
Abstract
The elderly population is the most susceptible to SARS-CoV-2 infection and develops the worst clinical phenotype with severe pneumonia and cardiac complications. Older COVID-19 patients are also at higher risk of sudden death, mainly attributable to electrolyte disorders and to an uncontrolled inflammatory response. After the identification of ACE 2 as the receptor of SARS-CoV-2 in human cells, several research studies have focused on the role of the activation of Renin Angiotensin System in COVID-19 clinical course. In the present opinion paper, we discuss the role of hyperaldosteronism in the increasing risk of cardiac complications in COVID-19 older patients. In particular, we focus on the immunoregulatory activity of aldosterone, as the last mediator of the Renin Angiotensin System cascade, in activating the innate and adaptive immune response related to SARS-CoV-2 infection in the elderly. Aldosterone may stimulate dendritic cells and the recruitment of monocytes/macrophages in the endothelium of coronary vessels, favoring the production of pro-inflammatory mediators and T-cells response. Higher basal levels of aldosterone together with SARS-CoV-2-induced production may explain the unfavorable course of COVID-19 in the elderly.
Similar content being viewed by others


Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection emerged in China in late December 2019 and, to date, its rapid spread has not yet been stopped despite the largest vaccination campaign in the world of the last century. The high rate of hospitalization related to coronavirus disease (COVID-19) pandemic has resulted in a global emergency with difficulties in accessing primary health care [1,2,3]. Thus, a great number of patients have faced this recent coronavirus disease isolated at home. Elderly patients represent the most affected population by COVID-19, with the highest mortality [4]. In these patients, frailty and multiple comorbidities may partially explain the severe clinical presentation, although the mechanisms of worse prognosis remain largely unexplained [1, 2, 4].
In the lockdown period, several evidences indicate an increased rate of out-of-hospital cardiac arrest and sudden death in the elders [1, 2, 4]. It has been widely ascertained that SARS-CoV-2 infection may exponentially increase the risk of cardiac arrhythmias through different mechanisms [5]. Among them, the increased rate of myocarditis and acute myocardial infarction with cardiac injury or fibrosis, the induction of a prothrombotic phenotype, and the QT prolongation derived mostly by multidrug therapy and sepsis have been identified as the main arrhythmogenic triggers [1, 2, 4, 5]. In addition, in these patients, the risk of proarrhythmogenic events is also due to their pre-COVID-19 clinical conditions (cardiovascular diseases, chronic kidney disease, diabetes mellitus, and cancer) which can, in turn, predispose to electrolyte disorders. As a matter of fact, electrolyte alterations are common findings in older compared to younger individuals and they are associated with the use of medications such as diuretics, insulin, bronchodilators, and anti-depressants, that are all potentially associated with the risk of hypokalemia and hyponatremia [5,6,7]. Moreover, electrolyte imbalance may be due to malnutrition with deficiency of vitamins/minerals, as well as to frailty or physical limitations associated with loss of skeletal mass.
