
COVID-19 and the Immune Response: A Multi-Phasic Approach to the Treatment of COVID-19

 , and
, and
Abstract
:1. Introduction
2. Innate Immune Response
2.1. Complement Activation
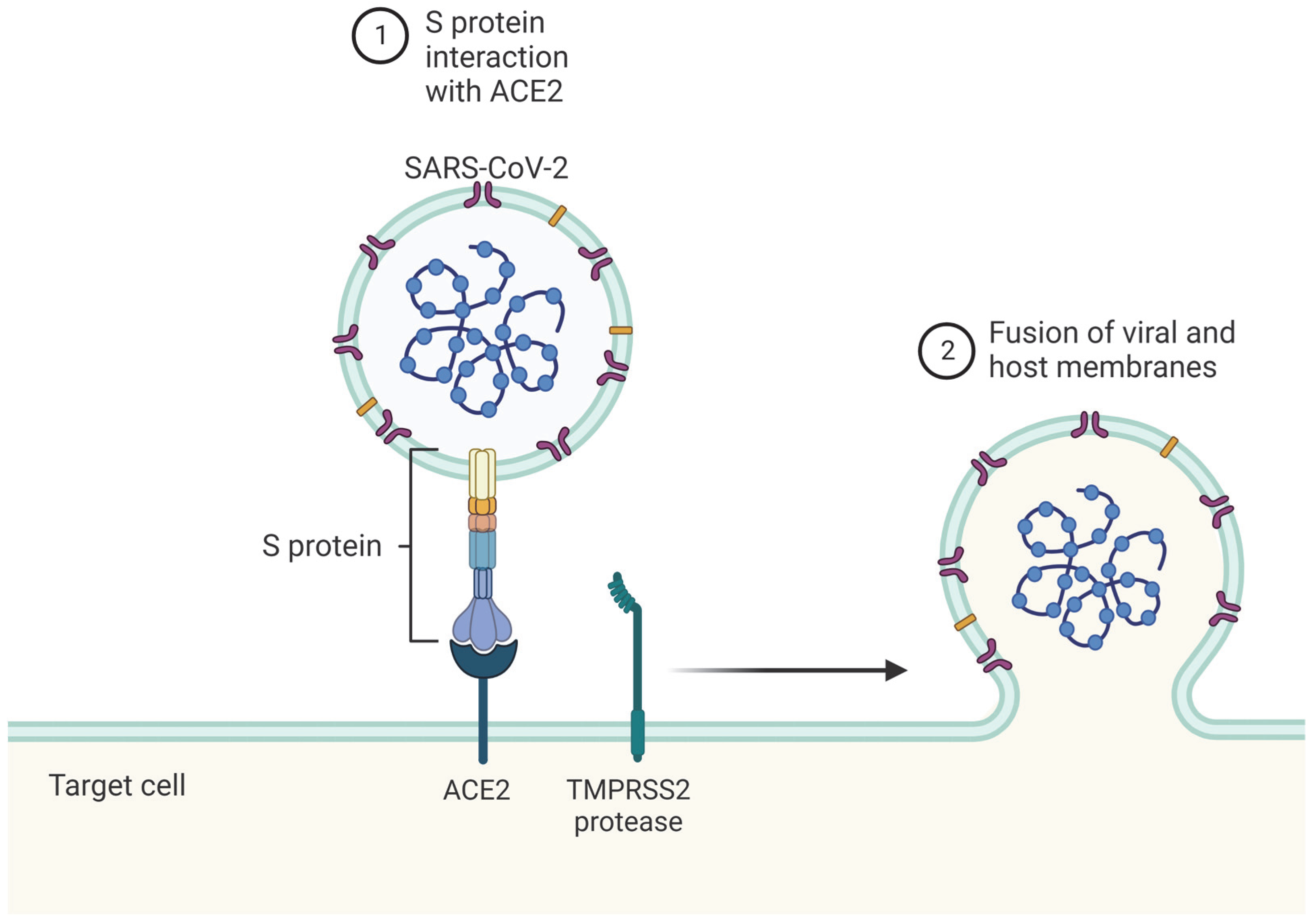
2.2. Immune Detection of SARS-CoV-2
2.3. SARS-CoV-2 Avoids Innate Immunity
2.4. IFN and IL Response
3. Adaptive Immune Response during COVID
3.1. Adaptive Immune Response Time to COVID-19
3.2. Antibodies for COVID-19
3.3. CD8+ & CD4+ T-Cell Response for COVID-19
3.4. Natural Killer Cells’ Response to COVID-19
4. Immune Clearance of COVID-19
4.1. Immune Cells through Infection
4.2. Early Infection vs. Late Infection
