
Predicting Potential SARS-COV-2 Drugs—In Depth Drug Database Screening Using Deep Neural Network Framework SSnet, Classical Virtual Screening and Docking


Abstract
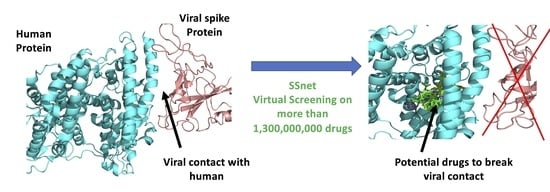
:
{kind=link}
{kind=link}
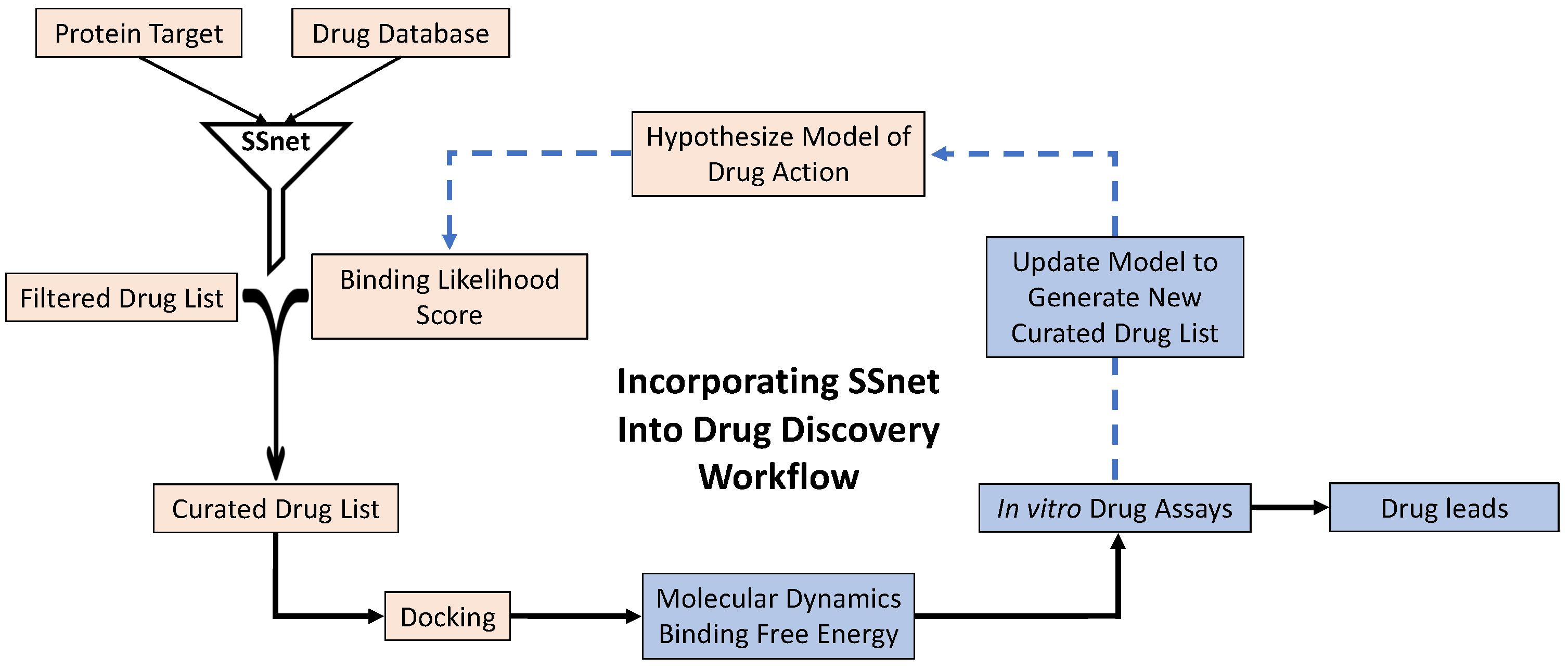{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
