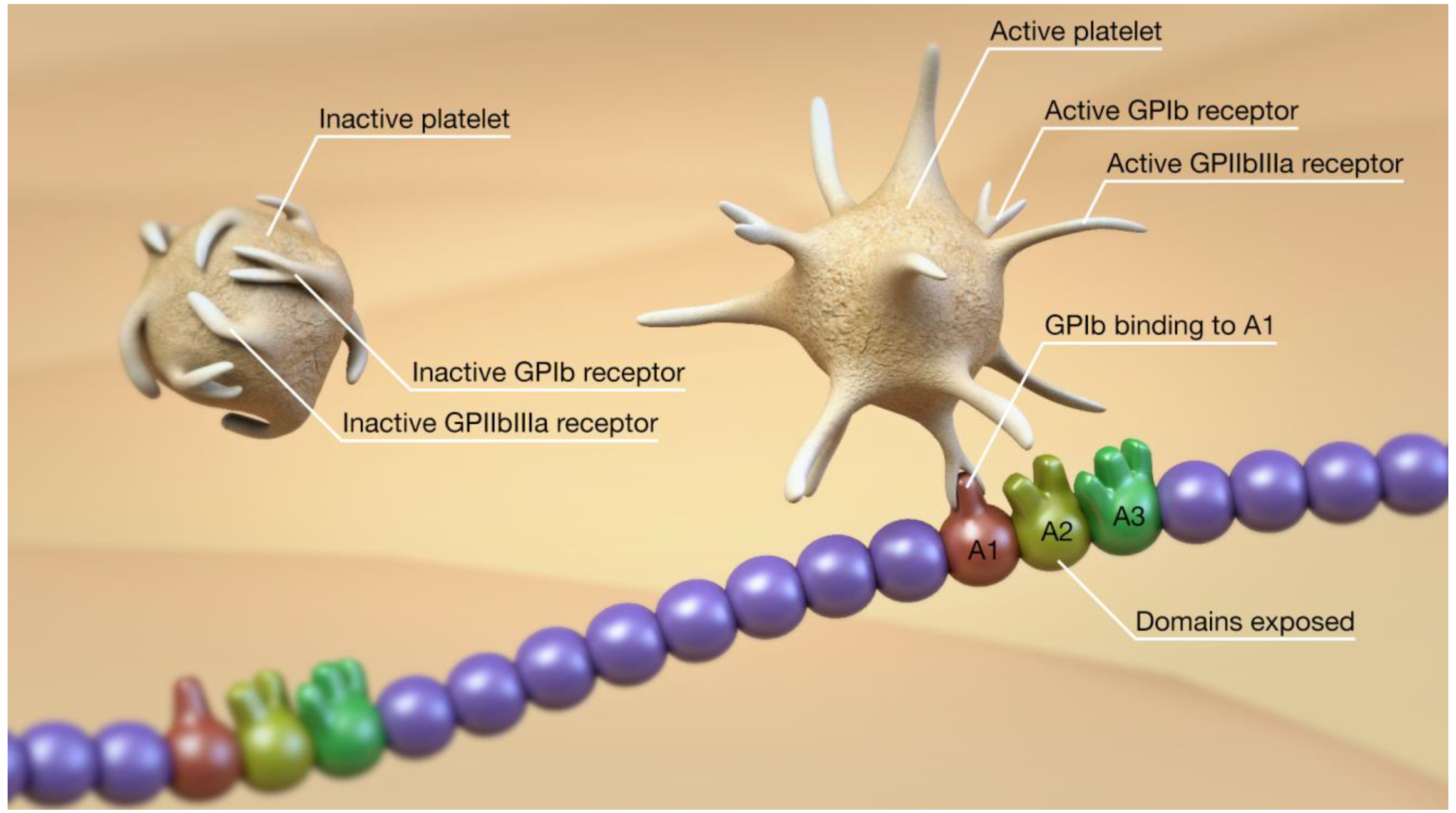
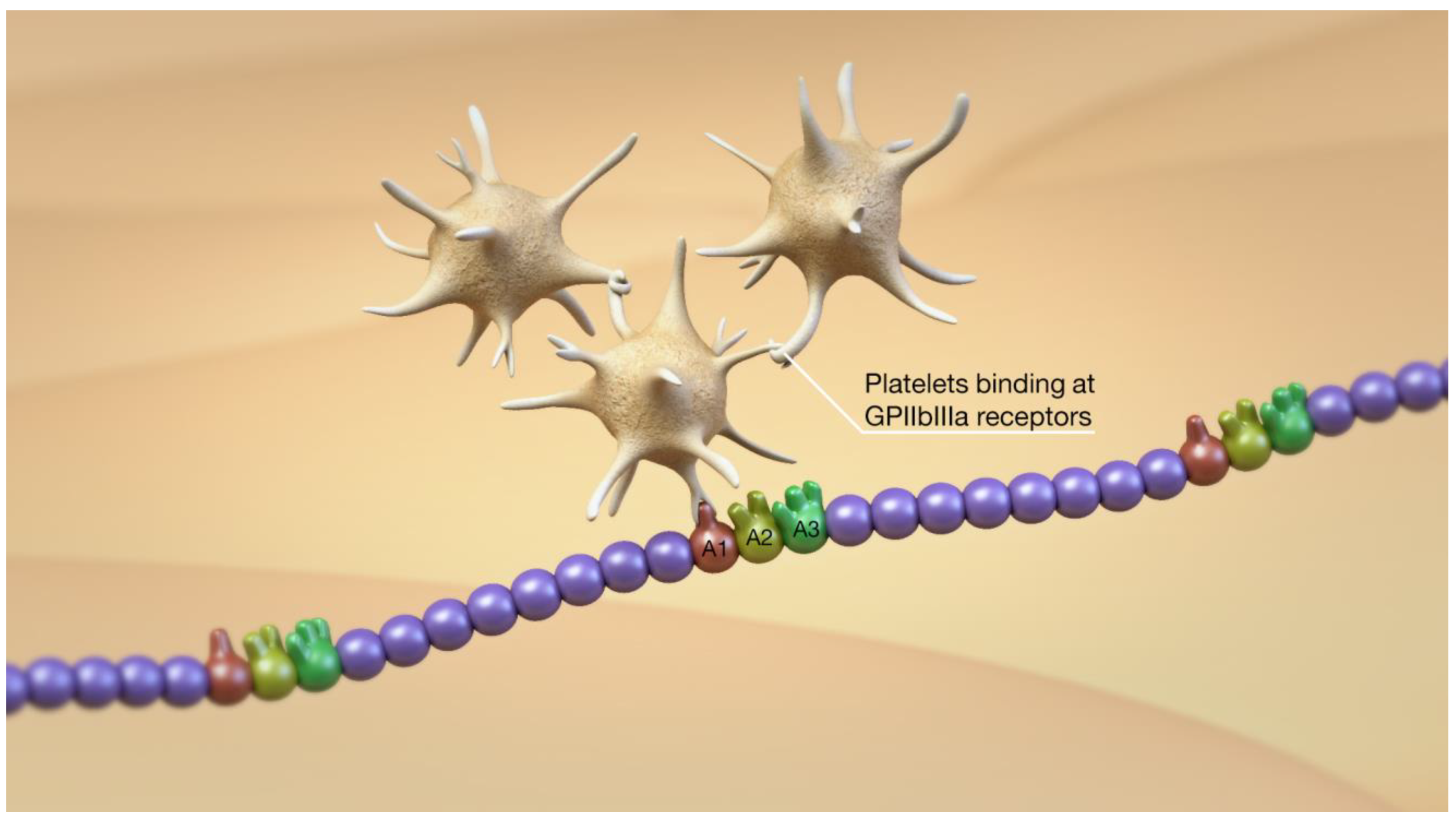
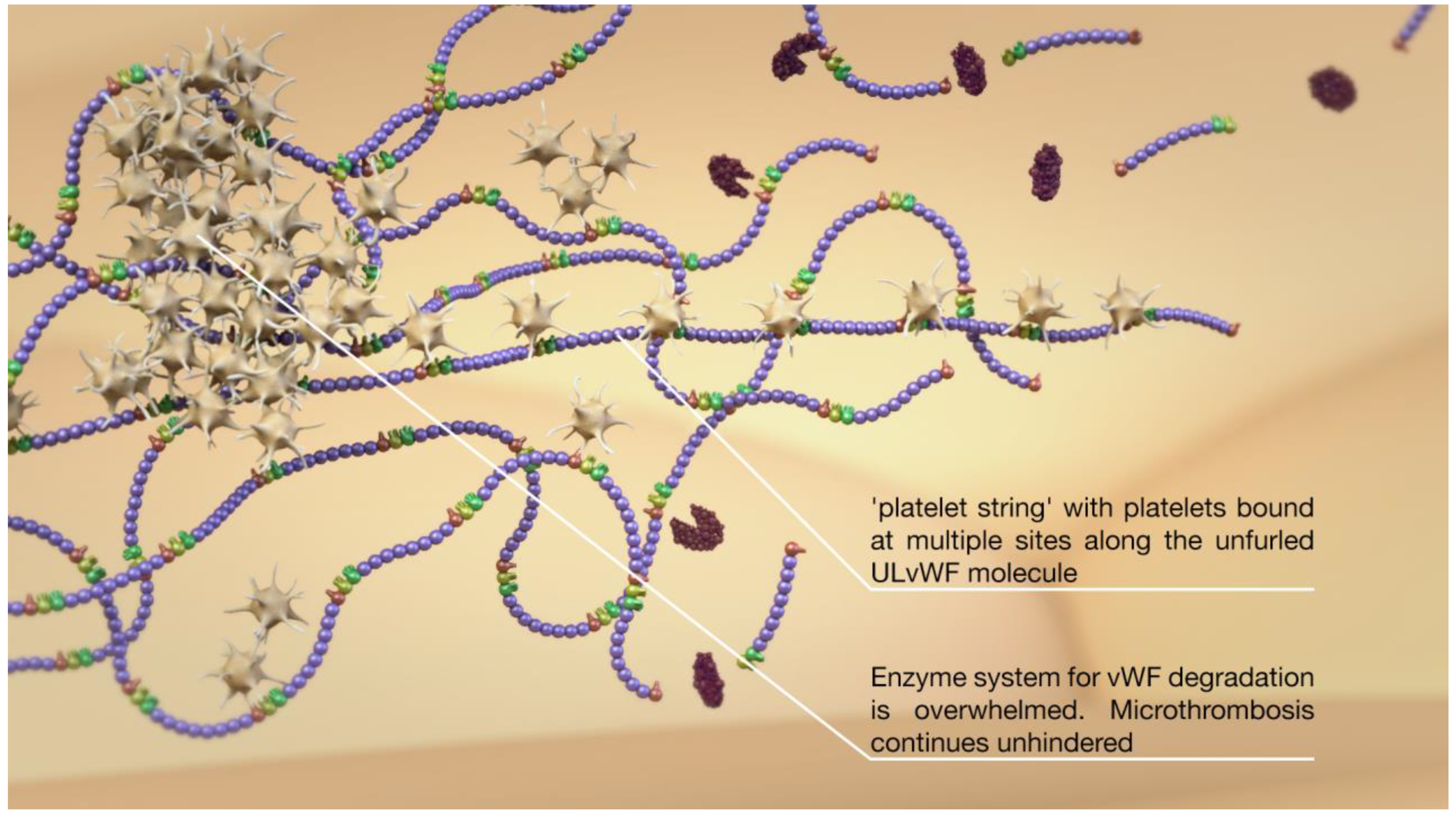
2. Overview of the vWF, ADAMTS13, Platelets, and Thromboinflammation
3. Evidence of Abnormalities in SAH and DCI
4. Evidence of Abnormalities in COVID-19
5. Potential Treatment Strategies
5.1. GPIb Receptor Antagonists
5.2. N-Acetyl Cysteine
5.3. Recombinant ADAMTS13
5.4. Plasma Exchange
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yamamoto, K.; de Waard, V.; Fearns, C.; Loskutoff, D.J. Tissue Distribution and Regulation of Murine von Willebrand Factor Gene Expression in Vivo. Blood 1998, 92, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Crawley, J.T.B.; de Groot, R.; Xiang, Y.; Luken, B.M.; Lane, D.A. Unraveling the Scissile Bond: How ADAMTS13 Recognizes and Cleaves von Willebrand Factor. Blood 2011, 118, 3212–3221. [Google Scholar] [CrossRef] [Green Version]
- Moake, J.L.; Chow, T.W. Increased von Willebrand Factor (VWf) Binding to Platelets Associated with Impaired VWf Breakdown in Thrombotic Thrombocytopenic Purpura. J. Clin. Apher. 1998, 13, 126–132. [Google Scholar] [CrossRef]
- Kumar, R.A.; Moake, J.L.; Nolasco, L.; Bergeron, A.L.; Sun, C.; Dong, J.; McIntire, L.V. Enhanced Platelet Adhesion and Aggregation by Endothelial Cell-Derived Unusually Large Multimers of von Willebrand Factor. Biorheology 2006, 43, 681–691. [Google Scholar]
- Sporn, L.A.; Marder, V.J.; Wagner, D.D. Inducible Secretion of Large, Biologically Potent von Willebrand Factor Multimers. Cell 1986, 46, 185–190. [Google Scholar] [CrossRef]
- Arya, M.; Anvari, B.; Romo, G.M.; Cruz, M.A.; Dong, J.-F.; McIntire, L.V.; Moake, J.L.; López, J.A. Ultralarge Multimers of von Willebrand Factor Form Spontaneous High-Strength Bonds with the Platelet Glycoprotein Ib-IX Complex: Studies Using Optical Tweezers. Blood 2002, 99, 3971–3977. [Google Scholar] [CrossRef]
- De Ceunynck, K.; De Meyer, S.F.; Vanhoorelbeke, K. Unwinding the von Willebrand Factor Strings Puzzle. Blood 2013, 121, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.J.; Lillicrap, D. A Sticky Proposition: The Endothelial Glycocalyx and von Willebrand Factor. J. Thromb. Haemost. JTH 2020, 18, 781–785. [Google Scholar] [CrossRef]
