
Molecular Docking and Dynamics Investigations for Identifying Potential Inhibitors of the 3-Chymotrypsin-like Protease of SARS-CoV-2: Repurposing of Approved Pyrimidonic Pharmaceuticals for COVID-19 Treatment


Abstract
:1. Introduction
2. Materials and Methods
2.1. The Pyrimidonic Pharmaceuticals (PPs)
2.2. Generation and Energy Minimization of the PPs and 3CLpro
2.3. Molecular Docking
2.4. Molecular Dynamics Simulations
2.5. The Binding Free Energies
3. Results and Discussion
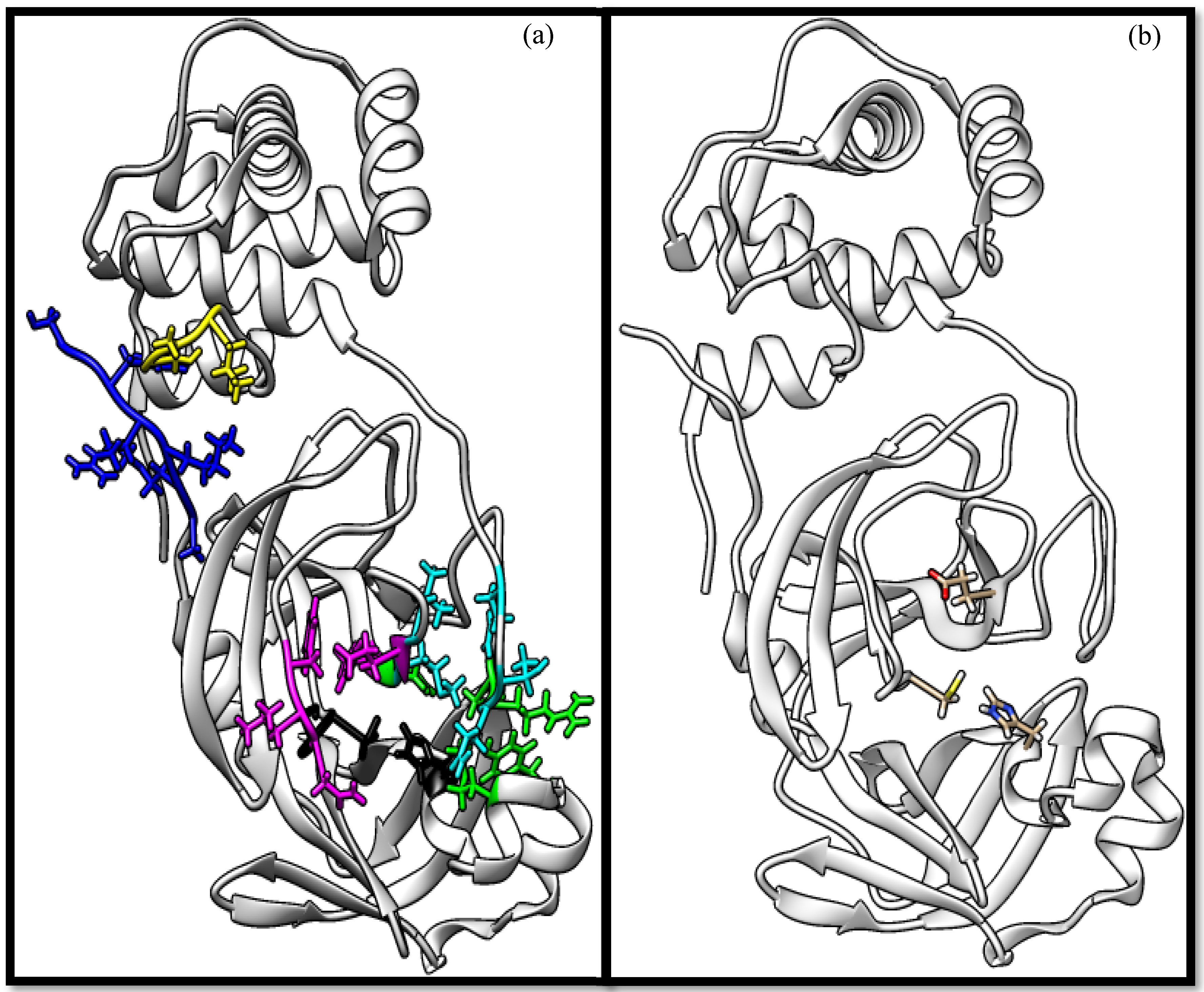