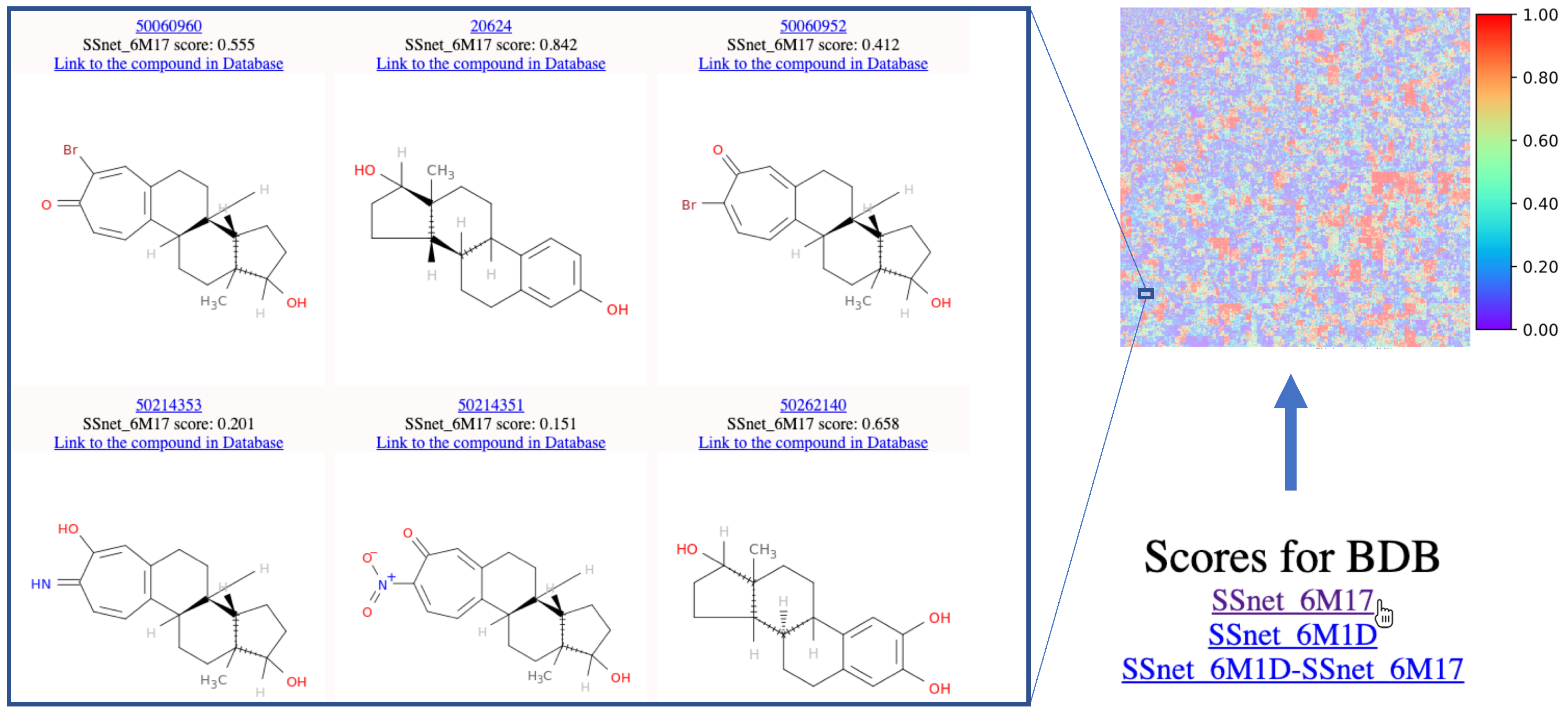{kind=link}
{kind=link}
1. Introduction
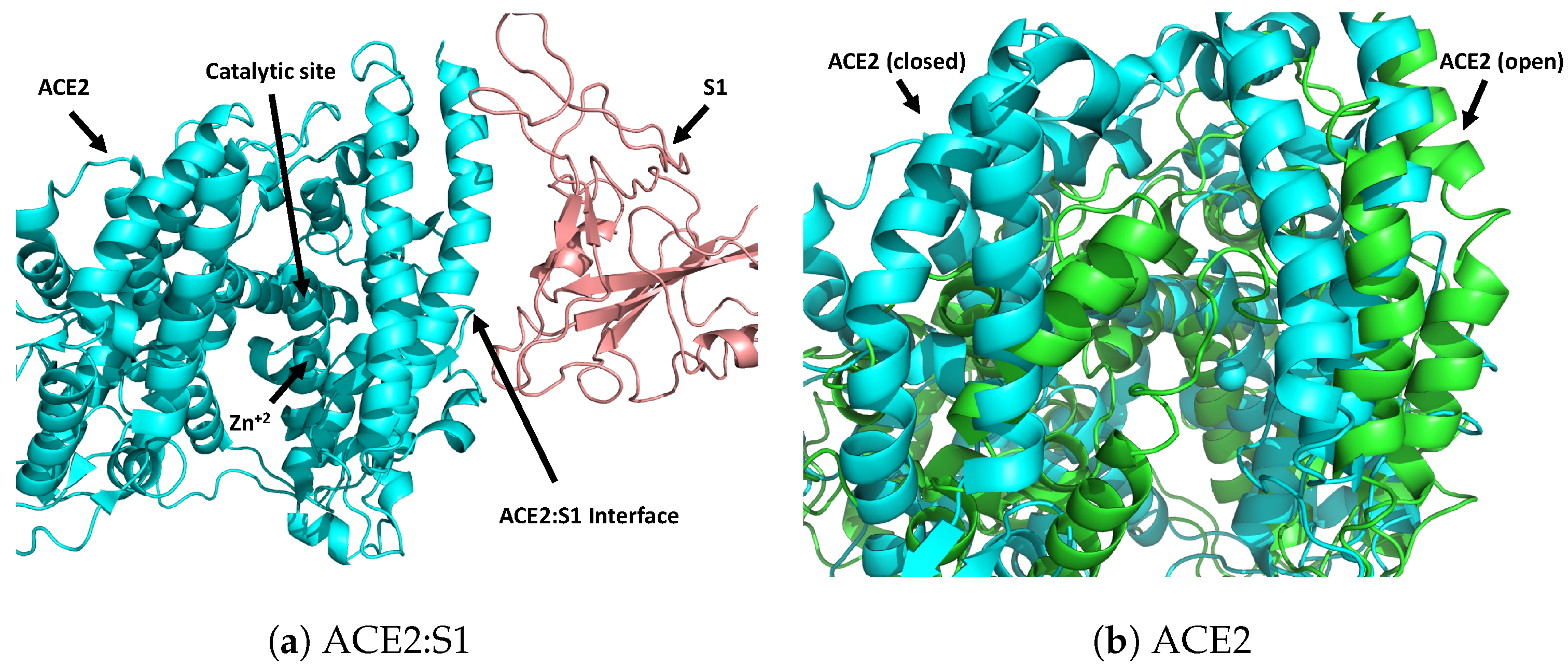
2. Materials and Methods
2.1. Dataset
2.2. SSnet
2.3. Ligand Preparation for Docking
2.4. Virtual Screening and Docking