4.3. Factors Necessary for Viral Clearence
4.4. Viral Clearance of Different SARS-CoV-2 Strains
4.5. Immune Exhaustion
4.6. Tissue-Dependent Responses to COVID-19
4.7. Future Research
5. Post-Acute Sequelae of COVID-19
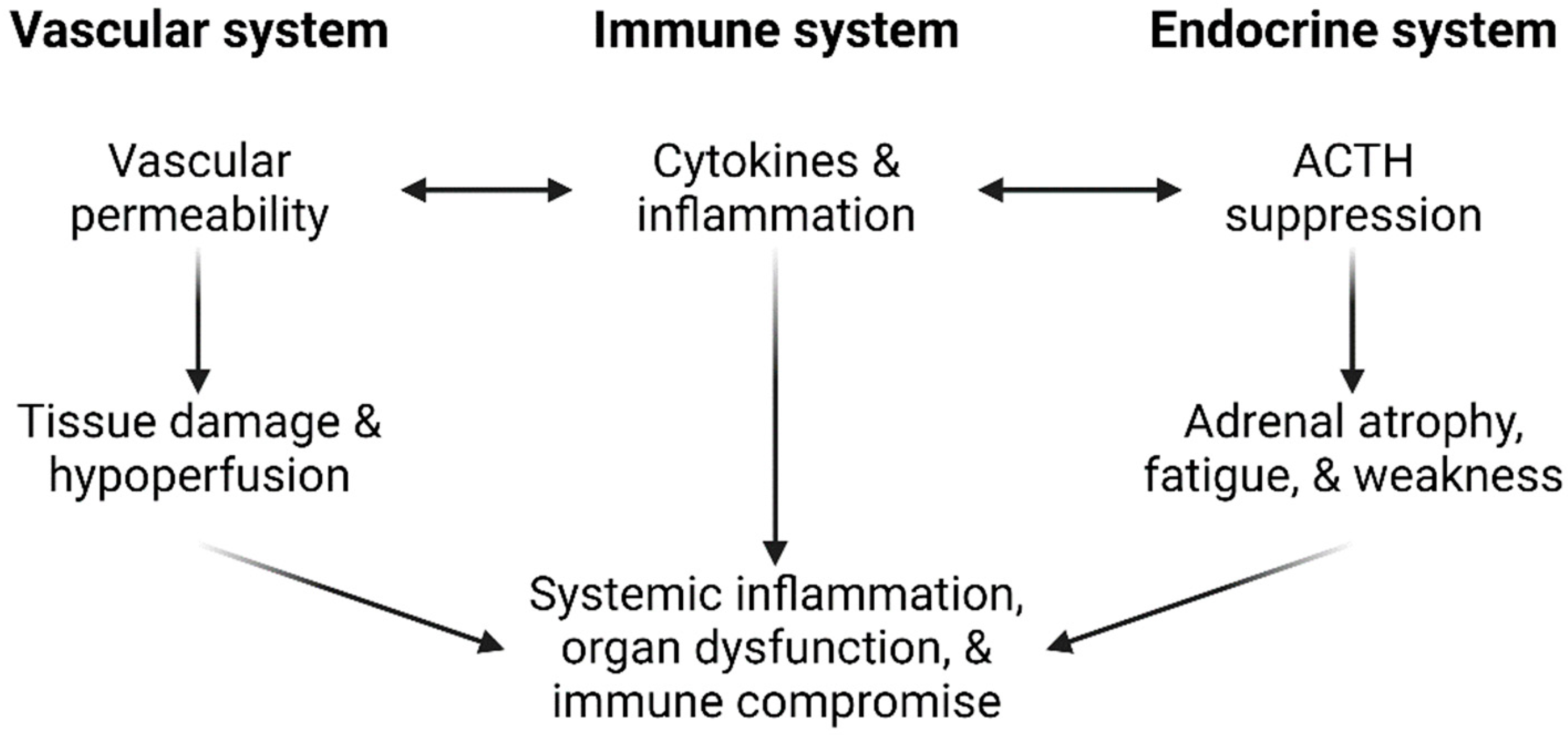
5.1. Potential Causes
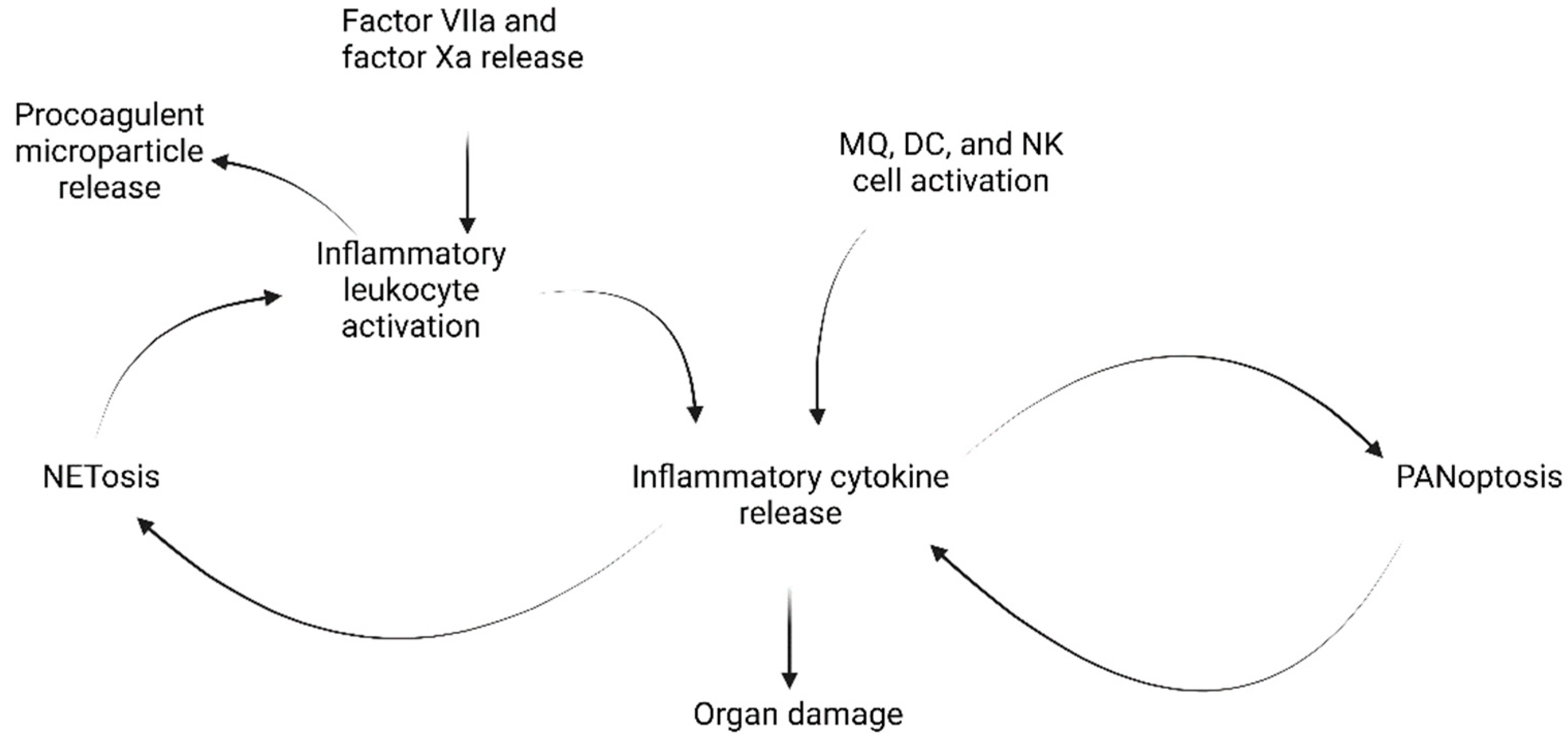
5.2. The Cytokine Storm and Hyperinflammation
- Perpetuated activation of lymphocytes and macrophages causing immune dysregulation;
- Large secretions of cytokines caused by such perpetuated activation;
- Overwhelming systemic inflammation and multi-organ failure with high mortality.