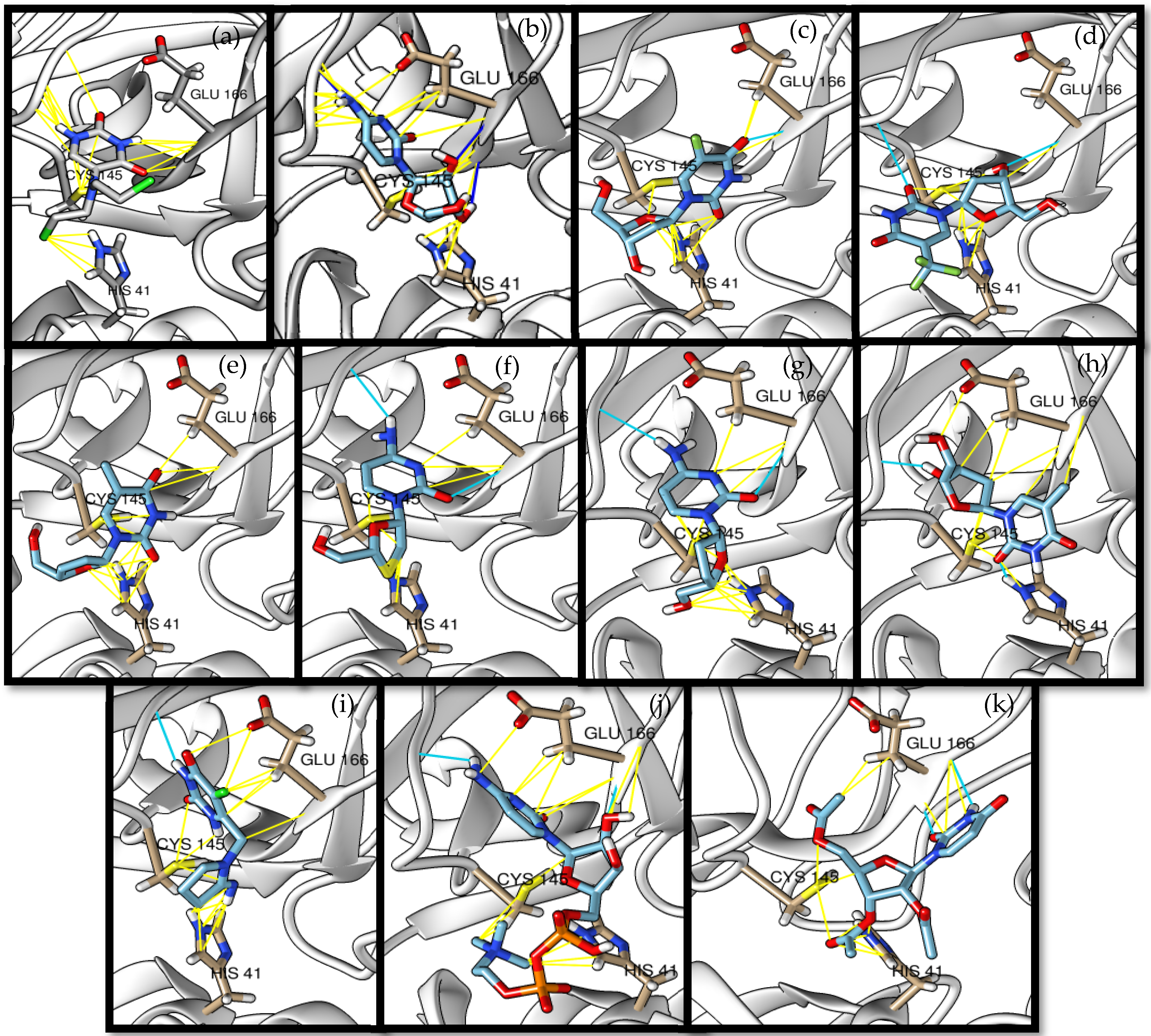
3.1. Molecular Docking
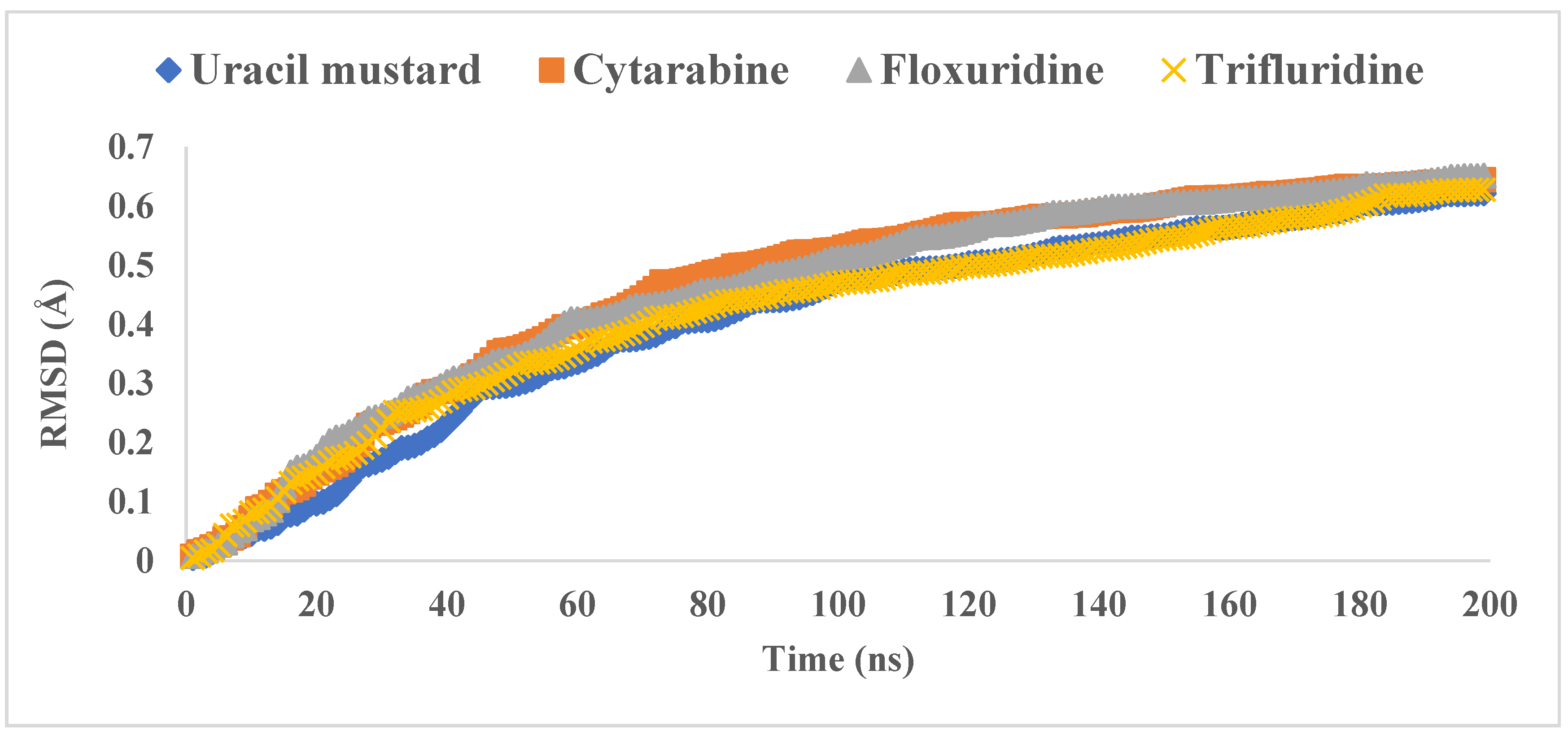
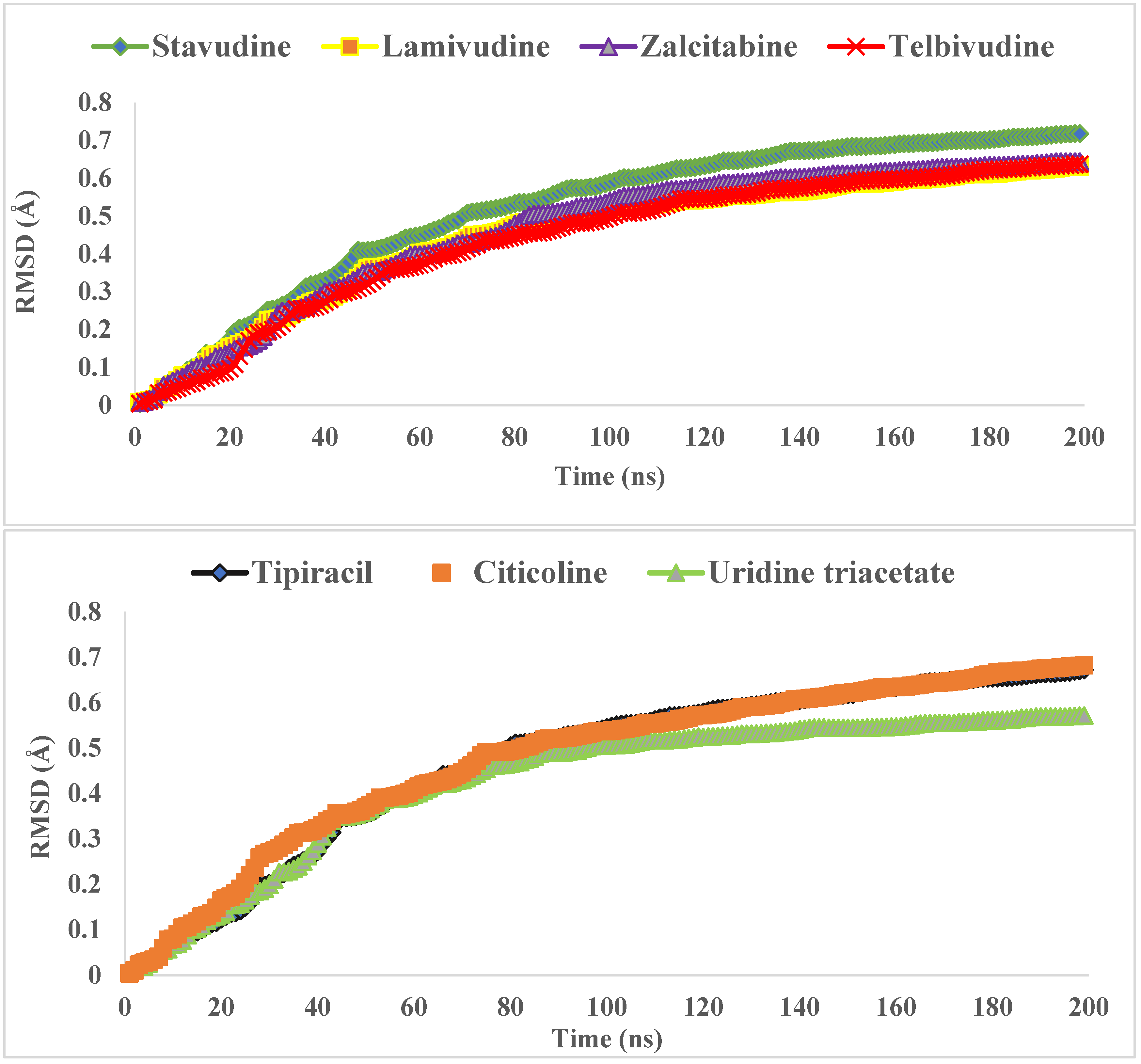
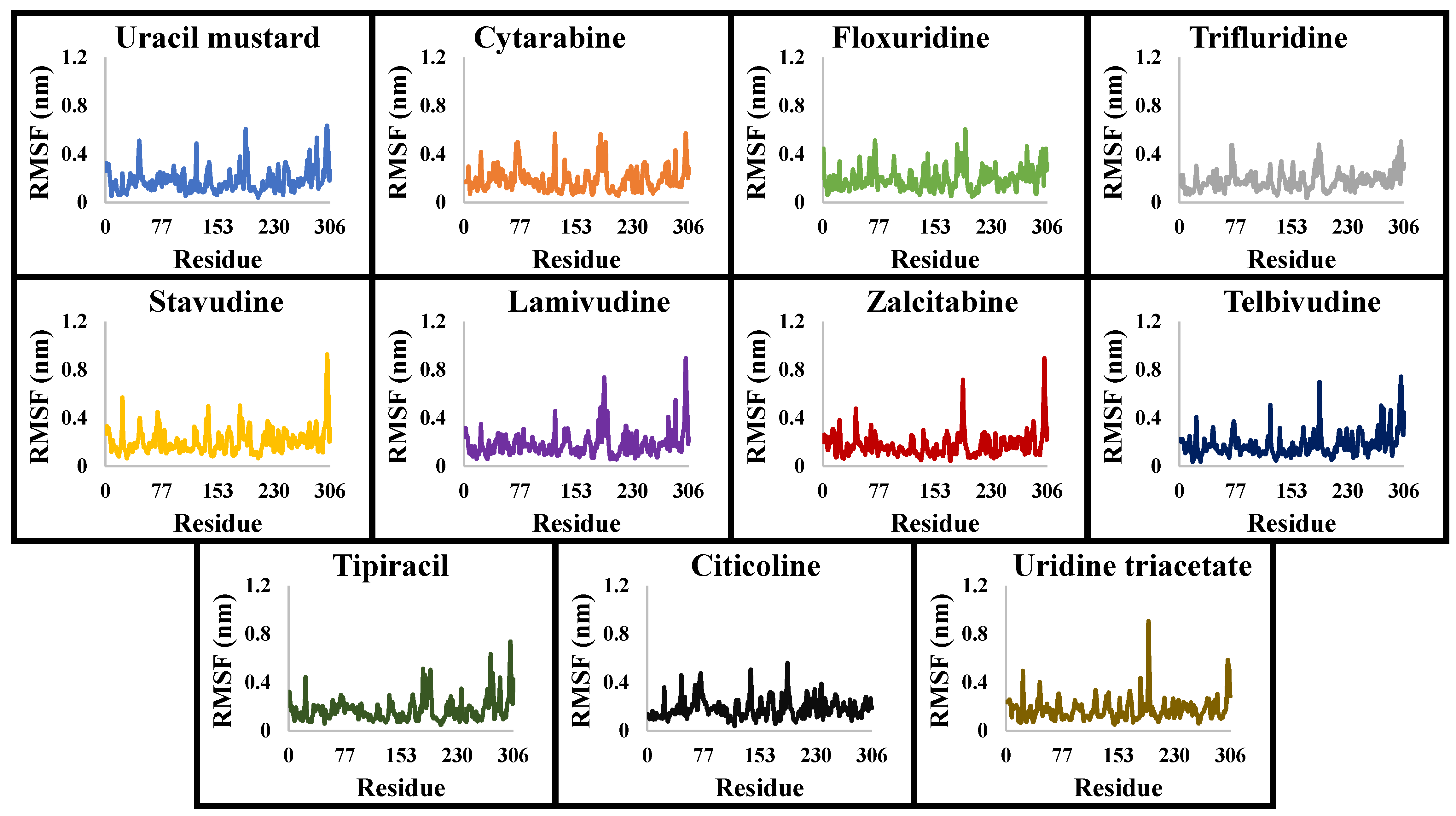
3.2. Molecular Dynamics Simulations
3.3. The Binding Free Energies
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ledford, H.; Cyranoski, D.; Van Noorden, R. The UK has approved a COVID vaccine-here’s what scientists now want to know. Nature 2020, 588, 205–206. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: UK approves Moderna vaccine to be given as two doses 28 days apart. BMJ 2021, 372. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Covid-19: Moderna applies for US and EU approval as vaccine trial reports 94.1% efficacy. BMJ 2020, 371. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Remdesivir: First approval. Drugs 2020, 80, 1355–1363. [Google Scholar] [CrossRef]
- Rubin, D.; Chan-Tack, K.; Farley, J.; Sherwat, A. FDA approval of remdesivir—A step in the right direction. N. Engl. J. Med. 2020, 383, 2598–2600. [Google Scholar] [CrossRef]
- Rivero-Segura, N.A.; Gomez-Verjan, J.C. In Silico Screening of Natural Products Isolated from Mexican Herbal Medicines against COVID-19. Biomolecules 2021, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Coronavirus Disease 2019 (COVID-19): Situation Report, 94. 2020. Available online: https://apps.who.int/iris/handle/10665/331865 (accessed on 27 November 2021).
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. COVID-19 Weekly Epidemiological Update. 2020. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed on 2 December 2021).