2.5. Chemical Sorting
2.6. Data Visualization
3. Results
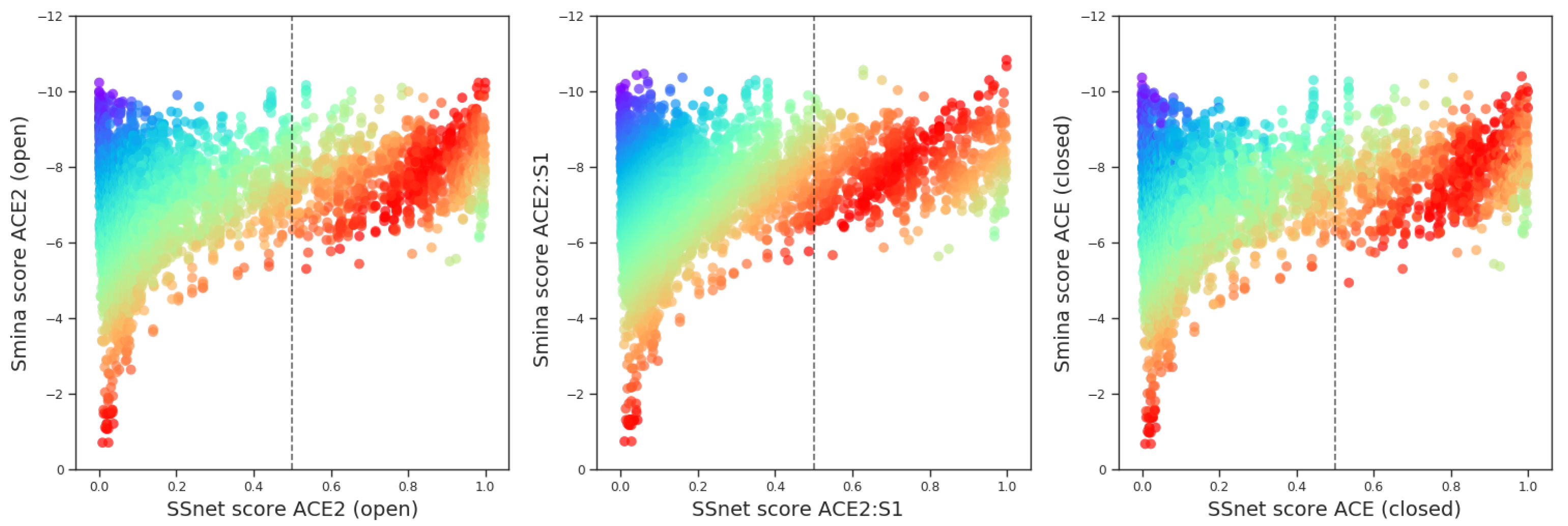
3.1. SSnet Predicts Ligands with Low Smina Binding Affinities
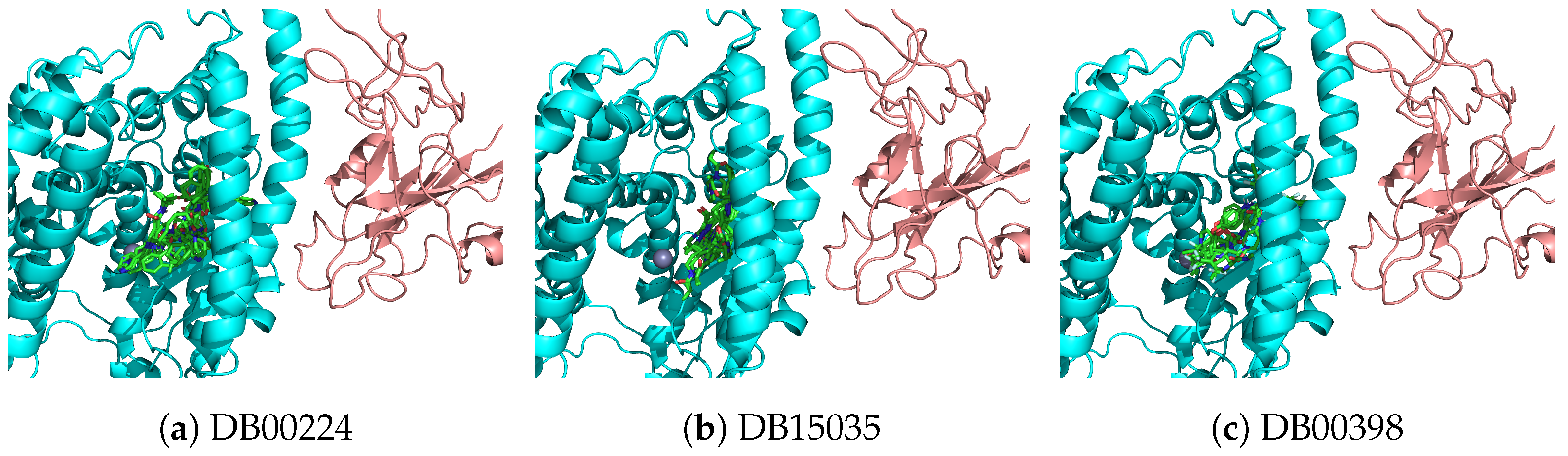
3.2. Ligands with High Binding Affinity Scores
Top Scores for both Smina and SSnet