- André, P.; Denis, C.V.; Ware, J.; Saffaripour, S.; Hynes, R.O.; Ruggeri, Z.M.; Wagner, D.D. Platelets Adhere to and Translocate on von Willebrand Factor Presented by Endothelium in Stimulated Veins. Blood 2000, 96, 3322–3328. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Moake, J.L.; Nolasco, L.; Bernardo, A.; Arceneaux, W.; Shrimpton, C.N.; Schade, A.J.; McIntire, L.V.; Fujikawa, K.; López, J.A. ADAMTS-13 Rapidly Cleaves Newly Secreted Ultralarge von Willebrand Factor Multimers on the Endothelial Surface under Flowing Conditions. Blood 2002, 100, 4033–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilla, A.; Moake, J.L.; Bernardo, A.; Ball, C.; Wang, Y.; Arya, M.; Nolasco, L.; Turner, N.; Berndt, M.C.; Anvari, B.; et al. P-Selectin Anchors Newly Released Ultralarge von Willebrand Factor Multimers to the Endothelial Cell Surface. Blood 2004, 103, 2150–2156. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Roth, R.; Heuser, J.E.; Sadler, J.E. Integrin Alpha(v)Beta(3) on Human Endothelial Cells Binds von Willebrand Factor Strings under Fluid Shear Stress. Blood 2009, 113, 1589–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, L.B.; Conner, E.R.; Matthay, M.A. Von Willebrand Factor Antigen Is an Independent Marker of Poor Outcome in Patients with Early Acute Lung Injury. Crit. Care Med. 2001, 29, 2325–2331. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Eisner, M.D.; Thompson, B.T.; Parsons, P.E.; Matthay, M.A. Significance of von Willebrand Factor in Septic and Nonseptic Patients with Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2004, 170, 766–772. [Google Scholar] [CrossRef]
- Hasegawa, T.; Watanabe, H.; Ishii, S. Studies of Intravascular Components in Cerebral Vasospasm Following Subarachnoid Hemorrhage. Neurosurg. Rev. 1980, 3, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T. Studies of intravascular components with cerebral vasospasm following subarachnoid hemorrhages with particular regard to platelet and blood coagulation functions (author’s transl). Neurol. Med. Chir. 1980, 20, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, S.; Suzuki, M.; Iwabuchi, T.; Kamata, Y. Role of Multiple Cerebral Microthrombosis in Symptomatic Cerebral Vasospasm: With a Case Report. Neurosurgery 1983, 13, 199–203. [Google Scholar] [CrossRef]