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorganic Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Amin, S.A.; Banerjee, S.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery: What we have learned from the past SARS-CoV inhibitors? Eur. J. Med. Chem. 2021, 113294. [Google Scholar] [CrossRef]
- Steuten, K.; Kim, H.; Widen, J.C.; Babin, B.M.; Onguka, O.; Lovell, S.; Bolgi, O.; Cerikan, B.; Neufeldt, C.J.; Cortese, M. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS Infect. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Beck, B.R.; Shin, B.; Choi, Y.; Park, S.; Kang, K. Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model. Comput. Struct. Biotechnol. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Qamar, M.T.U.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Gomez, C.R.; Espinoza, I.; Faruke, F.S.; Hasan, M.; Rahman, K.M.; Walker, L.A.; Muhammad, I. Therapeutic intervention of COVID-19 by natural products: A population-specific survey directed approach. Molecules 2021, 26, 1191. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Tung, Y.-A.; Lee, K.-H.; Chen, T.-F.; Hsiao, Y.-C.; Chang, H.-C.; Hsieh, T.-T.; Su, C.-H.; Wang, S.-S.; Yu, J.-Y. Potential therapeutic agents for COVID-19 based on the analysis of protease and RNA polymerase docking. Preprints 2020. [Google Scholar] [CrossRef] [Green Version]
- Terrier, O.; Dilly, S.; Pizzorno, A.; Chalupska, D.; Humpolickova, J.; Bouřa, E.; Berenbaum, F.; Quideau, S.; Lina, B.; Fève, B. Antiviral Properties of the NSAID Drug Naproxen Targeting the Nucleoprotein of SARS-CoV-2 Coronavirus. Molecules 2021, 26, 2593. [Google Scholar] [CrossRef]
- Le, N.P.K.; Herz, C.; Gomes, J.V.D.; Förster, N.; Antoniadou, K.; Mittermeier-Kleßinger, V.K.; Mewis, I.; Dawid, C.; Ulrichs, C.; Lamy, E. Comparative Anti-Inflammatory Effects of Salix Cortex Extracts and Acetylsalicylic Acid in SARS-CoV-2 Peptide and LPS-Activated Human In Vitro Systems. Int. J. Mol. Sci. 2021, 22, 6766. [Google Scholar] [CrossRef]
- Akhter, S.; Batool, A.I.; Selamoglu, Z.; Sevindik, M.; Eman, R.; Mustaqeem, M.; Akram, M.S.; Kanwal, F.; Lu, C.; Aslam, M. Effectiveness of Natural Antioxidants against SARS-CoV-2? Insights from the In-Silico World. Antibiotics 2021, 10, 1011. [Google Scholar]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef]
- Elzupir, A.O. Inhibition of SARS-CoV-2 main protease 3CLpro by means of α-ketoamide and pyridone-containing pharmaceuticals using in silico molecular docking. J. Mol. Struct. 2020, 1222, 128878. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapovalov, M.V.; Dunbrack Jr, R.L. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Elzupir, A.O. Caffeine and caffeine-containing pharmaceuticals as promising inhibitors for 3-chymotrypsin-like protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–8. [Google Scholar] [CrossRef]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE-Antechamber python parser interface. BMC Res. Notes 2012, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kalé, L.V.; Skeel, R.D.; Schulten, K. NAMD: A parallel, object-oriented molecular dynamics program. Int. J. Supercomput. Appl. High Perform. Comput. 1996, 10, 251–268. [Google Scholar] [CrossRef]
- De Leeuw, S.W.; Perram, J.W.; Smith, E.R. Simulation of electrostatic systems in periodic boundary conditions. I. Lattice sums and dielectric constants. Proc. R. Soc. Lond. A Math. Phys. Sci. 1980, 373, 27–56. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Miller, B.R., III; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA. py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Numata, J.; Wan, M.; Knapp, E.-W. Conformational entropy of biomolecules: Beyond the quasi-harmonic approximation. Genome Inform. 2007, 18, 192–205. [Google Scholar] [PubMed]
- Lim, L.; Shi, J.; Mu, Y.; Song, J. Dynamically-driven enhancement of the catalytic machinery of the SARS 3C-like protease by the S284-T285-I286/A mutations on the extra domain. PLoS ONE 2014, 9, e101941. [Google Scholar] [CrossRef] [PubMed]
- Saver, J.L. Citicoline: Update on a promising and widely available agent for neuroprotection and neurorepair. Rev. Neurol. Dis. 2008, 5, 167–177. [Google Scholar]
