
Comprehensive Virtual Screening of the Antiviral Potentialities of Marine Polycyclic Guanidine Alkaloids against SARS-CoV-2 (COVID-19)

 ,
,  ,
, 
 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Docking Studies
2.2. ADMET
2.3. Toxicity
2.4. Isolation and Characterization of Compounds 1–15
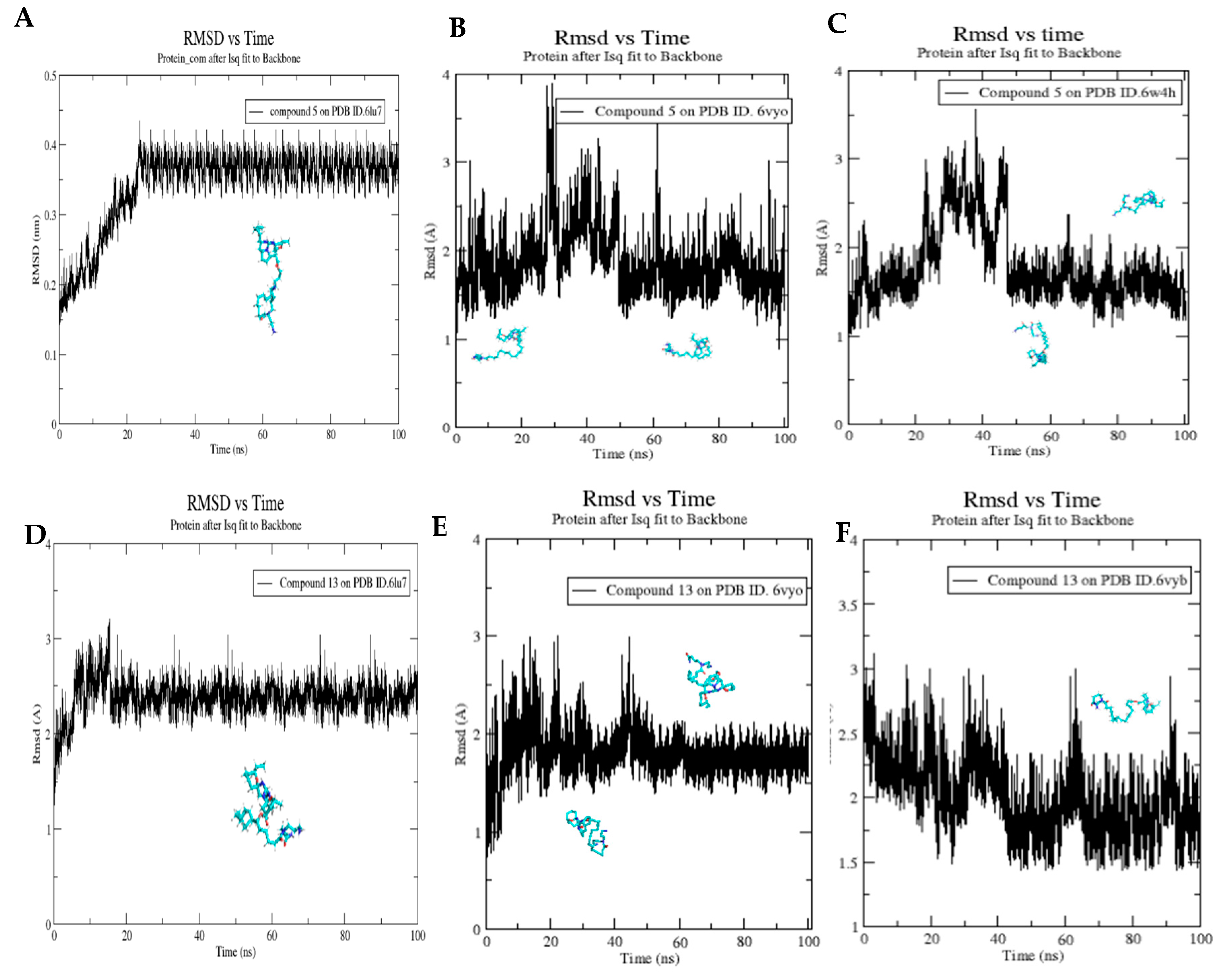