- Suzuki, S.; Kimura, M.; Souma, M.; Ohkima, H.; Shimizu, T.; Iwabuchi, T. Cerebral Microthrombosis in Symptomatic Cerebral Vasospasm—A Quantitative Histological Study in Autopsy Cases. Neurol. Med. Chir. 1990, 30, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Stein, S.C.; Browne, K.D.; Chen, X.-H.; Smith, D.H.; Graham, D.I. Thromboembolism and Delayed Cerebral Ischemia after Subarachnoid Hemorrhage: An Autopsy Study. Neurosurgery 2006, 59, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Sabri, M.; Ai, J.; Lakovic, K.; D’abbondanza, J.; Ilodigwe, D.; Macdonald, R.L. Mechanisms of Microthrombi Formation after Experimental Subarachnoid Hemorrhage. Neuroscience 2012, 224, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, G.; Zhu, W.-W.; Bian, J.-Y.; Shen, X.-O.; Zhou, D. Influence of Simvastatin on Microthrombosis in the Brain after Subarachnoid Hemorrhage in Rats: A Preliminary Study. Ann. Clin. Lab. Sci. 2010, 40, 32–42. [Google Scholar]
- Sehba, F.A.; Bederson, J.B. Mechanisms of Acute Brain Injury after Subarachnoid Hemorrhage. Neurol. Res. 2006, 28, 381–398. [Google Scholar] [CrossRef] [PubMed]
- Sehba, F.A.; Mostafa, G.; Friedrich, V.; Bederson, J.B. Acute Microvascular Platelet Aggregation after Subarachnoid Hemorrhage. J. Neurosurg. 2005, 102, 1094–1100. [Google Scholar] [CrossRef]
- Friedrich, V.; Flores, R.; Muller, A.; Sehba, F.A. Luminal Platelet Aggregates in Functional Deficits in Parenchymal Vessels after Subarachnoid Hemorrhage. Brain Res. 2010, 1354, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muroi, C.; Fujioka, M.; Mishima, K.; Irie, K.; Fujimura, Y.; Nakano, T.; Fandino, J.; Keller, E.; Iwasaki, K.; Fujiwara, M. Effect of ADAMTS-13 on Cerebrovascular Microthrombosis and Neuronal Injury after Experimental Subarachnoid Hemorrhage. J. Thromb. Haemost. JTH 2014, 12, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergouwen, M.D.I.; Knaup, V.L.; Roelofs, J.J.T.H.; de Boer, O.J.; Meijers, J.C.M. Effect of Recombinant ADAMTS-13 on Microthrombosis and Brain Injury after Experimental Subarachnoid Hemorrhage. J. Thromb. Haemost. JTH 2014, 12, 943–947. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Wang, Y.; Ai, J.; Brathwaite, S.; Ni, H.; Macdonald, R.L.; Hol, E.M.; Meijers, J.C.M.; Vergouwen, M.D.I. Role of von Willebrand Factor and ADAMTS-13 in Early Brain Injury after Experimental Subarachnoid Hemorrhage. J. Thromb. Haemost. JTH 2018, 16, 1413–1422. [Google Scholar] [CrossRef] [Green Version]
- Kumar, M.; Cao, W.; McDaniel, J.K.; Pham, H.P.; Raju, D.; Nawalinski, K.; Frangos, S.; Kung, D.; Zager, E.; Kasner, S.E.; et al. Plasma ADAMTS13 Activity and von Willebrand Factor Antigen and Activity in Patients with Subarachnoid Haemorrhage. Thromb. Haemost. 2017, 117, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Vergouwen, M.D.I.; Bakhtiari, K.; van Geloven, N.; Vermeulen, M.; Roos, Y.B.W.E.M.; Meijers, J.C.M. Reduced ADAMTS13 Activity in Delayed Cerebral Ischemia after Aneurysmal Subarachnoid Hemorrhage. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 1734–1741. [Google Scholar] [CrossRef]
- Bell, J.D.; Rhind, S.G.; Di Battista, A.P.; Macdonald, R.L.; Baker, A.J. Biomarkers of Glycocalyx Injury Are Associated with Delayed Cerebral Ischemia Following Aneurysmal Subarachnoid Hemorrhage: A Case Series Supporting a New Hypothesis. Neurocrit. Care 2017, 26, 339–347. [Google Scholar] [CrossRef]
- Jung, F.; Krüger-Genge, A.; Franke, R.P.; Hufert, F.; Küpper, J.-H. COVID-19 and the Endothelium. Clin. Hemorheol. Microcirc. 2020, 75, 7–11. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-Associated Coagulopathy: Evidence from a Single-Centre, Cross-Sectional Study. Lancet Haematol. 2020. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Escher, R.; Breakey, N.; Lämmle, B. Severe COVID-19 Infection Associated with Endothelial Activation. Thromb. Res. 2020, 190, 62. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial Cell Infection and Endotheliitis in COVID-19. Lancet 2020. [Google Scholar] [CrossRef]