- Dinsdale, J.; Griffiths, G.; Rowlands, C.; Castelló, J.; Ortiz, J.; Maddock, J.; Aylward, M. Pharmacokinetics of 14C CDP-choline. Arzneim. Forsch. 1983, 33, 1066–1070. [Google Scholar]
- Anwar, M.U.; Adnan, F.; Abro, A.; Khan, M.R.; Rehman, A.U.; Osama, M.; Javed, S.; Baig, A.; Shabbir, M.R.; Assir, M.Z. Combined Deep Learning and Molecular Docking Simulations Approach Identifies Potentially Effective FDA Approved Drugs for Repurposing Against SARS-CoV-2. Comput. Biol. Med. 2021. [Google Scholar] [CrossRef]
- Prajapat, M.; Shekhar, N.; Sarma, P.; Avti, P.; Singh, S.; Kaur, H.; Bhattacharyya, A.; Kumar, S.; Sharma, S.; Prakash, A. Virtual screening and molecular dynamics study of approved drugs as inhibitors of spike protein S1 domain and ACE2 interaction in SARS-CoV-2. J. Mol. Graph. Model. 2020, 101, 107716. [Google Scholar] [CrossRef]
- Elfiky, A.A. Ribavirin, Remdesivir, Sofosbuvir, Galidesivir, and Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study. Life Sci. 2020, 253, 117592. [Google Scholar] [CrossRef]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide analogues as inhibitors of SARS-CoV-2 polymerase, a key drug target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef]
- Nar, H.; Schnapp, G.; Hucke, O.; Hardman, T.C.; Klein, T. Action of dipeptidyl peptidase-4 inhibitors on SARS-CoV-2 main protease. ChemMedChem 2021, 16, 1425–1426. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, J.; Liu, G.; Lu, Q.; Dong, R.; Tian, G.; Yang, J.; Peng, L. Probing antiviral drugs against SARS-CoV-2 through virus-drug association prediction based on the KATZ method. Genomics 2020, 112, 4427–4434. [Google Scholar] [CrossRef]
- Zhang, Y.-N.; Zhang, Q.-Y.; Li, X.-D.; Xiong, J.; Xiao, S.-Q.; Wang, Z.; Zhang, Z.-R.; Deng, C.-L.; Yang, X.-L.; Wei, H.-P. Gemcitabine, lycorine and oxysophoridine inhibit novel coronavirus (SARS-CoV-2) in cell culture. Emerg. Microbes Infect. 2020, 9, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Groaz, E.; Snoeck, R.; De Jonghe, S.; Herdewijn, P.; Andrei, G. Influence of 4′-Substitution on the Activity of Gemcitabine and Its ProTide against VZV and SARS-CoV-2. ACS Med. Chem. Lett. 2020, 12, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Aggarwal, R. Repurposing potential of FDA-approved and investigational drugs for COVID-19 targeting SARS-CoV-2 spike and main protease and validation by machine learning algorithm. Chem. Biol. Drug Des. 2021, 97, 836–853. [Google Scholar] [CrossRef] [PubMed]
- Borbone, N.; Piccialli, G.; Roviello, G.N.; Oliviero, G. Nucleoside analogs and nucleoside precursors as drugs in the fight against SARS-CoV-2 and other coronaviruses. Molecules 2021, 26, 986. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Yoo, J.; Kim, M.-S.; Shin, D.H. A Study of 3CLpros as Promising Targets against SARS-CoV and SARS-CoV-2. Microorganisms 2021, 9, 756. [Google Scholar] [CrossRef] [PubMed]
- Pillaiyar, T.; Meenakshisundaram, S.; Manickam, M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today 2020, 25, 668–688. [Google Scholar] [CrossRef]
- Maurya, S.K.; Maurya, A.K.; Mishra, N.; Siddique, H.R. Virtual screening, ADME/T, and binding free energy analysis of anti-viral, anti-protease, and anti-infectious compounds against NSP10/NSP16 methyltransferase and main protease of SARS CoV-2. J. Recept. Signal Transduct. 2020, 40, 605–612. [Google Scholar] [CrossRef]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020, 251, 117627. [Google Scholar] [CrossRef]
- Choi, R.; Zhou, M.; Shek, R.; Wilson, J.W.; Tillery, L.; Craig, J.K.; Salukhe, I.A.; Hickson, S.E.; Kumar, N.; James, R.M. High-throughput screening of the ReFRAME, Pandemic Box, and COVID Box drug repurposing libraries against SARS-CoV-2 nsp15 endoribonuclease to identify small-molecule inhibitors of viral activity. PLoS ONE 2021, 16, e0250019. [Google Scholar] [CrossRef]
- Sharanya, C.; Abhithaj, J.; Sadasivan, C. Drug repurposing to identify therapeutics against COVID 19 with SARS-Cov-2 spike glycoprotein and main protease as targets: An in silico study. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Fadlalla, M. COVID19 Approved Drug Repurposing: Pocket Similarity Approach. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H. Virtual screening of natural products against type II transmembrane serine protease (TMPRSS2), the priming agent of coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1PPs | 2aPPs | 2bPPs | 3PPs |