2.5. Molecular Dynamics Simulation for Compounds 5 and 13
3. Results
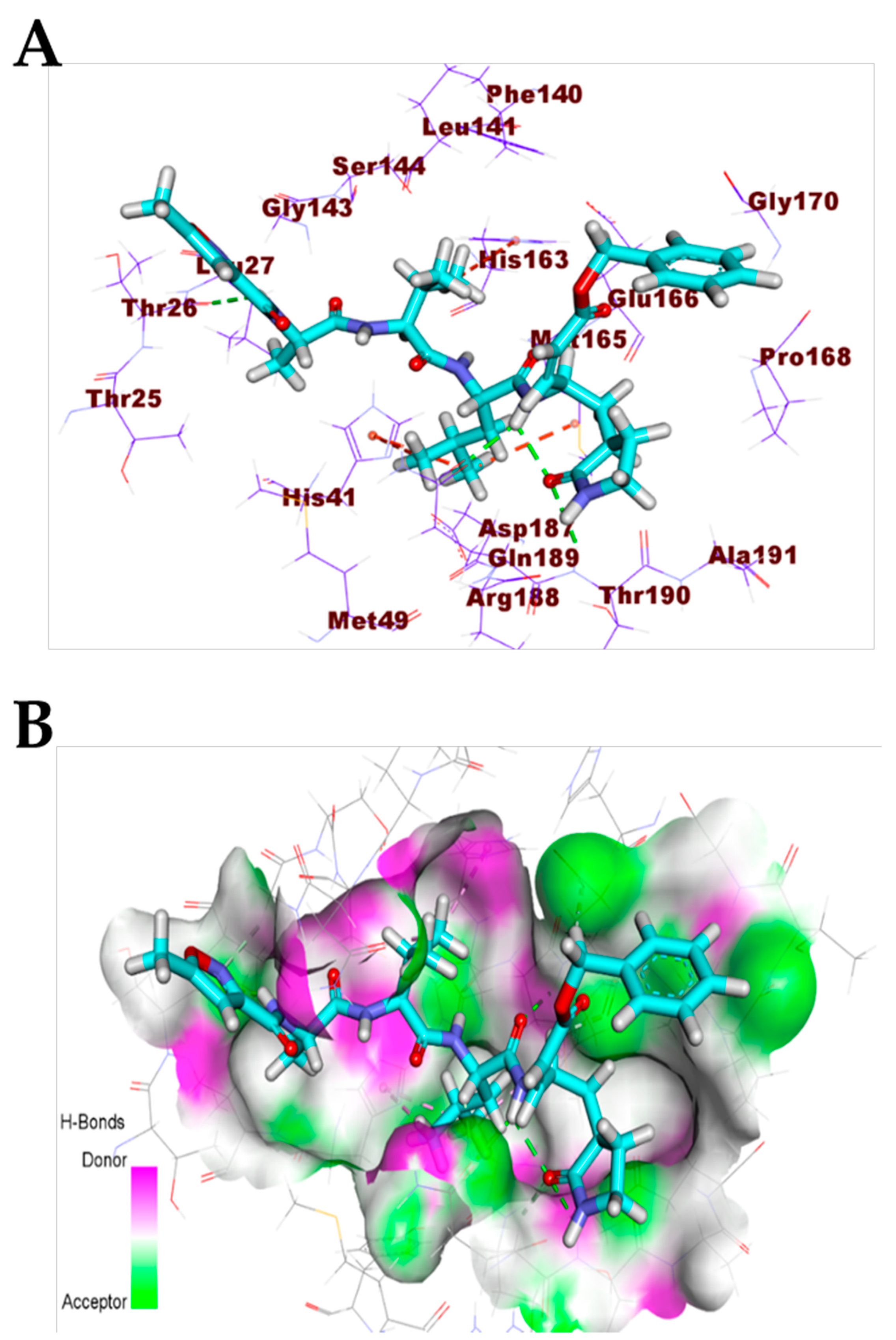