- Ladikou, E.E.; Sivaloganathan, H.; Milne, K.M.; Arter, W.E.; Ramasamy, R.; Saad, R.; Stoneham, S.M.; Philips, B.; Eziefula, A.C.; Chevassut, T. Von Willebrand Factor (VWF): Marker of Endothelial Damage and Thrombotic Risk in COVID-19? Clin. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.; Wong, A.; Yusuff, T.; Rao, D.; Sidhu, P. Contrast-Enhanced Ultrasound (CEUS) of the Lung Reveals Multiple Areas of Microthrombi in a COVID-19 Patient. Intensive Care Med. 2020. [Google Scholar] [CrossRef]
- Jung, E.M.; Stroszczynski, C.; Jung, F. Contrast Enhanced Ultrasonography (CEUS) to Detect Abdominal Microcirculatory Disorders in Severe Cases of COVID-19 Infection: First Experience. Clin. Hemorheol. Microcirc. 2020, 74, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Brown, J.Q.; Heide, R.S.V. Pulmonary and Cardiac Pathology in Covid-19: The First Autopsy Series from New Orleans. medRxiv 2020. [Google Scholar] [CrossRef]
- Ciceri, F.; Beretta, L.; Scandroglio, A.M.; Colombo, S.; Landoni, G.; Ruggeri, A.; Peccatori, J.; D’Angelo, A.; De Cobelli, F.; Rovere-Querini, P.; et al. Microvascular COVID-19 Lung Vessels Obstructive Thromboinflammatory Syndrome (MicroCLOTS): An Atypical Acute Respiratory Distress Syndrome Working Hypothesis. Crit. Care Resusc. J. Australas. Acad. Crit. Care Med. 2020, 22, 95–97. [Google Scholar]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary Post-Mortem Findings in a Series of COVID-19 Cases from Northern Italy: A Two-Centre Descriptive Study. Lancet Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Kirschenbaum, D.; Imbach, L.L.; Rushing, E.J.; Frauenknecht, K.B.M.; Gascho, D.; Ineichen, B.V.; Keller, E.; Kohler, S.; Lichtblau, M.; Reimann, R.R.; et al. Intracerebral Endotheliitis and Microbleeds Are Neuropathological Features of COVID-19. Neuropathol. Appl. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Keller, E.; Brandi, G.; Winklhofer, S.; Imbach, L.L.; Kirschenbaum, D.; Frontzek, K.; Steiger, P.; Dietler, S.; Haeberlin, M.; Willms, J.; et al. Large and Small Cerebral Vessel Involvement in Severe COVID-19: Detailed Clinical Workup of a Case Series. Stroke 2020, 51, 3719–3722. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.P.; Le Quesne, J.; Officer-Jones, L.; Teodòsio, A.; Thaventhiran, J.; Ficken, C.; Goddard, M.; Smith, C.; Menon, D.; Allinson, K.S.J. Neuropathological Findings in Two Patients with Fatal COVID-19. Neuropathol. Appl. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Hernández-Fernández, F.; Sandoval Valencia, H.; Barbella-Aponte, R.A.; Collado-Jiménez, R.; Ayo-Martín, Ó.; Barrena, C.; Molina-Nuevo, J.D.; García-García, J.; Lozano-Setién, E.; Alcahut-Rodriguez, C.; et al. Cerebrovascular Disease in Patients with COVID-19: Neuroimaging, Histological and Clinical Description. Brain 2020, 143, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High Risk of Thrombosis in Patients with Severe SARS-CoV-2 Infection: A Multicenter Prospective Cohort Study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S.; et al. Pulmonary Embolism in Patients With COVID-19: Awareness of an Increased Prevalence. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 Patients in Intensive Care Unit. A Report of Thromboelastography Findings and Other Parameters of Hemostasis. J. Thromb. Haemost. JTH 2020. [Google Scholar] [CrossRef]
- Rauch, A.; Labreuche, J.; Lassalle, F.; Goutay, J.; Caplan, M.; Charbonnier, L.; Rohn, A.; Jeanpierre, E.; Dupont, A.; Duhamel, A.; et al. Coagulation Biomarkers Are Independent Predictors of Increased Oxygen Requirements in COVID-19. J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- South, K.; Roberts, L.; Morris, L.V.; Mann, E.; Menon, M.; Knight, S.; Konkel, J.E.; Ustianowsk, A.; Bakerly, N.D.; Dark, P.M.; et al. Severity-Stratified and Longitudinal Analysis of VWF/ADAMTS13 Imbalance, Altered Fibrin Crosslinking and Inhibition of Fibrinolysis as Contributors to COVID-19 Coagulopathy. medRxiv 2020. [Google Scholar] [CrossRef]
- Philippe, A.; Chocron, R.; Gendron, N.; Bory, O.; Beauvais, A.; Peron, N.; Khider, L.; Guerin, C.L.; Goudot, G.; Levasseur, F.; et al. Circulating Von Willebrand Factor and High Molecular Weight Multimers as Markers of Endothelial Injury Predict COVID-19 in-Hospital Mortality. Angiogenesis 2021, 1–13. [Google Scholar] [CrossRef]