|---|---|---|---|
 |  |  |  |
 |  |  |  |
 |  |  |  |
 |  |  |  |
| - |  |  |  |
| - |  |  |  |
| - |  |  |  |
| - |  |  |  |
| - | - |  |  |
| - | - |  | - |
| - | - |  | - |
| Pharmaceutical Name | Binding Percentage a | Score ± SD (kcal/mol) b | RMSD | Hydrogen Bond (Number of Bonds/Number of Conformations) | Van Der Waals (Distance) (Number of Bonds/Number of Conformations) |
|---|---|---|---|---|---|
| Uracil mustard | a. 100 * c. 89 d. 100 e. 100 All. 100 | a. −4.6 ± 0.14 c. −4.6 ± 0.14 d. −4.6 ± 0.14 e. −4.6 ± 0.14 | a. 0.00–7.13 c. 0.00–7.13 d. 0.00–7.13 e. 0.00–7.13 | a. HIS 163 (5/5), GLU 166 (3/3), LEU 141 (3/3), ASN 142 (1/1) | a. HIS 163 (31/5), GLU 166 (40/8), LEU 141 (19/5), ASN 142 (50/7), PHE 140 (8/5) c. MET 49 (52/7), HIS 164 (12/4) d. GLN 189 (32/9), MET 165 (28/8) e. HIS 41 (53/9), SER 144 (10/5), GLY 143 (4/1), CYS 145 (28/9) |
| Cytarabine | a. 67 c. 67 d. 67 e. 67 All. 67 | a. −5.4 ± 0.26 c. −5.4 ± 0.26 d. −5.4 ± 0.26 e. −5.4 ± 0.26 | a. 0.00–6.07 c. 0.00–6.07 d. 0.00–6.07 e. 0.00–6.07 | a. GLU 166 (3/3), LEU 141 (2/2), HIS 163 (1/1), PHE 140 (2/2), ASN 142 (1/1) c. HIS 164 (2/2) d. GLN 189 (1/1) e. GLY 143 (1/1) | a. GLU 166 (44/5), LEU 141 (18/3), HIS 163 (18/4), PHE 140 (19/3), ASN 142 (27/6) c. HIS 164 (10/4), MET 49 (20/6) d. MET 165 (28/4), GLN 189 (4/2) e. GLY 143 (11/2), SER 144 (8/2), CYS 145 (15/6), HIS 41 (21/5) |
| Floxuridine | a. 44 c. 44 d. 44 e. 44 All. 56 | a. 5.5 ± 0.13 c. 5.6 ± 0.25 d. 5.6 ± 0.25 e. 5.6 ± 0.19 | a. 3.24–8.28 c. 0.00–8.28 d. 0.00–8.28 e. 0.00–8.28 | a. ASN 142 (1/1), HIS 163 (2/2), GLU 166 (2/2), PHE 140 (1/1), LEU 141 (1/1) e. HIS 41 (1/1) | a. GLU 166 (23/4), LEU 141 (6/2), PHE 140 (8/2), HIS 163 (5/2), ASN 142 (19/4) c. MET 49 (9/2), HIS 164 (3/3) d. MET 165 (5/3), GLN 189 (4/1) e. GLY 143 (6/1), SER 144 (4/1), CYS 145 (5/3), HIS 41 (16/3) |
| Trifluridine | a. 44 c. 44 d. 44 e. 44 f. 11 All. 56 | a. −6.03 ± 0.17 c. −6.03 ± 0.17 d. −6.03 ± 0.17 e. −6.03 ± 0.17 f. −5.7 | a. 0.00–5.58 c. 0.00–5.58 d. 0.00–5.58 e. 0.00–5.58 f. 28.34–30.21 | a. GLU 166 (2/2), ASN 142 (1/1). c. HIS 164 (2/2) e. GLY 143 (1/1) | a. GLU 166 (18/4), ASN 142 (15/3), HIS 163 (2/1), LEU 141 (2/1) b. c. HIS 164 (10/3), MET 49 (17/4) d. MET 165 (19/4), GLN 189 (1/1). e. CYS 145 (13/4), GLY 143 (10/2), HIS 41 (10/4) f. SER 284 (9/1) |
| Stavudine | a. 44 c. 56 d. 44 e. 56 All. 56 | a. −5.6 ± 0.28 c. −5.6 ± 0.28 d. −5.6 ± 0.28 e. −5.6 ± 0.28 | a. 27.12–32.34 c. 27.12–35.03 d. 27.12–32.34 e. 27.12–35.03 | a. GLU 166 (1/1) | a. ASN 142 (13/3), GLU 166 (14/4), HIS 163 (2/1), LEU 141 (1/1) c. HIS 164 (6/3), MET 49 (20/5) d. MET 165 (12/4), GLN 189 (1/1) e. HIS 41 (39/5), GLY 143 (9/2), CYS 145 (7/3) |
| Lamivudine | a. 56 c. 44 d. 44 e. 56 f. 11 All. 67 | a. −5.4 ± 0.24 c. −5.4 ± 0.28 d. −5.4 ± 0.25 e. −5.4 ± 0.24 f. −5.2 | a. 0.00–4.77 c. 0.00–3.38 d. 0.00–4.77 e. 0.00–4.77 f. 26.28–28.62 | a. HIS 163 (3/3), ASN 142 (1/1), PHE 140 (3/3), LEU 141 (2/2), GLU 166 (2/2) d. GLN 189 (1/1) e. SER 144 (2/2) | a. HIS 163 (26/5), ASN 142 (19/2), PHE 140 (31/5), LEU 141 (15/5), GLU 166 (46/5) c. MET 49 (12/4), HIS 164 (1/1) d. GLN 189 (12/2), MET 165 (10/4), LEU 167 (1/1) e. SER 144 (18/4), HIS 41 (5/2), CYS 145 (7/3) f. LEU 286 (1/1) |
| Zalcitabine | a. 44 b. 11 c. 33 d. 44 e. 44 f. 22 All. 67 | a. −5.5 ± 0.29 b. −5.1 c. −5.4 ± 0.35 d. −5.5 ± 0.29 e. −5.5 ± 0.29 f. −5.1 ± 0.00 | a. 0.00–6.39 b. 28.02–29.48 c. 0.00–6.39 d. 0.00–6.39 e. 0.00–6.39 f. 28.02–31.88 | a. PHE 140 (3/3), LEU 141 (1/1), GLU 166 (3/3), ASN 142 (1/1) c. GLN 189 (1/1) | a. HIS 163 (15/3), PHE 140 (16/3), LEU 141 (9/3), GLU 166 (25/4), ASN 142 (11/3) b. LYS 5 (6/1), ARG 4 (12/1), PHE 3 (4/1) c. MET 49 (16/2), HIS 164 (5/3) d. GLN 189 (6/2), MET 165 (15/4) e. SER 144 (7/2), HIS 41 (13/2), CYS 145 (6/3) f. SER 284 (4/1), LEU 286 (4/1) |