3.3. Top Scores with SSnet
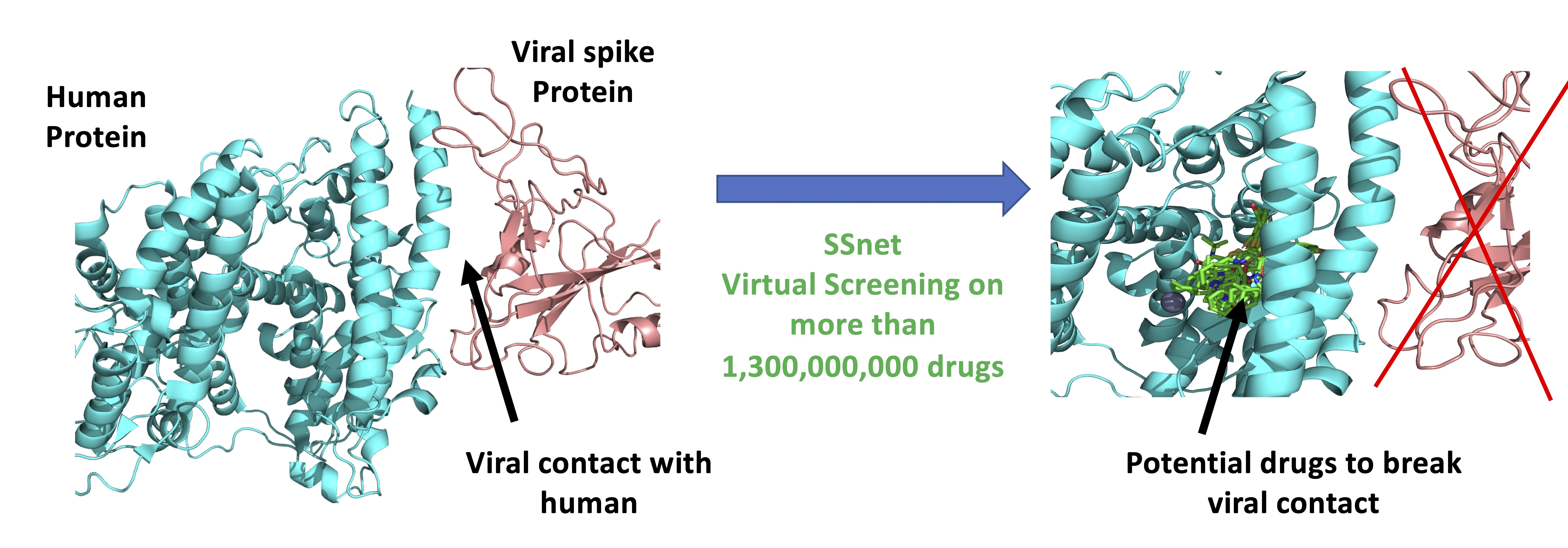
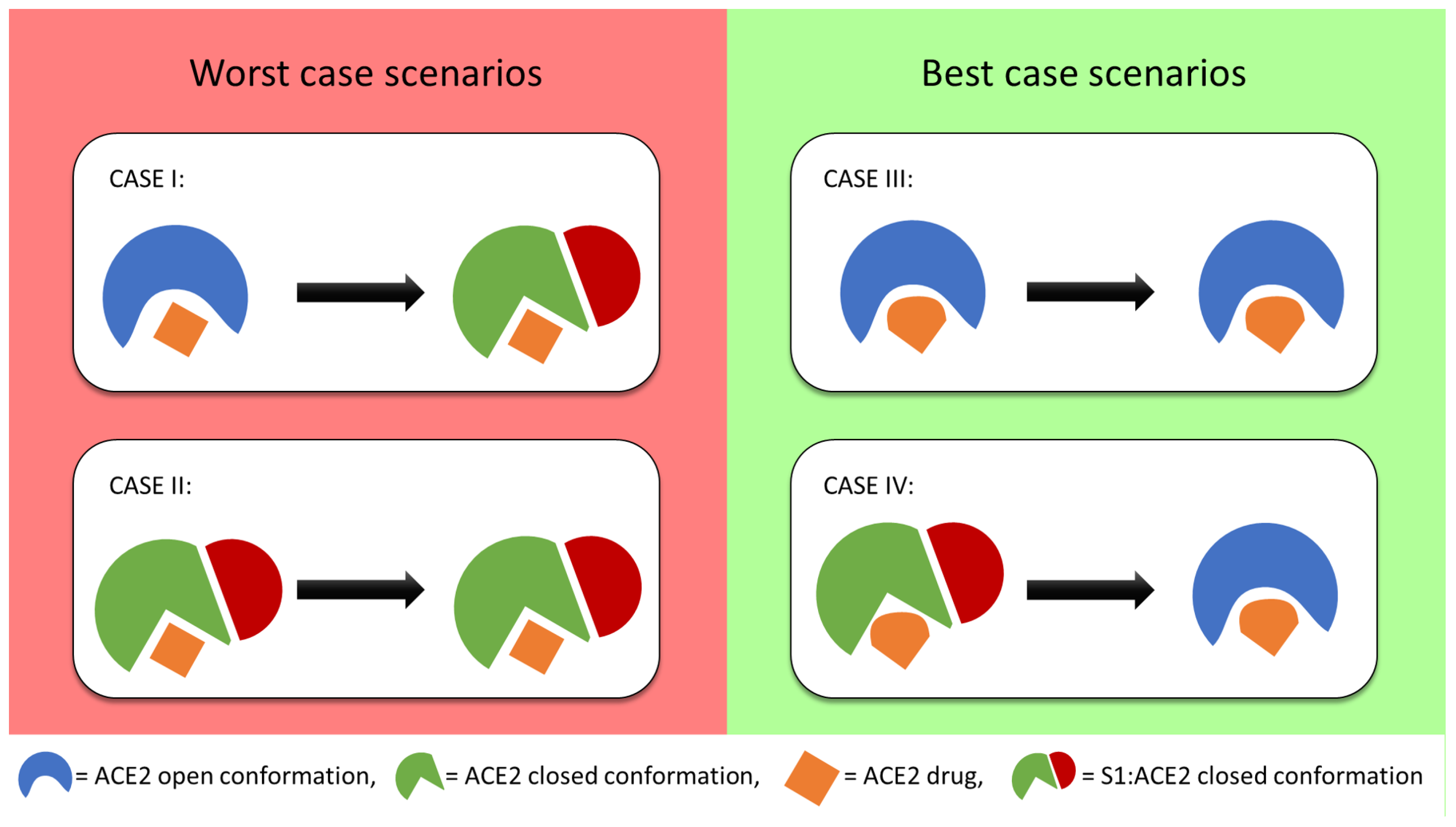
3.4. Model of Drug Action
- Case-I: The ligand binds to the open conformation but does not prevent the protein from exploring the closed conformation. This would render the drug ineffective.