- Huisman, A.; Beun, R.; Sikma, M.; Westerink, J.; Kusadasi, N. Involvement of ADAMTS13 and von Willebrand Factor in Thromboembolic Events in Patients Infected with SARS-CoV-2. Int. J. Lab. Hematol. 2020, 42, e211–e212. [Google Scholar] [CrossRef]
- Mancini, I.; Baronciani, L.; Artoni, A.; Colpani, P.; Biganzoli, M.; Cozzi, G.; Novembrino, C.; Boscolo Anzoletti, M.; De Zan, V.; Pagliari, M.T.; et al. The ADAMTS13-von Willebrand Factor Axis in COVID-19 Patients. J. Thromb. Haemost. JTH 2021, 19, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Doevelaar, A.A.N.; Bachmann, M.; Hölzer, B.; Seibert, F.S.; Rohn, B.J.; Bauer, F.; Witzke, O.; Dittmer, U.; Bachmann, M.; Yilmaz, S.; et al. Von Willebrand Factor Multimer Formation Contributes to Immunothrombosis in Coronavirus Disease 2019. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Knöbl, P.; Cataland, S.; De Beuf, K.; Callewaert, F.; De Winter, H.; Zeldin, R.K. Caplacizumab Reduces the Frequency of Major Thromboembolic Events, Exacerbations and Death in Patients with Acquired Thrombotic Thrombocytopenic Purpura. J. Thromb. Haemost. JTH 2017, 15, 1448–1452. [Google Scholar] [CrossRef] [PubMed]
- Scully, M.; Cataland, S.R.; Peyvandi, F.; Coppo, P.; Knöbl, P.; Kremer Hovinga, J.A.; Metjian, A.; de la Rubia, J.; Pavenski, K.; Callewaert, F.; et al. Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2019, 380, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, P.; Jensen, M.; Collins, G.; Spooner, O.; Makalanda, L.; Hart, D.; Jaffer, O. Letter in Response to: Coagulation Markers Are Independent Predictors of Increased Oxygen Requirements and Thrombosis in COVID-19. J. Thromb. Haemost. 2020, 18, 3382–3384. [Google Scholar] [CrossRef]
- De Lizarrondo, S.M.; Gakuba, C.; Herbig, B.A.; Repessé, Y.; Ali, C.; Denis, C.V.; Lenting, P.J.; Touzé, E.; Diamond, S.L.; Vivien, D.; et al. Potent Thrombolytic Effect of N-Acetylcysteine on Arterial Thrombi. Circulation 2017, 136, 646–660. [Google Scholar] [CrossRef]