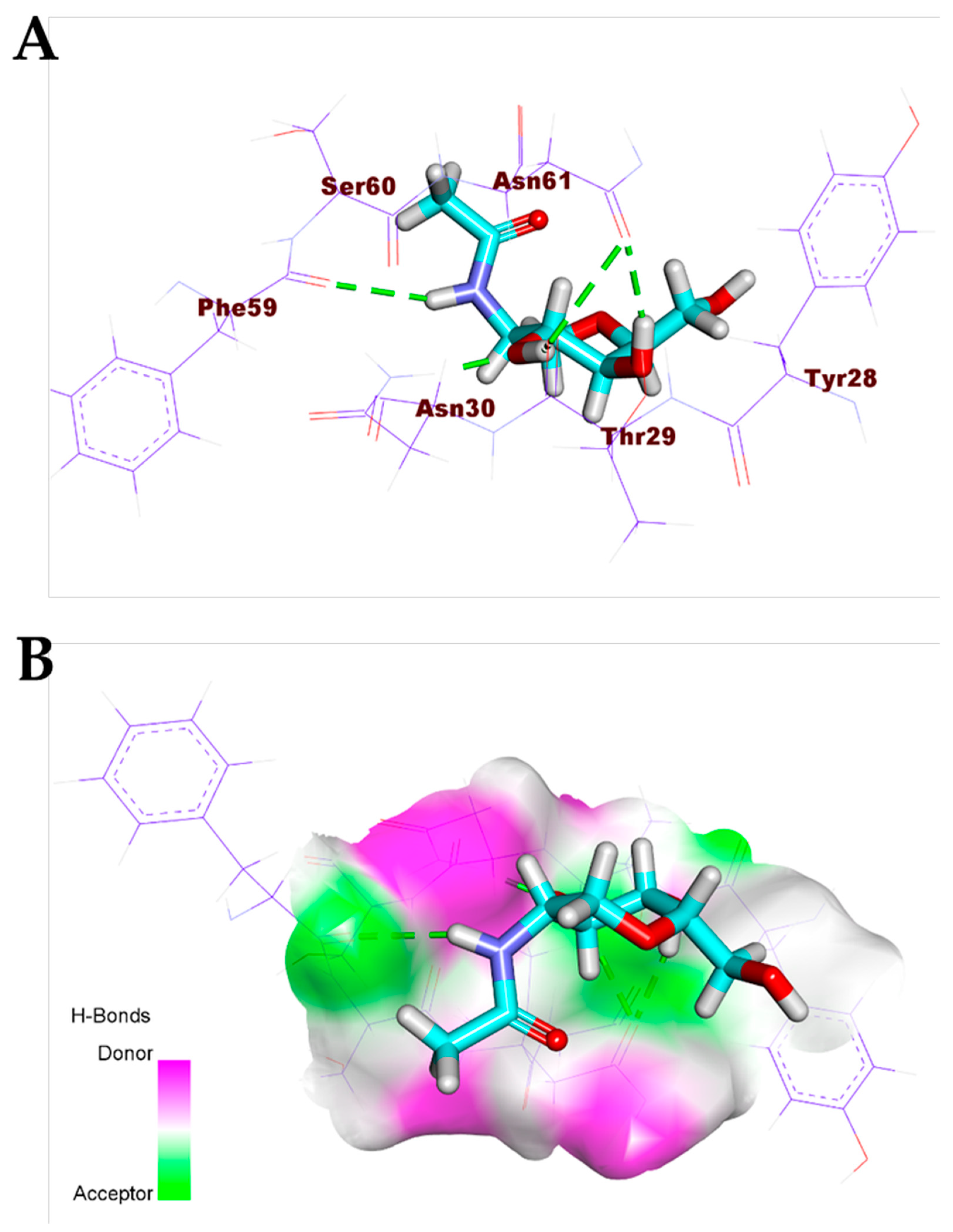
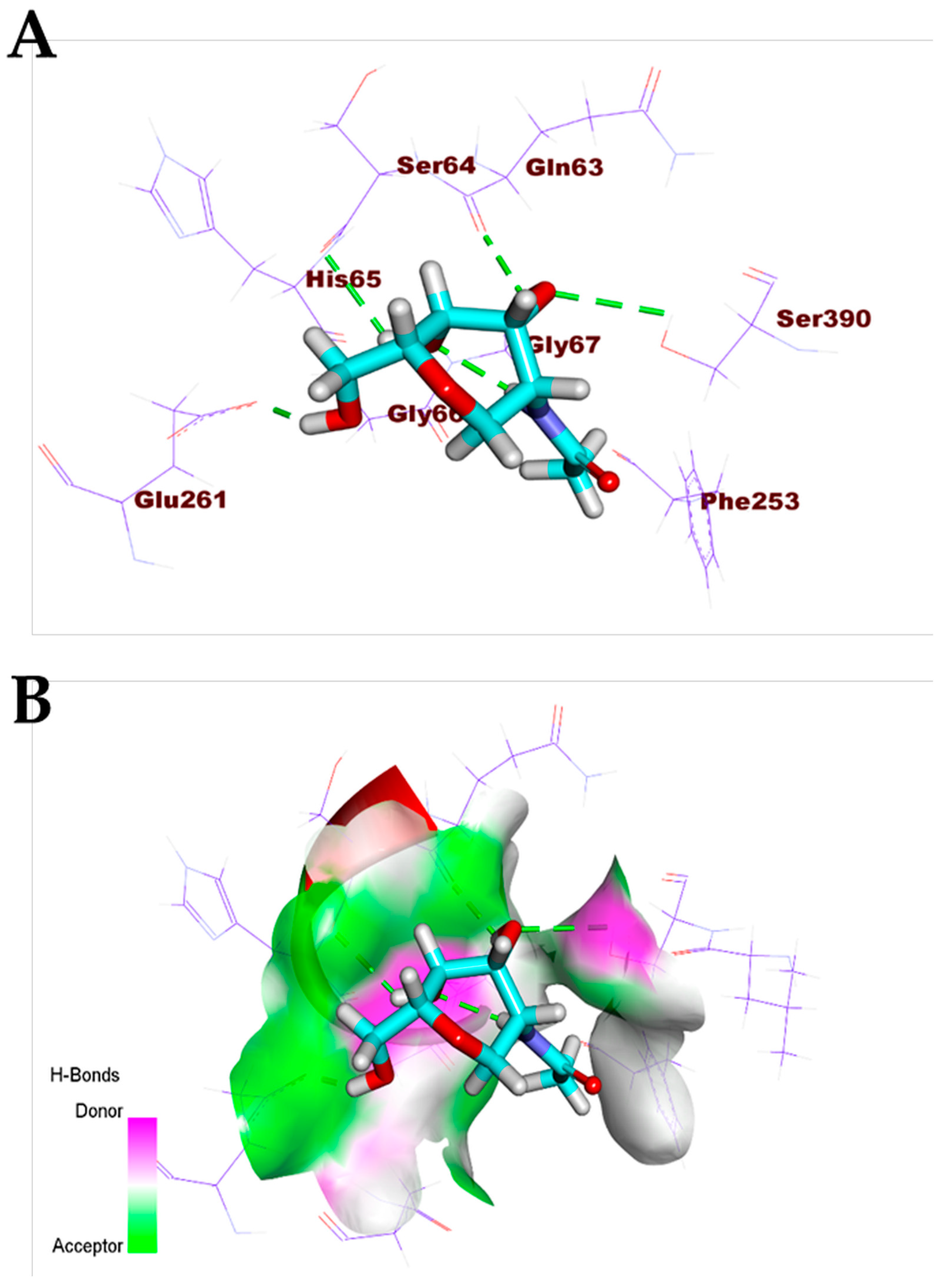