- Case-II: The ligand binds to and stabilizes the closed conformation. This may make the drug counter-productive by making the receptor more susceptible to the docking of the viral S protein.
- Case-III: The ligand binds to and stabilizes the open conformation. This would prevent the docking of the viral S protein since the closed conformation is no longer explored.
- Case-IV: The ligand binds to the closed conformation with or without the viral S1 protein and biases the receptor towards an open conformation. This would either prevent or disrupt the viral docking.
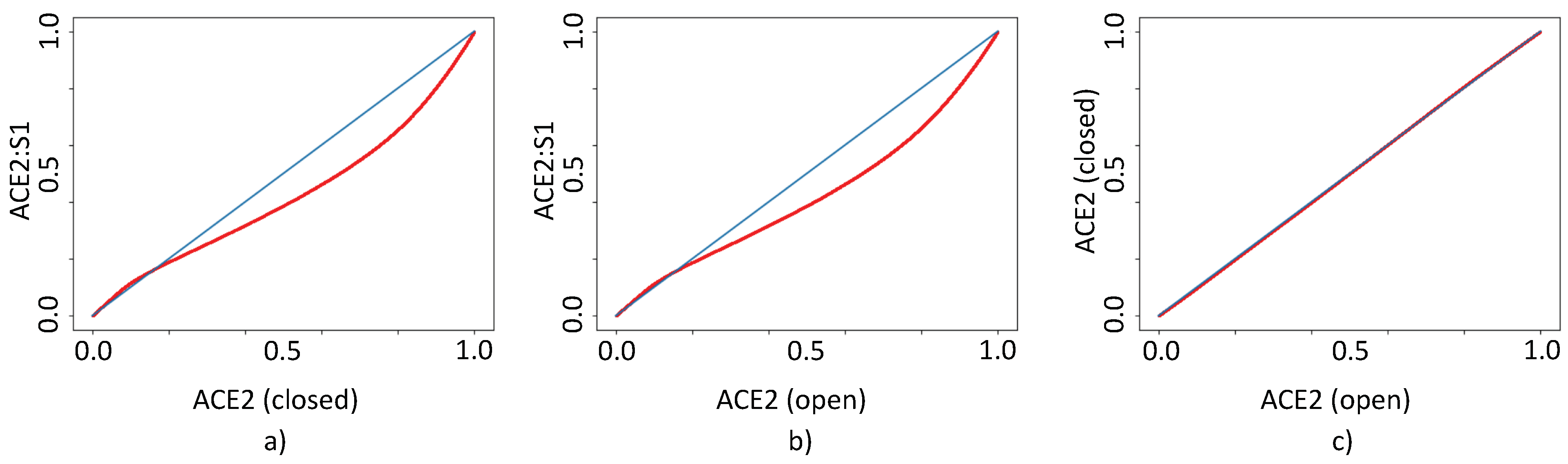
3.5. Top Scores for SSnet ACE2 (Open)-ACE2:S1 (Closed)
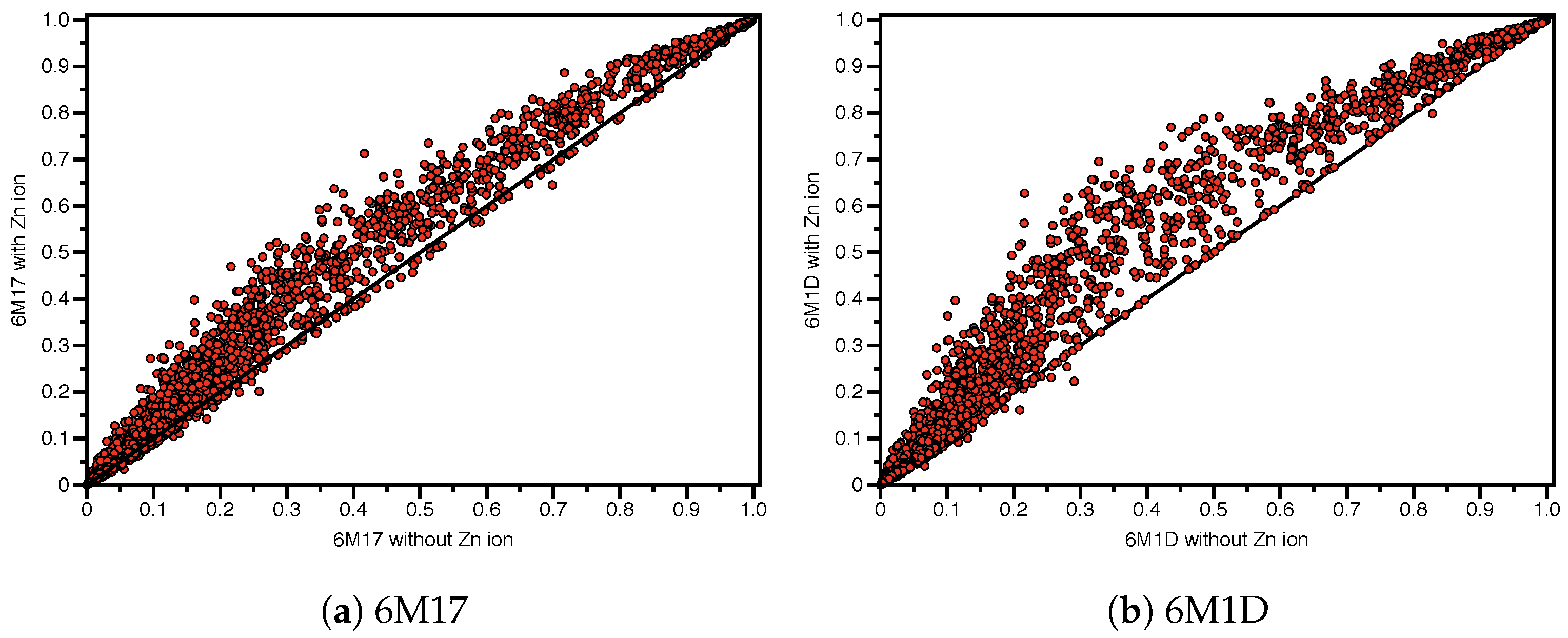
3.6. Zinc Effect on SSnet Binding Probabilities
3.7. Natural Compounds and Large Drug Molecules
3.8. SSnet as Tool to Aid the Discovery of De Novo Compounds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aberth, J. Plagues in World History; Exploring World History, Rowman & Littlefield Publishers: Lanham, MD, USA, 2011. [Google Scholar]
- Fair, R.J.; Tor, Y. Antibiotics and Bacterial Resistance in the 21st Century. Perspect. Med. Chem. 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldstone, M.B.A. Viruses, Plagues, and History: Past, Present, and Future, 3rd ed.; Oxford University Press: New York, NY, USA, 2020. [Google Scholar]
- WTO. Draft Landscape of COVID-19 Candidate Vaccines 30 September 2020; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Thomas, K.; LaFraniere, S.; Weiland, N.; Goodnough, A.; Haberman, M. Covid-19: F.D.A. Clears Pfizer Vaccine and Millions of Doses Will Be Shipped Right Away. The New York Times, 11 December 2020. [Google Scholar]
- Gallagher, T.M.; Buchmeier, M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology 2001, 279, 371–374. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Zmora, P.; Gierer, S.; Heurich, A.; Pöhlmann, S. Proteolytic activation of the SARS-coronavirus spike protein: Cutting enzymes at the cutting edge of antiviral research. Antivir. Res. 2013, 100, 605–614. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [Green Version]
- Hiscox, J.A. The interaction of animal cytoplasmic RNA viruses with the nucleus to facilitate replication. Virus Res. 2003, 95, 13–22. [Google Scholar] [CrossRef]
- Qu, X.X.; Hao, P.; Song, X.J.; Jiang, S.M.; Liu, Y.X.; Wang, P.G.; Rao, X.; Song, H.D.; Wang, S.Y.; Zuo, Y.; et al. Identification of two critical amino acid residues of the severe acute respiratory syndrome coronavirus spike protein for its variation in zoonotic tropism transition via a double substitution strategy. J. Biol. Chem. 2005, 280, 29588–29595. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.J. ACE2 Cell Biology, Regulation, and Physiological Functions. In The Protective Arm of the Renin Angiotensin System (RAS); Elsevier: Amsterdam, The Netherlands, 2015; pp. 185–189. [Google Scholar] [CrossRef]
- Turner, A.J.; Hiscox, J.A.; Hooper, N.M. ACE2: From vasopeptidase to SARS virus receptor. Trends Pharmacol. Sci. 2004, 25, 291–294. [Google Scholar] [CrossRef]