5.3. Myalgic Encephalomyelitis
5.4. Other Explanations for PASC
6. Treatments of COVID-19
6.1. Treatments of Acute COVID-19 and Early Phases of Disease
6.2. Treatment of Post-Acute Sequelae of COVID-19 and Late Phases of Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, M. Track the Coronavirus Outbreak on Johns Hopkins Live Dashboard. Available online: https://www.medpagetoday.com/infectiousdisease/publichealth/84698 (accessed on 29 July 2022).
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Kim, S. COVID-19 Drug Development. J. Microbiol. Biotechnol. 2022, 32, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef]
- Alene, M.; Yismaw, L.; Assemie, M.A.; Ketema, D.B.; Gietaneh, W.; Birhan, T.Y. Serial interval and incubation period of COVID-19: A systematic review and meta-analysis. BMC Infect. Dis. 2021, 21, 257. [Google Scholar] [CrossRef]
- Griffin, D.O.; Brennan-Rieder, D.; Ngo, B.; Kory, P.; Confalonieri, M.; Shapiro, L.; Iglesias, J.; Dube, M.; Nanda, N.; In, G.K.; et al. The Importance of Understanding the Stages of COVID-19 in Treatment and Trials. AIDS Rev. 2021, 23, 40–47. [Google Scholar] [CrossRef]
- Sahu, A.K.; Mathew, R.; Bhat, R.; Malhotra, C.; Nayer, J.; Aggarwal, P.; Galwankar, S. Steroids use in non-oxygen requiring COVID-19 patients: A systematic review and meta-analysis. QJM 2021, 114, 455–463. [Google Scholar] [CrossRef]
- Matricardi, P.M.; Dal Negro, R.W.; Nisini, R. The first, holistic immunological model of COVID-19: Implications for prevention, diagnosis, and public health measures. Pediatr. Allergy Immunol. 2020, 31, 454–470. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Stasi, C.; Fallani, S.; Voller, F.; Silvestri, C. Treatment for COVID-19: An overview. Eur. J. Pharmacol. 2020, 889, 173644. [Google Scholar] [CrossRef]
- Sudre, C.H.; Murray, B.; Varsavsky, T.; Graham, M.S.; Penfold, R.S.; Bowyer, R.C.; Pujol, J.C.; Klaser, K.; Antonelli, M.; Canas, L.S.; et al. Attributes and predictors of long COVID. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef] [PubMed]
- Duan, K.; Liu, B.; Li, C.; Zhang, H.; Yu, T.; Qu, J.; Zhou, M.; Chen, L.; Meng, S.; Hu, Y.; et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc. Natl. Acad. Sci. USA 2020, 117, 9490–9496. [Google Scholar] [CrossRef] [Green Version]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5, e140711. [Google Scholar] [CrossRef] [PubMed]
- Gralinski, L.E.; Sheahan, T.P.; Morrison, T.E.; Menachery, V.D.; Jensen, K.; Leist, S.R.; Whitmore, A.; Heise, M.T.; Baric, R.S. Complement Activation Contributes to Severe Acute Respiratory Syndrome Coronavirus Pathogenesis. mBio 2018, 9, e01753-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Zhao, G.; Song, N.; Li, P.; Chen, Y.; Guo, Y.; Li, J.; Du, L.; Jiang, S.; Guo, R.; et al. Blockade of the C5a-C5aR axis alleviates lung damage in hDPP4-transgenic mice infected with MERS-CoV. Emerg. Microbes Infect. 2018, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Hu, M.; Zhang, X.; Li, H.; Zhu, L.; Liu, H.; Dong, Q.; Zhang, Z.; Wang, Z.; Hu, Y.; et al. Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. medRxiv 2020. [Google Scholar] [CrossRef]
- Ramlall, V.; Thangaraj, P.M.; Meydan, C.; Foox, J.; Butler, D.; Kim, J.; May, B.; De Freitas, J.K.; Glicksberg, B.S.; Mason, C.E.; et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609–1615. [Google Scholar] [CrossRef]
- Cugno, M.; Meroni, P.L.; Gualtierotti, R.; Griffini, S.; Grovetti, E.; Torri, A.; Panigada, M.; Aliberti, S.; Blasi, F.; Tedesco, F.; et al. Complement activation in patients with COVID-19: A novel therapeutic target. J. Allergy Clin. Immunol. 2020, 146, 215–217. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Xiang, P.; Pu, L.; Xiong, H.; Li, C.; Zhang, M.; Tan, J.; Xu, Y.; Song, R.; et al. Neutrophil-to-lymphocyte ratio predicts critical illness patients with 2019 coronavirus disease in the early stage. J. Transl. Med. 2020, 18, 206. [Google Scholar] [CrossRef]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef]
- Bortolotti, D.; Gentili, V.; Rizzo, S.; Schiuma, G.; Beltrami, S.; Strazzabosco, G.; Fernandez, M.; Caccuri, F.; Caruso, A.; Rizzo, R. TLR3 and TLR7 RNA Sensor Activation during SARS-CoV-2 Infection. Microorganisms 2021, 9, 1820. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Petruk, G.; Puthia, M.; Petrlova, J.; Samsudin, F.; Stromdahl, A.C.; Cerps, S.; Uller, L.; Kjellstrom, S.; Bond, P.J.; Schmidtchen, A.A. SARS-CoV-2 spike protein binds to bacterial lipopolysaccharide and boosts proinflammatory activity. J. Mol. Cell Biol. 2020, 12, 916–932. [Google Scholar] [CrossRef]