- Derhaschnig, U.; Pachinger, C.; Jilma, B. Variable Inhibition of High-Shear-Induced Platelet Plug Formation by Eptifibatide and Tirofiban under Conditions of Platelet Activation and High von Willebrand Release: A Randomized, Placebo-Controlled, Clinical Trial. Am. Heart J. 2004, 147, E17. [Google Scholar] [CrossRef]
- Van den Bergh, W.M.; MASH Study Group; Algra, A.; Dorhout Mees, S.M.; van Kooten, F.; Dirven, C.M.F.; van Gijn, J.; Vermeulen, M.; Rinkel, G.J.E. Randomized Controlled Trial of Acetylsalicylic Acid in Aneurysmal Subarachnoid Hemorrhage: The MASH Study. Stroke 2006, 37, 2326–2330. [Google Scholar] [CrossRef] [Green Version]
- Nagahama, Y.; Allan, L.; Nakagawa, D.; Zanaty, M.; Starke, R.M.; Chalouhi, N.; Jabbour, P.; Brown, R.D.; Derdeyn, C.P.; Leira, E.C.; et al. Dual Antiplatelet Therapy in Aneurysmal Subarachnoid Hemorrhage: Association with Reduced Risk of Clinical Vasospasm and Delayed Cerebral Ischemia. J. Neurosurg. 2018, 129, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COVID-19 | SAH | |
|---|---|---|
| Raised vWF levels | Y | Y |
| Raised ULvWF levels | Y | Not investigated |
| Abnormal ADAMTS13 level | Y | Y |
| Evidence of microthrombosis | Y | Y |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhogal, P.; Makalanda, L.; Hassan, A.E.; Fiorella, D.; Andersson, T.; Ahmad, M.; Bäzner, H.; Jaffer, O.; Henkes, H. COVID-19 and Delayed Cerebral Ischemia—More in Common Than First Meets the Eye. J. Clin. Med. 2021, 10, 2646. https://doi.org/10.3390/jcm10122646
Bhogal P, Makalanda L, Hassan AE, Fiorella D, Andersson T, Ahmad M, Bäzner H, Jaffer O, Henkes H. COVID-19 and Delayed Cerebral Ischemia—More in Common Than First Meets the Eye. Journal of Clinical Medicine. 2021; 10(12):2646. https://doi.org/10.3390/jcm10122646
Chicago/Turabian StyleBhogal, Pervinder, Levansri Makalanda, Ameer E. Hassan, Dave Fiorella, Tommy Andersson, Muhammad Ahmad, Hansjörg Bäzner, Ounali Jaffer, and Hans Henkes. 2021. "COVID-19 and Delayed Cerebral Ischemia—More in Common Than First Meets the Eye" Journal of Clinical Medicine 10, no. 12: 2646. https://doi.org/10.3390/jcm10122646