- Imig, J.D. ACE Inhibition and Bradykinin-Mediated Renal Vascular Responses: EDHF Involvement. Hypertension 2004, 43, 533–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Barros, E.P.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Wang, Y.; Fallon, L.; Raguette, L.; Belfon, K.; Simmerling, C.; Amaro, R.E. The Flexibility of ACE2 in the Context of SARS-CoV-2 Infection. Biophys. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Teralı, K.; Baddal, B.; Gülcan, H.O. Prioritizing potential ACE2 inhibitors in the COVID-19 pandemic: Insights from a molecular mechanics-assisted structure-based virtual screening experiment. J. Mol. Graph. Model. 2020, 100, 107697. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, K.; Sarma, P.; Rana, S.; Medhi, B.; Naithani, M. Emerging role of artificial intelligence in therapeutics for COVID-19: A systematic review. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Francés-Monerris, A.; Hognon, C.; Miclot, T.; García-Iriepa, C.; Iriepa, I.; Terenzi, A.; Grandemange, S.; Barone, G.; Marazzi, M.; Monari, A. Molecular Basis of SARS-CoV-2 Infection and Rational Design of Potential Antiviral Agents: Modeling and Simulation Approaches. J. Proteome Res. 2020, 19, 4291–4315. [Google Scholar] [CrossRef]
- Choudhary, S.; Malik, Y.S.; Tomar, S.; Tomar, S. Identification of SARS-CoV-2 Cell Entry Inhibitors by Drug Repurposing Using in Silico Structure-Based Virtual Screening Approach. ChemRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Smith, J.C. Repurposing therapeutics for COVID-19: Supercomputer-based docking to the SARS-CoV-2 viral spike protein and viral spike protein-human ACE2 interface. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; Jesus, P.D.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Xu, Z.; Yao, H.; Shen, J.; Wu, N.; Xu, Y.; Lu, X.; Zhu, W.; Li, L.J. Nelfinavir Is Active Against SARS-CoV-2 in Vero E6 Cells. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved Drug Ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Abbasi, J. Drug Repurposing Study Pinpoints Potential COVID-19 Antivirals. JAMA 2020, 324, 928. [Google Scholar] [CrossRef]
- Frediansyah, A.; Tiwari, R.; Sharun, K.; Dhama, K.; Harapan, H. Antivirals for COVID-19: A critical review. Clin. Epidemiol. Glob. Health 2021, 9, 90–98. [Google Scholar] [CrossRef]
- Mancilla-Galindo, J.; García-Méndez, J.Ó.; Márquez-Sánchez, J.; Reyes-Casarrubias, R.E.; Aguirre-Aguilar, E.; Rocha-González, H.I.; Kammar-García, A. Use of antivirals and antibiotics for COVID-19 in Mexico City: A Real-World Multicenter Cohort Study. medRxiv 2020. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Qu, X.; Trozzi, F.; Elsaied, M.; Tao, Y.; Larson, E.C.; Kraka, E. SSnet-Secondary Structure based End-to-End Learning model for Protein-Ligand Interaction Prediction. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Hatherley, R.; Brown, D.K.; Musyoka, T.M.; Penkler, D.L.; Faya, N.; Lobb, K.A.; Özlem Tastan Bishop. SANCDB: A South African natural compound database. J. Cheminform. 2015, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Pilon, A.C.; Valli, M.; Dametto, A.C.; Pinto, M.E.F.; Freire, R.T.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. NuBBEDB: An updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 2017, 7, 725. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public Database for Medicinal Chemistry, Computational Chemistry and Systems Pharmacology. Nucleic Acids Res. 2015, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- Riniker, S.; Landrum, G.A. Open-source Platform to Benchmark Fingerprints for Ligand-based Virtual Screening. J. Cheminf. 2013, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef] [Green Version]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound–protein Interaction Prediction with End-to-end Learning of Neural Networks for Graphs and Sequences. Bioinformatics 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, G. RDKit: Open-Source Cheminformatics. 2020. Available online: http://www.rdkit.org (accessed on 1 March 2020).