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Maglorius Renkilaraj, M.R.L.; Zhang, P.; Meertens, L.; Bolze, A.; Materna, M.; et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348. [Google Scholar] [CrossRef]
- Mattoo, S.U.; Kim, S.J.; Ahn, D.G.; Myoung, J. Escape and Over-Activation of Innate Immune Responses by SARS-CoV-2: Two Faces of a Coin. Viruses 2022, 14, 530. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045e1039. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339.e1321. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e117. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Wang, X.; Sahu, K.K.; Cerny, J. Coagulopathy, endothelial dysfunction, thrombotic microangiopathy and complement activation: Potential role of complement system inhibition in COVID-19. J. Thromb. Thrombolysis 2021, 51, 657–662. [Google Scholar] [CrossRef]
- Triggle, C.R.; Bansal, D.; Ding, H.; Islam, M.M.; Farag, E.; Hadi, H.A.; Sultan, A.A. A Comprehensive Review of Viral Characteristics, Transmission, Pathophysiology, Immune Response, and Management of SARS-CoV-2 and COVID-19 as a Basis for Controlling the Pandemic. Front. Immunol. 2021, 12, 631139. [Google Scholar] [CrossRef]
- Kurahashi, Y.; Sutandhio, S.; Furukawa, K.; Tjan, L.H.; Iwata, S.; Sano, S.; Tohma, Y.; Ohkita, H.; Nakamura, S.; Nishimura, M.; et al. Cross-Neutralizing Breadth and Longevity Against SARS-CoV-2 Variants After Infections. Front. Immunol. 2022, 13, 773652. [Google Scholar] [CrossRef]
- Muecksch, F.; Weisblum, Y.; Barnes, C.O.; Schmidt, F.; Schaefer-Babajew, D.; Wang, Z.; JC, C.L.; Flyak, A.I.; DeLaitsch, A.T.; Huey-Tubman, K.E.; et al. Affinity maturation of SARS-CoV-2 neutralizing antibodies confers potency, breadth, and resilience to viral escape mutations. Immunity 2021, 54, 1853–1868.e1857. [Google Scholar] [CrossRef]
- Moriyama, S.; Adachi, Y.; Sato, T.; Tonouchi, K.; Sun, L.; Fukushi, S.; Yamada, S.; Kinoshita, H.; Nojima, K.; Kanno, T.; et al. Temporal maturation of neutralizing antibodies in COVID-19 convalescent individuals improves potency and breadth to circulating SARS-CoV-2 variants. Immunity 2021, 54, 1841–1852.e1844. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.e2379. [Google Scholar] [CrossRef] [PubMed]
- Barda, N.; Dagan, N.; Cohen, C.; Hernan, M.A.; Lipsitch, M.; Kohane, I.S.; Reis, B.Y.; Balicer, R.D. Effectiveness of a third dose of the BNT162b2 mRNA COVID-19 vaccine for preventing severe outcomes in Israel: An observational study. Lancet 2021, 398, 2093–2100. [Google Scholar] [CrossRef]
- Pellicano, C.; Campagna, R.; Oliva, A.; Leodori, G.; Miglionico, M.; Colalillo, A.; Mezzaroma, I.; Mastroianni, C.M.; Turriziani, O.; Rosato, E. Antibody response to BNT162b2 SARS-CoV-2 mRNA vaccine in adult patients with systemic sclerosis. Clin. Rheumatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Boyarsky, B.J.; Werbel, W.A.; Avery, R.K.; Tobian, A.A.R.; Massie, A.B.; Segev, D.L.; Garonzik-Wang, J.M. Immunogenicity of a Single Dose of SARS-CoV-2 Messenger RNA Vaccine in Solid Organ Transplant Recipients. JAMA 2021, 325, 1784–1786. [Google Scholar] [CrossRef]
- Painter, M.M.; Mathew, D.; Goel, R.R.; Apostolidis, S.A.; Pattekar, A.; Kuthuru, O.; Baxter, A.E.; Herati, R.S.; Oldridge, D.A.; Gouma, S.; et al. Rapid induction of antigen-specific CD4(+) T cells is associated with coordinated humoral and cellular immunity to SARS-CoV-2 mRNA vaccination. Immunity 2021, 54, 2133–2142.e2133. [Google Scholar] [CrossRef] [PubMed]
- Pavan Kumar, N.; Moideen, K.; Nancy, A.; Selvaraj, N.; Renji, R.M.; Munisankar, S.; Thangaraj, J.W.V.; Muthusamy, S.K.; Kumar, C.P.G.; Bhatnagar, T.; et al. Enhanced SARS-CoV-2-Specific CD4(+) T Cell Activation and Multifunctionality in Late Convalescent COVID-19 Individuals. Viruses 2022, 14, 511. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J.; Brokstad, K.A. Not just antibodies: B cells and T cells mediate immunity to COVID-19. Nat. Rev. Immunol. 2020, 20, 581–582. [Google Scholar] [CrossRef]