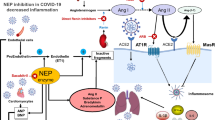
Chen et al. have described hypokalemia as a hallmark of the most severe forms of COVID-19 and suggested that it was the result of the COVID-19-related activation of Renin Angiotensin Aldosterone System (RAAS), with the consequent increase of aldosterone levels [8]. This evidence suggests that high levels of aldosterone may be associated with increased mortality due to cardiac arrest in COVID-19. Aldosterone represents the last hormone in the RAAS cascade, with its main role being the regulation of plasma volume and pressure by stimulating renal reabsorption of sodium and excretion of potassium. The role of RAAS has been largely investigated in COVID-19 pathophysiology, since the first phase of the pandemic, when it was demonstrated that SARS-CoV-2 infects cells via binding to Angiotensin-Converting Enzyme 2 (ACE 2) [9]. Hence, a brief description of the other RAAS mediators seems to be necessary for understanding the pivotal role of aldosterone in COVID-19. In juxtaglomerular cells, a reduction of blood flow is the main trigger for the conversion of prorenin into renin, that, in turn, hydrolyzes angiotensinogen, an α-2-globulin synthesized in the liver, in Angiotensin I (Ang I). Ang I appears to have poor biological effects; however, it is converted into Angiotensin II (Ang II) by the Angiotensin Converting Enzyme (ACE), which is predominantly localized on the endothelial cell membranes of the pulmonary vessels. Ang II is an octapeptide with potent vasoconstrictive proprieties and it stimulates inflammation, atherogenesis, cardiac remodeling/fibrosis, insulin resistance, and thrombosis by binding to the Angiotensin II Receptor-1 (AT-1). Ang II/AT-1 interaction directly stimulates the secretion of aldosterone in the adrenal cortex. However, a small amount of Ang II is hydrolyzed by ACE2 into Angiotensin 1-7 (Ang 1-7) which has anti-inflammatory and vasodilatory effects by binding to the MAS-related G-protein coupled receptor [9,10,11]. Therefore, ACE and ACE 2 seem to play an antithetical activity with negative feedback, giving that ACE/Ang II/AT1 pathway activation stimulates the cleavage/conversion of ACE 2 into its circulating form, unable to bind Ang 1-7 and MAS receptors pathway [9, 10].
Similarly, ACE 2 is down-regulated after SARS-CoV-2 internalization in human cells, with consequent RAAS overactivation [10, 11]. Consequently, the overproduction of Ang II/aldosterone leads to vasoconstriction, inflammation, oxidative stress, and fibrosis [11]. Moreover, ACE 2 is largely expressed in the endothelium [9, 10, 12], where SARS-CoV-2 may locally stimulate RAAS activation. The resulting increased local levels of Ang II stimulate aldosterone production [11]; nevertheless, low doses of aldosterone could also be produced directly in the vessels through the activity of aldosterone synthase [13]. Finally, high levels of Ang II/aldosterone also increase the circulating levels of ACE 2, that may act as a “competitive interceptor” of SARS-CoV-2 [9,10,11].
To the best of our knowledge, the role of aldosterone has not been extensively investigated in COVID-19. Other than negatively affecting serum potassium levels, aldosterone may increase cardiovascular mortality through its pro-inflammatory and pro-atherosclerotic activities [9]. Although low aldosterone levels have been described in healthy aging [14], higher levels are frequently reported in the geriatric population affected by hypertension, diabetes, and cardiovascular diseases, despite treatment with RAAS inhibitors (RAASi) [14,15,16]. Indeed, the inhibition of aldosterone effects via administration of mineralocorticoid receptors (MRs) antagonists reduces the risk of cardiac arrest and sudden cardiac death [17].
Aldosterone regulates the innate and adaptive immune response by promoting the activation of inflammatory mediators and cells such as monocytes, macrophages, dendritic cells (DCs), and lymphocytes [13]. The activation of these cells and the consequent cytokine storm correlates with the highest mortality and sudden death in COVID-19 geriatric patients [1, 2]. Therefore, in this opinion piece, we discuss the potential role of aldosterone in regulating immune cell activity and the response to SARS-CoV-2 infection in this at-risk population.