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva, F.P., Jr. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, LLC. The AxPyMOL Molecular Graphics Plugin for Microsoft PowerPoint, Version 1.8. 2015. Available online: http://www.pymol.org (accessed on 1 March 2020).
- Klann, K.; Bojkova, D.; Tascher, G.; Ciesek, S.; Münch, C.; Cinatl, J. Growth factor receptor signaling inhibition prevents SARS-CoV-2 replication. Mol. Cell. 2020, 80, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Lovetrue, B. The AI-discovered aetiology of COVID-19 and rationale of the irinotecan etoposide combination therapy for critically ill COVID-19 patients. Med. Hypotheses 2020, 144, 110180. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Petrini, M.; Baratè, C.; Ricci, F.; Balducci, S.; Grassi, S.; Guerrini, F.; Ciabatti, E.; Mechelli, S.; Paolo, A.D.; et al. Tyrosine Kinase Inhibitors Play an Antiviral Action in Patients Affected by Chronic Myeloid Leukemia: A Possible Model Supporting Their Use in the Fight Against SARS-CoV-2. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef]
- Fürstenau, M.; Langerbeins, P.; Silva, N.D.; Fink, A.M.; Robrecht, S.; von Tresckow, J.; Simon, F.; Hohloch, K.; Droogendijk, J.; van der Klift, M.; et al. COVID-19 among fit patients with CLL treated with venetoclax-based combinations. Leukemia 2020, 34, 2225–2229. [Google Scholar] [CrossRef]
- Guo, Y.; Zeng, J.; Li, Q.; Li, P.; Luo, F.; Zhang, W.; Lu, Y.; Wang, Q.; Zhang, W.; Zeng, Z.; et al. Preliminary clinical study of direct renin inhibitor aliskiren in the treatment of severe COVID-19 patients with hypertension. Zhonghua Nei Ke Za Zhi 2020, 59, E011. [Google Scholar]
- Gendrot, M.; Andreani, J.; Boxberger, M.; Jardot, P.; Fonta, I.; Bideau, M.L.; Duflot, I.; Mosnier, J.; Rolland, C.; Bogreau, H.; et al. Antimalarial drugs inhibit the replication of SARS-CoV-2: An in vitro evaluation. Travel Med. Infect Dis. 2020, 37, 101873. [Google Scholar] [CrossRef]
- Summa, V.; Ludmerer, S.W.; McCauley, J.A.; Fandozzi, C.; Burlein, C.; Claudio, G.; Coleman, P.J.; DiMuzio, J.M.; Ferrara, M.; Filippo, M.D.; et al. MK-5172, a Selective Inhibitor of Hepatitis C Virus NS3/4a Protease with Broad Activity across Genotypes and Resistant Variants. Antimicrob. Agents Chemother. 2012, 56, 4161–4167. [Google Scholar] [CrossRef] [Green Version]
- Seyfried, F.; Demir, S.; Hörl, R.L.; Stirnweiß, F.U.; Ryan, J.; Scheffold, A.; Villalobos-Ortiz, M.; Boldrin, E.; Zinngrebe, J.; Enzenmüller, S.; et al. Prediction of venetoclax activity in precursor B-ALL by functional assessment of apoptosis signaling. Cell Death Dis. 2019, 10, 571. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.I.; Tripathi, R.; Reisch, T.; Lu, L.; Middleton, T.; Hopkins, T.A.; Pithawalla, R.; Irvin, M.; Dekhtyar, T.; Krishnan, P.; et al. In Vitro Antiviral Activity and Resistance Profile of the Next-Generation Hepatitis C Virus NS3/4A Protease Inhibitor Glecaprevir. Antimicrob. Agents Chemother 2017, 62, e01620-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dana, P.M.; Sadoughi, F.; Hallajzadeh, J.; Asemi, Z.; Mansournia, M.A.; Yousefi, B.; Momen-Heravi, M. An insight into the sex differences in COVID-19 patients: What are the possible causes? Prehosp Disaster Med. 2020, 35, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Wambier, C.G.; Goren, A. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is likely to be androgen mediated. J. Am. Acad. Dermatol. 2020, 83, 308–309. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzilli, P.; Lenzi, A. Commentary: Testosterone, a key hormone in the context of COVID-19 pandemic. Metab. Clin. Exp. 2020, 108. [Google Scholar] [CrossRef] [PubMed]