- Bao, C.; Tao, X.; Cui, W.; Hao, Y.; Zheng, S.; Yi, B.; Pan, T.; Young, K.H.; Qian, W. Natural killer cells associated with SARS-CoV-2 viral RNA shedding, antibody response and mortality in COVID-19 patients. Exp. Hematol. Oncol. 2021, 10, 5. [Google Scholar] [CrossRef]
- Rydyznski Moderbacher, C.; Ramirez, S.I.; Dan, J.M.; Grifoni, A.; Hastie, K.M.; Weiskopf, D.; Belanger, S.; Abbott, R.K.; Kim, C.; Choi, J.; et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell 2020, 183, 996–1012.e1019. [Google Scholar] [CrossRef]
- Paces, J.; Strizova, Z.; Smrz, D.; Cerny, J. COVID-19 and the immune system. Physiol. Res. 2020, 69, 379–388. [Google Scholar] [CrossRef]
- Bolouri, H.; Speake, C.; Skibinski, D.; Long, S.A.; Hocking, A.M.; Campbell, D.J.; Hamerman, J.A.; Malhotra, U.; Buckner, J.H.; Benaroya Research Institute, C.-R.T. The COVID-19 immune landscape is dynamically and reversibly correlated with disease severity. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Rangchaikul, P.; Venketaraman, V. SARS-CoV-2 and the Immune Response in Pregnancy with Delta Variant Considerations. Infect. Dis. Rep. 2021, 13, 993–1008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T-cells and inflammatory monocytes incite inflammatory storms in severe COVID-19 patients. Natl. Sci. Rev. 2020, 7, 998–1002. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.; Li, J.; Venzon, D.J.; Berzofsky, J.A. SARS-CoV-2 Spike Protein Suppresses ACE2 and Type I Interferon Expression in Primary Cells From Macaque Lung Bronchoalveolar Lavage. Front. Immunol. 2021, 12, 658428. [Google Scholar] [CrossRef] [PubMed]
- Vardhana, S.A.; Wolchok, J.D. The many faces of the anti-COVID immune response. J. Exp. Med. 2020, 217, e20200678. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Kanneganti, T.D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef]
- Israelow, B.; Mao, T.; Klein, J.; Song, E.; Menasche, B.; Omer, S.B.; Iwasaki, A. Adaptive immune determinants of viral clearance and protection in mouse models of SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021, 34, 108728. [Google Scholar] [CrossRef] [PubMed]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef] [Green Version]
- Wastnedge, E.A.N.; Reynolds, R.M.; van Boeckel, S.R.; Stock, S.J.; Denison, F.C.; Maybin, J.A.; Critchley, H.O.D. Pregnancy and COVID-19. Physiol. Rev. 2021, 101, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.; Chen, H.; Williams, C.M. Identification of Transcription Factors Regulating SARS-CoV-2 Tropism Factor Expression by Inferring Cell-Type-Specific Transcriptional Regulatory Networks in Human Lungs. Viruses 2022, 14, 837. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein. J. Med. Virol. 2022, 94, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Li, X.; Zhang, L.; Wan, S.; Zhang, L.; Zhou, F. SARS-CoV-2 Omicron variant: Recent progress and future perspectives. Signal Transduct. Target Ther. 2022, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, A.C.; Buchan, S.A.; Daneman, N.; Brown, K.A. Estimates of SARS-CoV-2 Omicron Variant Severity in Ontario, Canada. JAMA 2022, 327, 1286–1288. [Google Scholar] [CrossRef] [PubMed]
- Veneti, L.; Boas, H.; Brathen Kristoffersen, A.; Stalcrantz, J.; Bragstad, K.; Hungnes, O.; Storm, M.L.; Aasand, N.; Ro, G.; Starrfelt, J.; et al. Reduced risk of hospitalisation among reported COVID-19 cases infected with the SARS-CoV-2 Omicron BA.1 variant compared with the Delta variant, Norway, December 2021 to January 2022. Euro Surveill. 2022, 27, 2200077. [Google Scholar] [CrossRef] [PubMed]
- Iuliano, A.D.; Brunkard, J.M.; Boehmer, T.K.; Peterson, E.; Adjei, S.; Binder, A.M.; Cobb, S.; Graff, P.; Hidalgo, P.; Panaggio, M.J.; et al. Trends in Disease Severity and Health Care Utilization During the Early Omicron Variant Period Compared with Previous SARS-CoV-2 High Transmission Periods—United States, December 2020–January 2022. MMWR Morb. Mortal Wkly. Rep. 2022, 71, 146–152. [Google Scholar] [CrossRef]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 And Novel Therapeutics Against Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Gandhi, R.T.; Lynch, J.B.; Del Rio, C. Mild or Moderate COVID-19. N. Engl. J. Med. 2020, 383, 1757–1766. [Google Scholar] [CrossRef]
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Lopez-Collazo, E.; Avendano-Ortiz, J.; Martin-Quiros, A.; Aguirre, L.A. Immune Response and COVID-19: A mirror image of Sepsis. Int. J. Biol. Sci. 2020, 16, 2479–2489. [Google Scholar] [CrossRef]