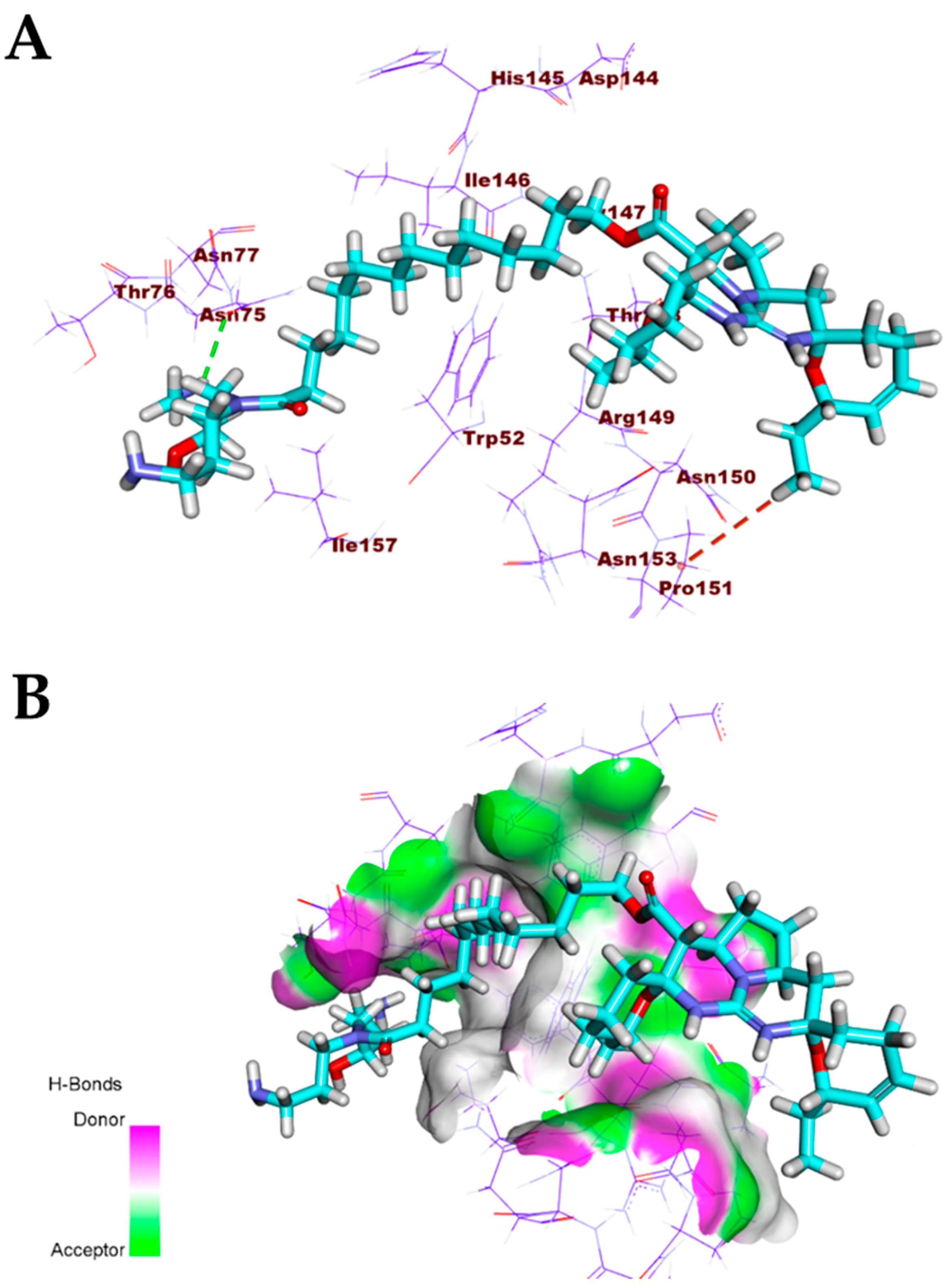
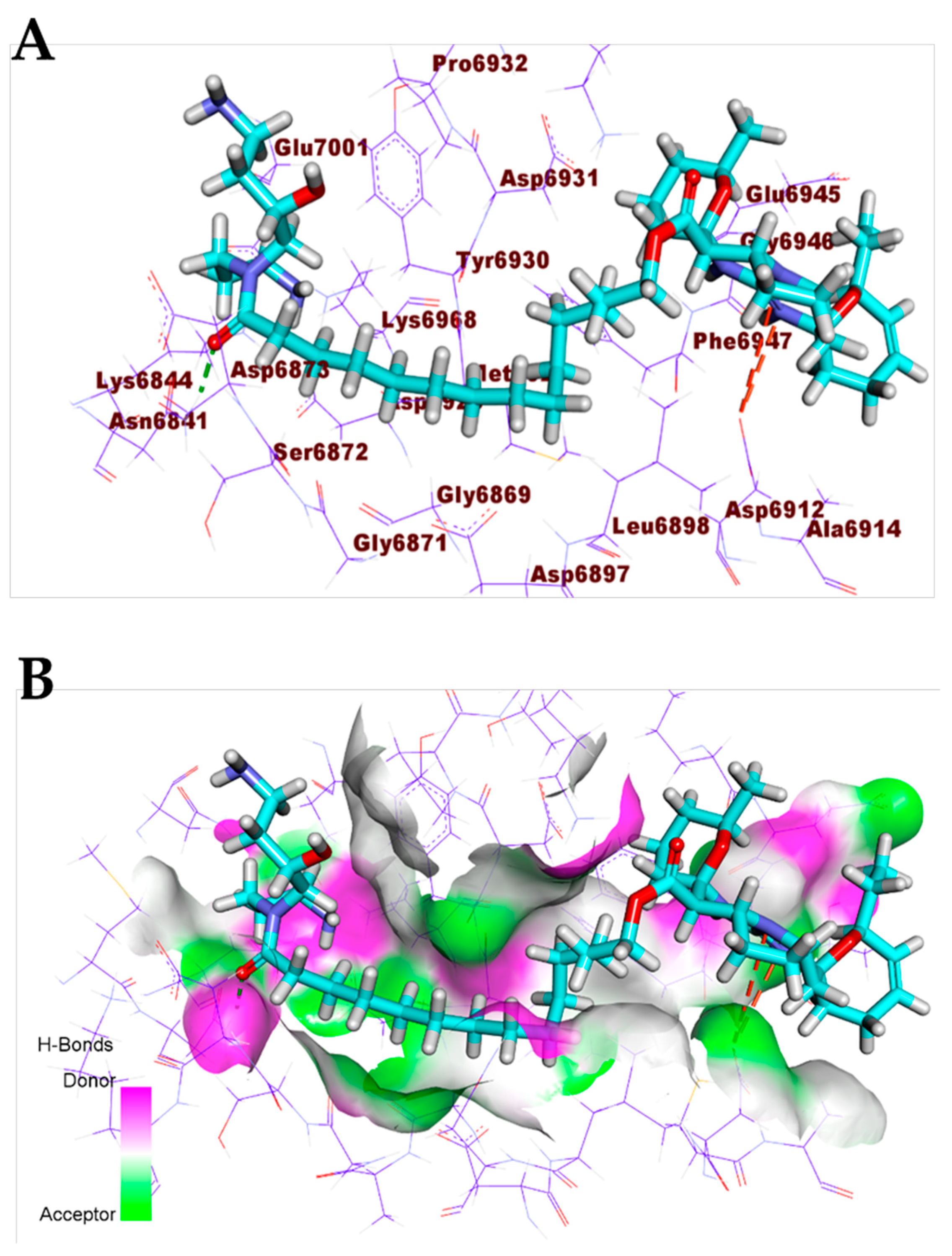