Monocytes/macrophages represent a specific target of aldosterone, which stimulates the production of pro-inflammatory mediators such as Transforming Growth Factor-beta-1 (TGF-beta 1), Interleukin 6 (IL-6), and Plasminogen Activator Inhibitor-1 in these cells [13]. In turn, it promotes the activation/migration of these cells in the endothelium by increasing the endothelial expression of VCAM-1 and ICAM-1 [13]. This may explain why, in SARS-CoV-2 infection, aldosterone favors the recruitment of polarized monocytes/macrophages in the endothelium of coronary vessels, thus promoting plaque rupture and myocardial infarction [18]. In this regard, it has been shown that aldosterone infusion causes the infiltration/activation of CD 68+ (a marker of M1 polarization) macrophages in coronary atherosclerotic plaques in mice. Moreover, hyperaldosteronism induces vascular and cardiac remodeling by enhancing the concentration of IL-6 produced by macrophages [18]. The detrimental role of aldosterone may also induce a monocyte shift towards CD 16 + pro-inflammatory phenotype which is the most common subpopulation of inflammatory cells isolated in severe forms of COVID-19. This phenomenon may be mediated by aldosterone-related oxidative stress through the activation of NADPH oxidase [13]. Noteworthy, CD 16 + monocytes also represent the typical cells of the “inflammaging” process that are characterized by an enhanced pro-inflammatory activity and reduced antigen-presenting function [19].
Another point of interest is represented by the potential role of aldosterone in exacerbating the activity of DCs in SARS-CoV-2 infection [13]. It is known that aldosterone may induce the activation of DCs which regulate both the innate and adaptive immunity responses. In fact, these cells recognize Pathogen Associated Molecular Patterns and Damage-associated Molecular Patterns induced by pathogens’ pyroptosis through their innate receptors, such as Toll-like receptors (TLRs) and NOD-like receptors, and largely enhance T-cell response in inflammatory sites and in lymph nodes [20].
In vivo, DCs pre-treated with aldosterone show an increased capacity to activate CD 8+ T cells response and to enhance Th 17 polarization of CD 4+ T cells [21]. Additionally, this hormone can reduce regulatory T cells (T-reg), that in physiological conditions act as important immune suppressors and can lead to a reduction of vascular damages induced by aldosterone [22]. The decrease and dysregulation of T-reg have been postulated in COVID-19 infection, especially in older hospitalized patients [23]. The latter evidence suggests that although the percentage of CD 4+ T-regs in peripheral blood is increased in elderly population compared to young individuals, the immune functions are partially impaired [24]. In fact, T-reg may inhibit IFN-γ-producing CD4+ T cells but they seem to fail in the suppression of IL-17-producing CD4+ T in elders. Interestingly, this mechanism could also explain the reduced response of elderly population to vaccination. In addition, T-regs can suppress DCs function by downregulating activation markers such as CD40, CD80, and CD86, resulting in a reduced antigen-presenting function [24]. In conclusion, in COVID-19 older patients, aldosterone may downregulate T-reg number, promoting vascular injury and, in turn, enhancing DCs function and a hyperinflammatory state. Importantly, DCs are able to maintain their activity and capacity to release cytokines despite aging and immunosenescence; thus, they could represent the most important APCs in the early phases of SARS-CoV-2 infection in elderly [20]. This mechanism could be used for the creation of new DC vaccines against COVID-19 with more effectiveness mostly in older patients with a poor antibody response to the vaccines currently present [25]. Moreover, aldosterone induces the secretion of IL-6 and TGF-beta in DCs by stimulating mitogen-activated protein kinase pathway [13] and, in primary hyperaldosteronism, the high levels of this hormone correlate with the highest production of IL-6 [26]. Hyperactivation of both innate and adaptive immunity is strongly involved in COVID-19 pneumonia and correlates with the worst prognosis [27].