- Shen, X.R.; Geng, R.; Li, Q.; Chen, Y.; Li, S.F.; Wang, Q.; Min, J.; Yang, Y.; Li, B.; Jiang, R.D.; et al. ACE2-independent infection of T lymphocytes by SARS-CoV-2. Signal Transduct. Target. Ther. 2022, 7, 83. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef]
- Shaaban, M.; Othman, H.; Ibrahim, T.; Ali, M.; Abdelmoaty, M.; Abdel-Kawi, A.R.; Mostafa, A.; El Nakeeb, A.; Emam, H.; Refaat, A. Immune Checkpoint Regulators: A New Era Toward Promising Cancer Therapy. Curr. Cancer Drug Targets 2020, 20, 429–460. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, R.; Carrette, F.; Barraza, M.L.; Otero, D.C.; Magana, J.; Bosenberg, M.W.; Swain, S.L.; Bradley, L.M. PSGL-1 Is an Immune Checkpoint Regulator that Promotes T Cell Exhaustion. Immunity 2016, 44, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezeshki, P.S.; Rezaei, N. Immune checkpoint inhibition in COVID-19: Risks and benefits. Expert Opin. Biol. Ther. 2021, 21, 1173–1179. [Google Scholar] [CrossRef]
- Shen, C.; Li, Q.; Wei, Y.; Li, Y.; Li, J.; Tao, J. Management of immune checkpoint therapy for patients with cancer in the face of COVID-19. J. Immunother. Cancer 2020, 8, e001593. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.; Goncalves, P.; Charbit, B.; Grzelak, L.; Beretta, M.; Planchais, C.; Bruel, T.; Rouilly, V.; Bondet, V.; Hadjadj, J.; et al. Distinct systemic and mucosal immune responses during acute SARS-CoV-2 infection. Nat. Immunol. 2021, 22, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Logue, J.K.; Franko, N.M.; McCulloch, D.J.; McDonald, D.; Magedson, A.; Wolf, C.R.; Chu, H.Y. Sequelae in Adults at 6 Months After COVID-19 Infection. JAMA Netw. Open 2021, 4, e210830. [Google Scholar] [CrossRef]
- Tenforde, M.W.; Billig Rose, E.; Lindsell, C.J.; Shapiro, N.I.; Files, D.C.; Gibbs, K.W.; Prekker, M.E.; Steingrub, J.S.; Smithline, H.A.; Gong, M.N.; et al. Characteristics of Adult Outpatients and Inpatients with COVID-19—11 Academic Medical Centers, United States, March–May 2020. MMWR Morb. Mortal Wkly. Rep. 2020, 69, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Carfi, A.; Bernabei, R.; Landi, F.; Gemelli Against, C.-P.-A.C.S.G. Persistent Symptoms in Patients After Acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Taquet, M.; Dercon, Q.; Luciano, S.; Geddes, J.R.; Husain, M.; Harrison, P.J. Incidence, co-occurrence, and evolution of long-COVID features: A 6-month retrospective cohort study of 273,618 survivors of COVID-19. PLoS Med. 2021, 18, e1003773. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Romieu, A.C.; Carton, T.W.; Saydah, S.; Azziz-Baumgartner, E.; Boehmer, T.K.; Garret, N.Y.; Bailey, L.C.; Cowell, L.G.; Draper, C.; Mayer, K.H.; et al. Prevalence of Select New Symptoms and Conditions Among Persons Aged Younger Than 20 Years and 20 Years or Older at 31 to 150 Days After Testing Positive or Negative for SARS-CoV-2. JAMA Netw. Open 2022, 5, e2147053. [Google Scholar] [CrossRef] [PubMed]
- Sykes, D.L.; Holdsworth, L.; Jawad, N.; Gunasekera, P.; Morice, A.H.; Crooks, M.G. Post-COVID-19 Symptom Burden: What is Long-COVID and How Should We Manage It? Lung 2021, 199, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Proal, A.D.; VanElzakker, M.B. Long COVID or Post-acute Sequelae of COVID-19 (PASC): An Overview of Biological Factors That May Contribute to Persistent Symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Khalil, B.A.; Shakartalla, S.B.; Goel, S.; Madkhana, B.; Halwani, R.; Maghazachi, A.A.; AlSafar, H.; Al-Omari, B.; Al Bataineh, M.T. Immune Profiling of COVID-19 in Correlation with SARS and MERS. Viruses 2022, 14, 164. [Google Scholar] [CrossRef]
- Chen, L.Y.C.; Quach, T.T.T. COVID-19 cytokine storm syndrome: A threshold concept. Lancet Microbe 2021, 2, e49–e50. [Google Scholar] [CrossRef]
- Wu, J.; Shen, J.; Han, Y.; Qiao, Q.; Dai, W.; He, B.; Pang, R.; Zhao, J.; Luo, T.; Guo, Y.; et al. Upregulated IL-6 Indicates a Poor COVID-19 Prognosis: A Call for Tocilizumab and Convalescent Plasma Treatment. Front. Immunol. 2021, 12, 598799. [Google Scholar] [CrossRef]
- Guirao, J.J.; Cabrera, C.M.; Jimenez, N.; Rincon, L.; Urra, J.M. High serum IL-6 values increase the risk of mortality and the severity of pneumonia in patients diagnosed with COVID-19. Mol. Immunol. 2020, 128, 64–68. [Google Scholar] [CrossRef]