| Telbivudine | a. 56 c. 44 d. 44 e. 56 f. 11 All. 67 | a. −5.6 ± 0.38 c. −5.7 ± 0.42 d. −5.7 ± 0.42 e. −5.6 ± 0.38 f. −5.3 | a. 0.00–7.50 c. 0.00–7.50 d. 0.00–7.50 e. 0.00–7.50 f. 22.85–24.17 | a. ASN 142 (2/1), HIS 163 (3/3), GLU 166 (1/1), PHE 140 (2/2), LEU 141 (1/1) c. HIS 164 (1/1) d. GLN 189 (1/1) e. HIS 41 (1/1) | a. GLU 166 (36/4), HIS 163 (11/3), PHE 140 (11/3), ASN 142 (27/5), LEU 141 (9/3) c. HIS 164 (6/3), MET 49 (27/3) d. GLN 189 (10/3), MET 165 (14/4), e. CYS 145 (11/4), SER 144 (8/2), GLY 143 (3/1), HIS 41 (12/5) f. LEU 286 (5/1) |
| Tipiracil | a. 56 b. 11 c. 44 d. 44 e. 44 f. 11 All. 67 | a. −5.8 ± 0.16 b. −5.7 c. −5.9 ± 0.17 d. −5.8 ± 0.08 e. −5.9 ± 0.17 f. −5.7 | a. 26.28–29.69 b. 19.12–20.08 c. 26.28–29.69 d. 26.67–29.69 e. 26.28–29.69 f. 19.12–20.08 | a. HIS 163 (2/2), GLU 166 (2/1), PHE 140 (2/2), ASN 142 (1/1) b. LYS 5 (1/1) c. HIS 164 (1/1) e. GLY 143 (1/1) | a. HIS 163 (14/3), GLU 166 (40/4), PHE 140 (8/3), ASN 142 (22/4), LEU 141 (10/3) b. PHE 3 (6/1), LYS 5 (9/1), ARG 4 (5/1) c. HIS 164 (8/4), MET 49 (22/3) d. MET 165 (9/3), GLN 189 (13/3) e. GLY 143 (7/2), HIS 41 (26/4), SER 144(2/1), CYS 145 (13/4) f. SER 284 (7/1), LEU 286 (2/1) |
| Citicoline | a. 56 * c. 56 d. 56 e. 56 All. 56 | a. −7.0 ± 0.19 c. −7.0 ± 0.19 d. −7.0 ± 0.19 e. −7.0 ± 0.19 | a. 0.00–8.01 c. 0.00–8.01 d. 0.00–8.01 e. 0.00–8.01 | a. PHE 140 (2/2), GLU 166 (4/4), HIS 163 (3/3), ASN 142 (2/1), LEU 141 (1/1) e. SER 144 (1/1) | a. PHE 140 (24/5), GLU 166 (57/5), HIS 163 (21/5), ASN 142 (34/5), LEU 141 (21/5) c. MET 49 (23/5), HIS 164 (1/1) d. MET 165 (29/5), GLN 189 (6/3) e. SER 144 (12/3), GLY 143 (4/1), CYS 145 (9/5), HIS 41 (21/5) |
| Uridine triacetate | a. 56 c. 56 d. 56 e. 56 f. 11 All. 67 | a. −6.2 ± 0.26 c. −6.2 ± 0.26 d. −6.2 ± 0.26 e. −6.2 ± 0.26 f. −6.4 | a. 0.00–6.79 c. 0.00–6.79 d. 0.00–6.79 e. 0.00–6.79 f. 24.97–28.01 | a. HIS 163 (2/2), GLU 166 (3/2) e. HIS 41 (1/1) | a. HIS 163 (5/2), GLU 166 (31/5), ASN 142 (30/5), PHE 140 (4/2), LEU 141 (6/2) c. MET 49 (18/5), HIS 164 (5/2) d. GLN 189 (15/4), MET 165 (18/4) e. CYS 145 (16/5), HIS 41 (35/5), SER 144 (4/2), GLY 143 (2/1) f. LEU 286 (1/1) |
| Pharmaceutical Name | 100 ns | 150 ns | 200 ns |
|---|---|---|---|
| Uracil mustard |  |  |  |
| Cytarabine |  |  |  |
| Floxuridine |  |  |  |
| Trifluridine |  |  |  |
| Stavudine |  |  |  |
| Lamivudine |  |  |  |
| Zalcitabine |  |  |  |
| Telbivudine |  |  |  |
| Tipiracil |  |  |  |
| Citicoline |  |  |  |
| Uridine triacetate |  |  |  |
| 3CLpro Complex Type | −TΔS | Evdw | MMGBSA | MMPBSA | ||||
|---|---|---|---|---|---|---|---|---|
| Ealac | Esol | Δg (kcal/mol) | Ealac | Esol | Δg (kcal/mol) | |||
| Uracil mustard | 22.58 | −23.76 | −16.08 | 20.46 | 3.20 | −0.80 | 0.94 | −1.05 |
| Cytarabine | 22.39 | −19.25 | −33.50 | 28.39 | −1.96 | −1.67 | 1.09 | 2.55 |
| Floxuridine | 22.42 | −24.54 | 0.00 | 3.70 | 1.58 | 0.00 | 0.39 | −1.73 |
| Trifluridine | 22.96 | −29.48 | 0.00 | 4.20 | −2.32 | 0.00 | 0.41 | −6.11 |
| Stavudine | 22.25 | −24.41 | −17.67 | 17.90 | −1.94 | −0.88 | 0.93 | −2.12 |
| Lamivudine | 22.26 | −20.58 | −35.11 | 28.94 | −4.50 | −1.76 | 1.06 | 0.98 |
| Zalcitabine | 22.12 | −24.34 | −30.10 | 26.50 | −5.81 | −1.50 | 1.19 | 2.53 |
| Telbivudine | 22.48 | −25.29 | −46.88 | 42.53 | −7.16 | −2.34 | 1.61 | −3.54 |
| Tipiracil | 22.54 | −28.79 | 0.00 | 5.07 | −1.18 | 0.00 | 0.44 | −5.81 |
| Citicoline | 24.17 | −54.50 | 0.00 | 4.80 | −25.53 | 0.00 | 0.64 | −29.69 |
| Uridine triacetate | 23.60 | −32.98 | −31.09 | 33.40 | −7.07 | −1.55 | 1.43 | −9.51 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elzupir, A.O. Molecular Docking and Dynamics Investigations for Identifying Potential Inhibitors of the 3-Chymotrypsin-like Protease of SARS-CoV-2: Repurposing of Approved Pyrimidonic Pharmaceuticals for COVID-19 Treatment. Molecules 2021, 26, 7458. https://doi.org/10.3390/molecules26247458
Elzupir AO. Molecular Docking and Dynamics Investigations for Identifying Potential Inhibitors of the 3-Chymotrypsin-like Protease of SARS-CoV-2: Repurposing of Approved Pyrimidonic Pharmaceuticals for COVID-19 Treatment. Molecules. 2021; 26(24):7458. https://doi.org/10.3390/molecules26247458
Chicago/Turabian StyleElzupir, Amin Osman. 2021. "Molecular Docking and Dynamics Investigations for Identifying Potential Inhibitors of the 3-Chymotrypsin-like Protease of SARS-CoV-2: Repurposing of Approved Pyrimidonic Pharmaceuticals for COVID-19 Treatment" Molecules 26, no. 24: 7458. https://doi.org/10.3390/molecules26247458