Finally, SARS-CoV-2 may directly increase the production of aldosterone through activation of RAAS, and we hypothesize that the main site for production might be in the endothelial cells of pulmonary vessel. Higher levels of aldosterone may induce severe forms of COVID-19, especially in older patients, by promoting the inflammatory response and inducing electrolyte disorders such as hypokalemia. Therefore, elderly COVID-19 patients are exposed to higher risk of complications and sudden death. Further studies are necessary to evaluate in vivo and in vitro effects of aldosterone on SARS-CoV-2 affected cells and to test the potential beneficial effects of mineralocorticoid receptors antagonists (MRAs) in COVID-19 and the related cardiac complications. Indeed, to date, the effects of MRAs in COVID-19 remain controversial [16, 29]. MRAs and the other RAASi such as ACE-inhibitors (ACE-I) and Angiotensin Receptor Blockers (ARBs) have been widely analyzed in COVID-19. During the first phase of COVID-19 pandemic, a great number of patients had improperly decided to discontinue RAASi treatment as these drugs may upregulate ACE 2 expression and levels. Conversely, many studies have demonstrated that there is no association between RAASi use and the risk of COVID-19 [16, 29, 30]. To date, only ACE-I and ARBs have shown a protective role by reducing risk of hospitalization and mortality for COVID-19 [29], while the therapeutic benefit of MRA treatment such as spironolactone in COVID-19 pneumonia has not been proven yet. Noteworthy, in these studies, the majority of patients on chronic treatment with MRAs coincided with the frailest population with multiple comorbidities, such as heart failure [29]. In addition, no research has tested de novo administration of MRAs in patients with COVID-19 pneumonia. The use of MRAs in COVID-19 may have some advantages compared to ACE-I and ARBs. Indeed, despite their greater effect in upregulating ACE 2 compared to ACE-I and ARBs, MRAs have demonstrated to increase circulating ACE 2 levels by stimulating ADAM metallopeptidase domain 17 protein and, thus, ACE 2 shedding. In turn, circulating ACE 2 might bind SARS-CoV-2 as a competitive interceptor, as explained above [29]. In addition, MRAs represent the most effective drugs in inhibiting aldosterone effects and levels since this hormone can persist elevated in patients on treatment with ACE-I and ARBs due to the phenomenon of “aldosterone escape” [15]. Noteworthy, MRAs such as spironolactone may have additional mechanism in SARS-CoV-2 inhibition due to their anti-androgenic activity. Indeed, these drugs can suppress the expression of type II transmembrane serine protease TMPRSS2, that represents an androgen-regulated protease involved in COVID-19 infection. Interestingly, TMPRSS2 has shown to increase viral uptake of SARS-CoV-2 in target cells by promoting membrane fusion of Spike glycoprotein through a proteolytic cleavage between the S1 and S2 subunits and, also, by cleaving ACE 2 that, in turn, activates cathepsin L-dependent pathway [31]. Therefore, the inhibition of these pathways induced by MRAs could suppress or reduce viral entry in human cells and might have beneficial effects in COVID-19 infection and in acute respiratory distress syndrome [27, 32]. In conclusion, a more comprehensive evaluation of aldosterone activity and its inhibition in COVID-19 pneumonia seems to be necessary, and this opinion piece could pave the way for new therapeutical strategies.
Data sharing is not applicable to this article because no new data were created or analyzed in this study.
References
Marijon E, Karam N, Jost D, Perrot D, Benoit B, Derkenne C et al. Out-of-hospital cardiac arrest during the COVID-19 pandemic in Paris, France: a population-based, observational study. Lancet Public Health. 2020. S2468-2667. 30117-1. https://doi.org/10.1016/S2468-2667(20)30117-1 .
Lai PH, Lancet EA, Weiden MD, Webber MP, Zeig-Owens R, Hall CB, Prezant DJ. Characteristics associated with out-of-hospital cardiac arrests and resuscitations during the novel coronavirus disease 2019 pandemic in New York city. JAMA Cardiol. 2020;10(1001). https://doi.org/10.1001/jamacardio.2020.2488.
Bagcchi S. The world’s largest COVID-19 vaccination campaign. Lancet Infect Dis. 2021 Mar;21(3):323. https://doi.org/10.1016/S1473-3099(21)00081-5.
Palmieri L, Vanacore N, Donfrancesco C, Lo Noce C, Canevelli M, Punzo O et al. Clinical characteristics of hospitalized individuals dying with covid-19 by age group in italy. J Gerontol A Biol Sci Med Sci. 2020. glaa146. https://doi.org/10.1093/gerona/glaa146.