- Chen, L.Y.C.; Hoiland, R.L.; Stukas, S.; Wellington, C.L.; Sekhon, M.S. Confronting the controversy: Interleukin-6 and the COVID-19 cytokine storm syndrome. Eur. Respir. J. 2020, 56, 2003006. [Google Scholar] [CrossRef]
- Webb, B.J.; Peltan, I.D.; Jensen, P.; Hoda, D.; Hunter, B.; Silver, A.; Starr, N.; Buckel, W.; Grisel, N.; Hummel, E.; et al. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: A cohort study. Lancet Rheumatol. 2020, 2, e754–e763. [Google Scholar] [CrossRef]
- Boehmer, T.K.; Kompaniyets, L.; Lavery, A.M.; Hsu, J.; Ko, J.Y.; Yusuf, H.; Romano, S.D.; Gundlapalli, A.V.; Oster, M.E.; Harris, A.M. Association Between COVID-19 and Myocarditis Using Hospital-Based Administrative Data—United States, March 2020–January 2021. MMWR Morb. Mortal Wkly. Rep. 2021, 70, 1228–1232. [Google Scholar] [CrossRef] [PubMed]
- Puntmann, V.O.; Carerj, M.L.; Wieters, I.; Fahim, M.; Arendt, C.; Hoffmann, J.; Shchendrygina, A.; Escher, F.; Vasa-Nicotera, M.; Zeiher, A.M.; et al. Outcomes of Cardiovascular Magnetic Resonance Imaging in Patients Recently Recovered From Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.; Ganigara, M.; Galeotti, C.; Burns, J.; Berganza, F.M.; Hayes, D.A.; Singh-Grewal, D.; Bharath, S.; Sajjan, S.; Bayry, J. Multisystem inflammatory syndrome in children and Kawasaki disease: A critical comparison. Nat. Rev. Rheumatol. 2021, 17, 731–748. [Google Scholar] [CrossRef]
- Jason, L.A.; Mirin, A.A. Updating the National Academy of Medicine ME/CFS prevalence and economic impact figures to account for population growth and inflation. Fatigue: Biomed. Health Behav. 2021, 9, 9–13. [Google Scholar] [CrossRef]
- Stanculescu, D.; Bergquist, J. Perspective: Drawing on Findings From Critical Illness to Explain Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Med. 2022, 9, 818728. [Google Scholar] [CrossRef] [PubMed]
- Vallet, B.; Wiel, E. Endothelial cell dysfunction and coagulation. Crit. Care Med. 2001, 29, S36–S41. [Google Scholar] [CrossRef] [PubMed]
- Boonen, E.; Bornstein, S.R.; Van den Berghe, G. New insights into the controversy of adrenal function during critical illness. Lancet Diabetes Endocrinol. 2015, 3, 805–815. [Google Scholar] [CrossRef]
- Boonen, E.; Langouche, L.; Janssens, T.; Meersseman, P.; Vervenne, H.; De Samblanx, E.; Pironet, Z.; Van Dyck, L.; Vander Perre, S.; Derese, I.; et al. Impact of duration of critical illness on the adrenal glands of human intensive care patients. J. Clin. Endocrinol. Metab 2014, 99, 4214–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatto, M.C.; Persi, A.; Tung, M.; Masi, R.; Canitano, S.; Kol, A. Bradyarrhythmias in patients with SARS-CoV-2 infection: A narrative review and a clinical report. Pacing Clin. Electrophysiol. 2021, 44, 1607–1615. [Google Scholar] [CrossRef]
- Kamau-Mitchell, C. GPs need awareness about post-covid ME/CFS. BMJ 2021, 374, n1995. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Marx, W.; O’Neil, A.; Athan, E.; Walder, K.; Berk, M.; Olive, L.; Carvalho, A.F.; et al. The cytokine storms of COVID-19, H1N1 influenza, CRS and MAS compared. Can one sized treatment fit all? Cytokine 2021, 144, 155593. [Google Scholar] [CrossRef]
- Singh, V.; Sharma, B.B.; Patel, V. Pulmonary sequelae in a patient recovered from swine flu. Lung India 2012, 29, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.U.; Parida, S.; Lingaraju, M.C.; Kesavan, M.; Kumar, D.; Singh, R.K. Drug repurposing approach to fight COVID-19. Pharmacol. Rep. 2020, 72, 1479–1508. [Google Scholar] [CrossRef] [PubMed]
- Chavda, V.P.; Kapadia, C.; Soni, S.; Prajapati, R.; Chauhan, S.C.; Yallapu, M.M.; Apostolopoulos, V. A global picture: Therapeutic perspectives for COVID-19. Immunotherapy 2022, 14, 351–371. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9, eaal3653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angamo, M.T.; Mohammed, M.A.; Peterson, G.M. Efficacy and safety of remdesivir in hospitalised COVID-19 patients: A systematic review and meta-analysis. Infection 2022, 50, 27–41. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Takes Actions to Expand Use of Treatment for Outpatients with Mild-to-Moderate COVID-19. Available online: https://www.fda.gov/news-events/press-announcements/fda-takes-actions-expand-use-treatment-outpatients-mild-moderate-covid-19 (accessed on 29 July 2022).