- Strope, J.D.; Chau, C.H.; Figg, W.D. Are sex discordant outcomes in COVID-19 related to sex hormones? Semin. Oncol. 2020, 47, 335–340. [Google Scholar] [CrossRef]
- Penna, C.; Mercurio, V.; Tocchetti, C.G.; Pagliaro, P. Sex-related differences in COVID-19 lethality. Br. J. Pharmacol. 2020, 177, 4375–4385. [Google Scholar] [CrossRef]
- Kalidhindi, R.S.R.; Borkar, N.A.; Ambhore, N.S.; Pabelick, C.M.; Prakash, Y.; Sathish, V. Sex steroids skew ACE2 expression in human airway: A contributing factor to sex differences in COVID-19? Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L843–L847. [Google Scholar] [CrossRef]
- Li, Y.; Jerkic, M.; Slutsky, A.S.; Zhang, H. Molecular mechanisms of sex bias differences in COVID-19 mortality. Crit Care 2020, 24, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Taneja, V. Sex hormones determine immune response. Front. Immunol. 2018, 9, 1931. [Google Scholar] [CrossRef]
- Cattrini, C.; Bersanelli, M.; Latocca, M.M.; Conte, B.; Vallome, G.; Boccardo, F. Sex hormones and hormone therapy during covid-19 pandemic: Implications for patients with cancer. Cancers 2020, 12, 2325. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Klein, S.L.; Levin, E.R. Estradiol, progesterone, immunomodulation, and COVID-19 outcomes. Endocrinology 2020, 161, bqaa127. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Zhang, J.; Wang, T.; Cui, P.; Chen, Z.; Jiang, J.; Zhou, S.; Dai, J.; Wang, B.; Yuan, S.; et al. Potential influence of menstrual status and sex hormones on female SARS-CoV-2 infection: A cross-sectional study from multicentre in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y. Comprehensive assessment of flexible-ligand docking algorithms: Current effectiveness and challenges. Brief. Bioinform. 2018, 19, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Vieira, T.F.; Sousa, S.F. Comparing AutoDock and Vina in Ligand/Decoy Discrimination for Virtual Screening. Appl. Sci. 2019, 9, 4538. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karki, N.; Verma, N.; Trozzi, F.; Tao, P.; Kraka, E.; Zoltowski, B. Predicting Potential SARS-COV-2 Drugs—In Depth Drug Database Screening Using Deep Neural Network Framework SSnet, Classical Virtual Screening and Docking. Int. J. Mol. Sci. 2021, 22, 1573. https://doi.org/10.3390/ijms22041573
Karki N, Verma N, Trozzi F, Tao P, Kraka E, Zoltowski B. Predicting Potential SARS-COV-2 Drugs—In Depth Drug Database Screening Using Deep Neural Network Framework SSnet, Classical Virtual Screening and Docking. International Journal of Molecular Sciences. 2021; 22(4):1573. https://doi.org/10.3390/ijms22041573
Chicago/Turabian StyleKarki, Nischal, Niraj Verma, Francesco Trozzi, Peng Tao, Elfi Kraka, and Brian Zoltowski. 2021. "Predicting Potential SARS-COV-2 Drugs—In Depth Drug Database Screening Using Deep Neural Network Framework SSnet, Classical Virtual Screening and Docking" International Journal of Molecular Sciences 22, no. 4: 1573. https://doi.org/10.3390/ijms22041573