Carpenter A, Chambers OJ, El Harchi A, Bond R, Hanington O, Harmer SC, et al. COVID-19 management and arrhythmia: risks and challenges for clinicians treating patients affected by SARS-CoV-2. Front Cardiovasc Med. 2020;7:85. https://doi.org/10.3389/fcvm.2020.00085.
Baldi E, Sechi GM, Mare C, Canevari F, Brancaglione A, Primi R et al. COVID-19 kills at home: the close relationship between the epidemic and the increase of out-of-hospital cardiac arrests. Eur Heart J. 2020. ehaa508. https://doi.org/10.1093/eurheartj/ehaa508.
Ferreira JP, Butler J, Rossignol P, Pitt B, Anker SD, Kosiborod M, et al. Abnormalities of potassium in heart failure: Jacc state-of-the-art review. J Am Coll Cardiol. 2020;75:2836–50. https://doi.org/10.1016/j.jacc.2020.04.021.
Chen D, Li X, Song Q, Hu C, Su F, Dai J, et al. Assessment of hypokalemia and clinical characteristics in patients with coronavirus disease 2019 in wenzhou, china. JAMA Netw Open. 2020;3:e2011122. https://doi.org/10.1001/jamanetworkopen.2020.11122.
Alexandre J, Cracowski JL, Richard V, Bouhanick B; ‘Drugs, COVID-19’ working group of the French Society of Pharmacology, Therapeutics. Renin-angiotensin-aldosterone system and COVID-19 infection. Ann Endocrinol (Paris). 2020 Jun;81(2-3):63-67. https://doi.org/10.1016/j.ando.2020.04.005.
Williams B, Zhang Y. Hypertension, renin-angiotensin-aldosterone system inhibition, and COVID-19. Lancet. 2020 May 30;395(10238):1671–3. https://doi.org/10.1016/S0140-6736(20)31131-4.
Brojakowska A, Narula J, Shimony R, Bander J. Clinical implications of sars-cov-2 interaction with renin angiotensin system: jacc review topic of the week. J Am Coll Cardiol. 2020;75:3085–95. https://doi.org/10.1016/j.jacc.2020.04.028.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–8. https://doi.org/10.1016/S0140-6736(20)30937-5.
Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nature reviews Nephrology. 2013;9:459–69. https://doi.org/10.1038/nrneph.2013.110.
Koch CA, Fulop T. Clinical aspects of changes in water and sodium homeostasis in the elderly. Rev Endocr Metab Disord. 2017;18:49–66. https://doi.org/10.1007/s11154-017-9420-5.
Schrier RW. Aldosterone ‘escape’ vs ‘breakthrough’. Nat Rev Nephrol. 2010;6:61. https://doi.org/10.1038/nrneph.2009.228.
Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-angiotensin-aldosterone system inhibitors in patients with Covid-19. N Engl J Med. 2020 Apr 23;382(17):1653–9. https://doi.org/10.1056/NEJMsr2005760.
Le HH, El-Khatib C, Mombled M, Guitarian F, Al-Gobari M, Fall M, et al. Impact of aldosterone antagonists on sudden cardiac death prevention in heart failure and post-myocardial infarction patients: a systematic review and meta-analysis of randomized controlled trials. PLoS One. 2016;11:e0145958. https://doi.org/10.1371/journal.pone.0145958.
Van der Heijden CDCC, Deinum J, Joosten LAB, Netea MG, Riksen NP. The mineralocorticoid receptor as a modulator of innate immunity and atherosclerosis. Cardiovasc Res. 2018;114:944–53. https://doi.org/10.1093/cvr/cvy092.
De Maeyer RPH, Chambers ES. The impact of ageing on monocytes and macrophages. Immunol Lett. 2021 Feb;230:1–10. https://doi.org/10.1016/j.imlet.2020.12.003.