- Low, Z.Y.; Yip, A.J.W.; Lal, S.K. Repositioning Ivermectin for COVID-19 treatment: Molecular mechanisms of action against SARS-CoV-2 replication. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166294. [Google Scholar] [CrossRef]
- Yuan, S.; Chan, C.C.; Chik, K.K.; Tsang, J.O.; Liang, R.; Cao, J.; Tang, K.; Cai, J.P.; Ye, Z.W.; Yin, F.; et al. Broad-Spectrum Host-Based Antivirals Targeting the Interferon and Lipogenesis Pathways as Potential Treatment Options for the Pandemic Coronavirus Disease 2019 (COVID-19). Viruses 2020, 12, 628. [Google Scholar] [CrossRef]
- Hwang, Y.C.; Lu, R.M.; Su, S.C.; Chiang, P.Y.; Ko, S.H.; Ke, F.Y.; Liang, K.H.; Hsieh, T.Y.; Wu, H.C. Monoclonal antibodies for COVID-19 therapy and SARS-CoV-2 detection. J. Biomed. Sci. 2022, 29, 1. [Google Scholar] [CrossRef] [PubMed]
- Jayk Bernal, A.; Gomes da Silva, M.M.; Musungaie, D.B.; Kovalchuk, E.; Gonzalez, A.; Delos Reyes, V.; Martin-Quiros, A.; Caraco, Y.; Williams-Diaz, A.; Brown, M.L.; et al. Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 2022, 386, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Gotte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Nirmatrelvir Plus Ritonavir: First Approval. Drugs 2022, 82, 585–591. [Google Scholar] [CrossRef]
- Pavan, M.; Bolcato, G.; Bassani, D.; Sturlese, M.; Moro, S. Supervised Molecular Dynamics (SuMD) Insights into the mechanism of action of SARS-CoV-2 main protease inhibitor PF-07321332. J. Enzyme Inhib. Med. Chem. 2021, 36, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Molhave, M.; Agergaard, J.; Wejse, C. Clinical Management of COVID-19 Patients—An Update. Semin. Nucl. Med. 2022, 52, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Billett, H.H.; Reyes-Gil, M.; Szymanski, J.; Ikemura, K.; Stahl, L.R.; Lo, Y.; Rahman, S.; Gonzalez-Lugo, J.D.; Kushnir, M.; Barouqa, M.; et al. Anticoagulation in COVID-19: Effect of Enoxaparin, Heparin, and Apixaban on Mortality. Thromb. Haemost. 2020, 120, 1691–1699. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Infection Phase | Clinical Presentation | Treatment Approach | Sources |
| Viral replication | Upper respiratory tract infection, fever, muscle fatigue, pain | Antiviral agents are used to decrease viral load, transmission, and prevent progression to the next phases of the disease | [10,11,12] |
| Immune hyperactivation | Dyspnea, pneumonia, vasculopathy, acute cardiac and renal damage, sepsis, secondary infections | Monoclonal antibodies, anti-coagulants, immunosuppressants, oxygen, antiviral drugs | |
| Post-Acute Sequelae of COVID-19 | Fatigue, headache, dyspnea, and anosmia | Immunosuppressants, convalescent plasma therapy | [13,14,15] |
| Toll-Like Receptor | Interaction | Source |
|---|---|---|
| TLR1 | Binds SARS-CoV-2 spike protein | [28] |
| TLR2 | Interacts with viral envelope protein to induce pro-inflammatory response | [26] |
| TLR3 | Recognizes double-stranded RNA and is activated within 24 h of SARS-CoV-2 infection | [27] |
| TLR4 | Binds SARS-CoV-2 spike protein | [28] |
| TLR6 | Binds SARS-CoV-2 spike protein | [28] |
| TLR7 | Detects single-stranded RNA | [27,29,30] |
| Severity of Infection | Time Post Infection | Protection Level Against VOCs * | Sources |
|---|---|---|---|
| Severe | 1–3 months | Low to high | [40,41,42] |
| 4–6 months | Medium to high | ||
| 6+ months | High | ||
| Mild | 1–3 months | Low to medium | |
| 4–6 months | Medium to high | ||
| 6+ months | Medium to high |
| Date | Study Size | Mean Population Age (Standard Deviation, When Provided) | Gender | Percent of Outpatients with Persisting Symptoms | Source |
|---|---|---|---|---|---|
| May 2020 | 350 | Median: 43 * | F: 53% M: 47% | 36% (14–21 days) | [81] |
| March 2021 | 177 | 48 | F: 57.1% M: 42.9% | 32% (median: 169 days) | [80] |
| April 2021 | 4182 | 45.97 (15.8) | F: 71.5% M: 28.5% | 13.3% (28+ days) | [13] |
| July 2020 | 143 | 56.5 (14.6) | F: 37% M: 63% | 87.4% (Mean 60.3 days [SD: 13.6]) | [82] |
| Sep 2021 | 106,578 | 39.4 (18.4) | F: 58.4% M: 41.6% | 36.55% (90–180 days) | [83] |
| Feb 2022 | 5,080,312 | See source ** | F: 61.2% M: 38.8% | 35% (31–150 days) | [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sapir, T.; Averch, Z.; Lerman, B.; Bodzin, A.; Fishman, Y.; Maitra, R. COVID-19 and the Immune Response: A Multi-Phasic Approach to the Treatment of COVID-19. Int. J. Mol. Sci. 2022, 23, 8606. https://doi.org/10.3390/ijms23158606
Sapir T, Averch Z, Lerman B, Bodzin A, Fishman Y, Maitra R. COVID-19 and the Immune Response: A Multi-Phasic Approach to the Treatment of COVID-19. International Journal of Molecular Sciences. 2022; 23(15):8606. https://doi.org/10.3390/ijms23158606
Chicago/Turabian StyleSapir, Tzuriel, Zaelig Averch, Brian Lerman, Abraham Bodzin, Yeshaya Fishman, and Radhashree Maitra. 2022. "COVID-19 and the Immune Response: A Multi-Phasic Approach to the Treatment of COVID-19" International Journal of Molecular Sciences 23, no. 15: 8606. https://doi.org/10.3390/ijms23158606