Janssen N, Derhovanessian E, Demuth I, Arnaout F, Steinhagen-Thiessen E, Pawelec G. Responses of dendritic cells to tlr-4 stimulation are maintained in the elderly and resist the effects of cmv infection seen in the young. J Gerontol A Biol Sci Med Sci. 2016;71:1117–23. https://doi.org/10.1093/gerona/glv119.
Herrada AA, Contreras FJ, Marini NP, Amador CA, González PA, Cortés CM, et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J Immunol. 2010;184:191–202. https://doi.org/10.4049/jimmunol.0802886.
Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, Neves MF, Laurant P, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012 Feb;59(2):324–30. https://doi.org/10.1161/HYPERTENSIONAHA.111.181123.
Meckiff BJ, Ramírez-Suástegui C, Fajardo V, Chee SJ, Kusnadi A, Simon H, Eschweiler S, Grifoni A, Pelosi E, Weiskopf D, Sette A, Ay F, Seumois G, Ottensmeier CH, Vijayanand P. Imbalance of regulatory and cytotoxic SARS-CoV-2-reactive CD4+ T cells in COVID-19. Cell. 2020 Nov 25;183(5):1340-1353.e16. https://doi.org/10.1016/j.cell.2020.10.001 .
Jagger A, Shimojima Y, Goronzy JJ, Weyand CM. Regulatory T cells and the immune aging process: a mini-review. Gerontology. 2014;60(2):130–7. https://doi.org/10.1159/000355303.
Zhou Q, Gu H, Sun S, Zhang Y, Hou Y, Li C, Zhao Y, Ma P, Lv L, Aji S, Sun S, Wang X, Zhan L. Large-sized graphene oxide nanosheets increase DC-T-cell synaptic contact and the efficacy of DC vaccines against SARS-CoV-2. Adv Mater. 2021 Aug;16:e2102528. https://doi.org/10.1002/adma.202102528.
Chou CH, Hung CS, Liao CW, Wei LH, Chen CW, Shun CT, et al. IL-6 trans-signalling contributes to aldosterone-induced cardiac fibrosis. Cardiovasc Res. 2018;114:690–702. https://doi.org/10.1093/cvr/cvy013.
Vardhana SA, Wolchok JD. The many faces of the anti-COVID immune response. J Exp Med. 2020;217:20200678. https://doi.org/10.1084/jem.20200678.
Liaudet L, Szabo C. Blocking mineralocorticoid receptor with spironolactone may have a wide range of therapeutic actions in severe COVID-19 disease. Crit Care. 2020;24:318. https://doi.org/10.1186/s13054-020-03055-6.
Savarese G, Benson L, Sundström J, Lund LH. Association between renin-angiotensin-aldosterone system inhibitor use and COVID-19 hospitalization and death: a 1.4 million patient nationwide registry analysis. Eur J Heart Fail. 2021 Mar;23(3):476–85. https://doi.org/10.1002/ejhf.2060.
Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Renin-angiotensin-aldosterone system blockers and the risk of Covid-19. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2006923.
Zipeto D, Palmeira JDF, Argañaraz GA, Argañaraz ER. ACE2/ADAM17/TMPRSS2 interplay may be the main risk factor for COVID-19. Front Immunol. 2020 Oct 7;11:576745. https://doi.org/10.3389/fimmu.2020.576745.
Campana P, Flocco V, Aruta F, Cacciatore F, Abete P. Can aldosterone increase interleukin-6 levels in Covid-19 pneumonia? J Med Virol. 2021 Feb;93(2):622–3. https://doi.org/10.1002/jmv.26382.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
All procedures performed in the study were in accordance with the ethical standards of the institutional or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards and conformed to the Declaration of Helsinki on human research.
Conflict of interest
The authors declare no competing interests.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Campana, P., Palaia, M.E., Conte, M. et al. The elderly at risk: aldosterone as modulator of the immune response to SARS-CoV-2 infection. GeroScience 44, 567–572 (2022). https://doi.org/10.1007/s11357-021-00481-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-021-00481-4