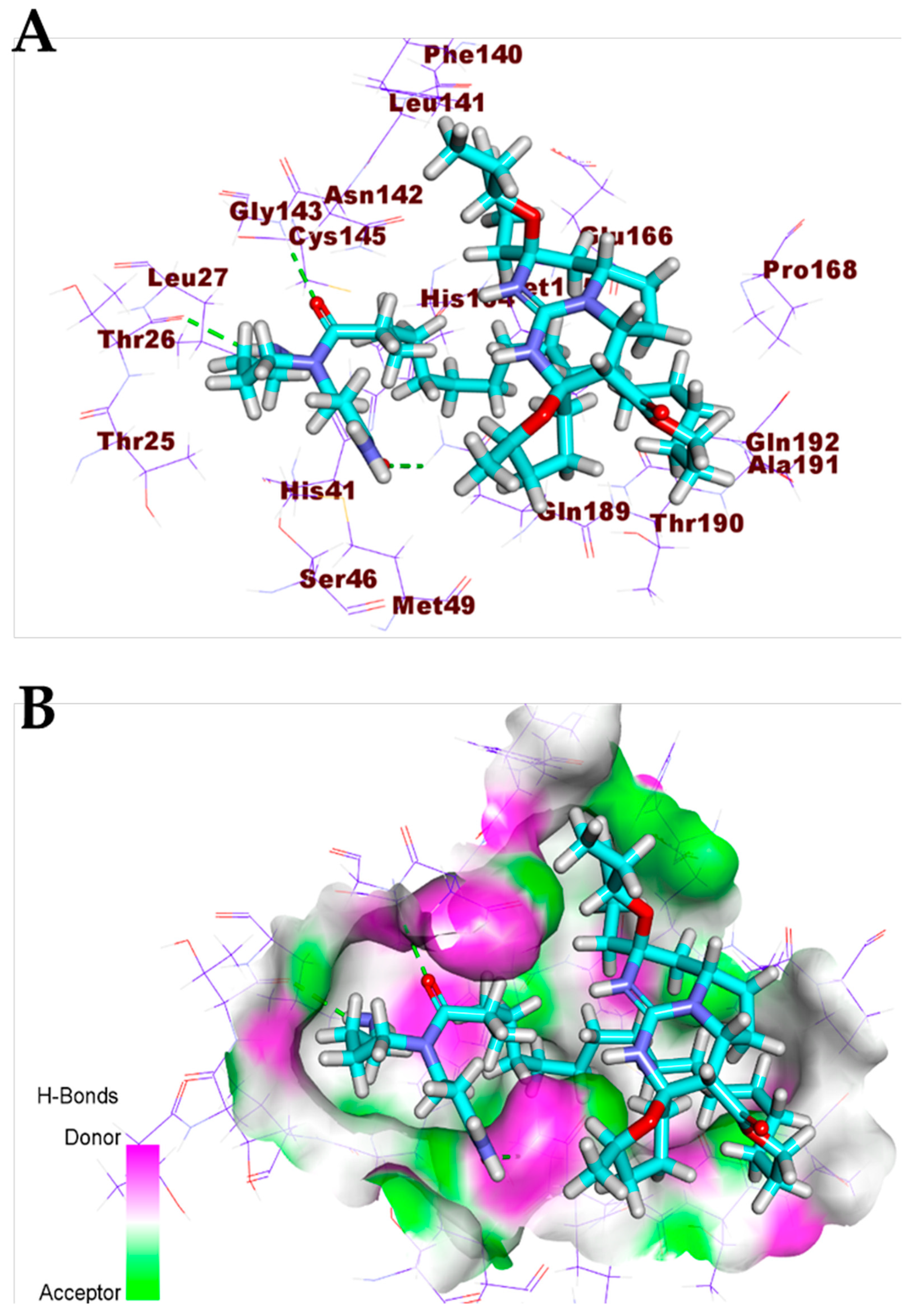
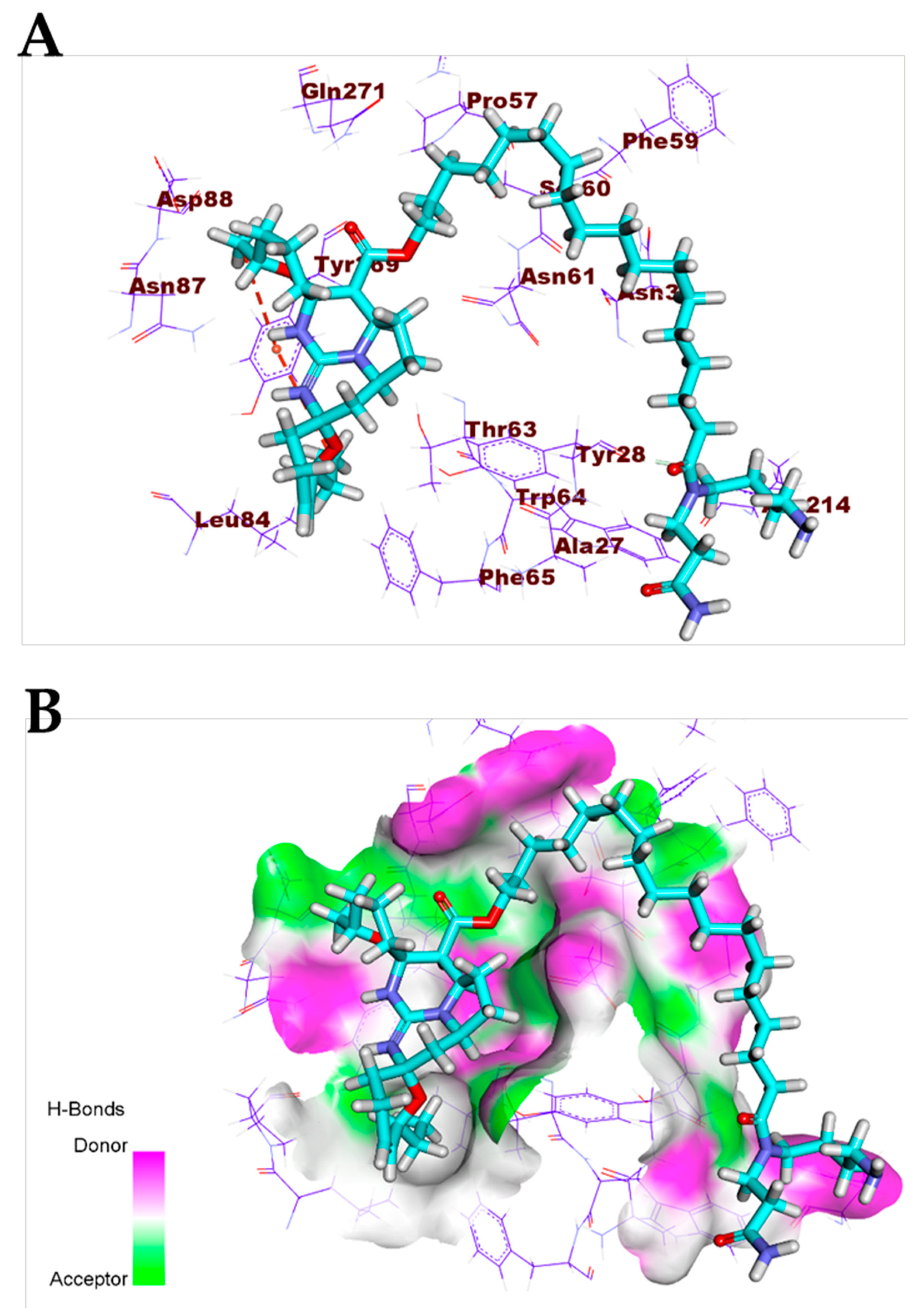
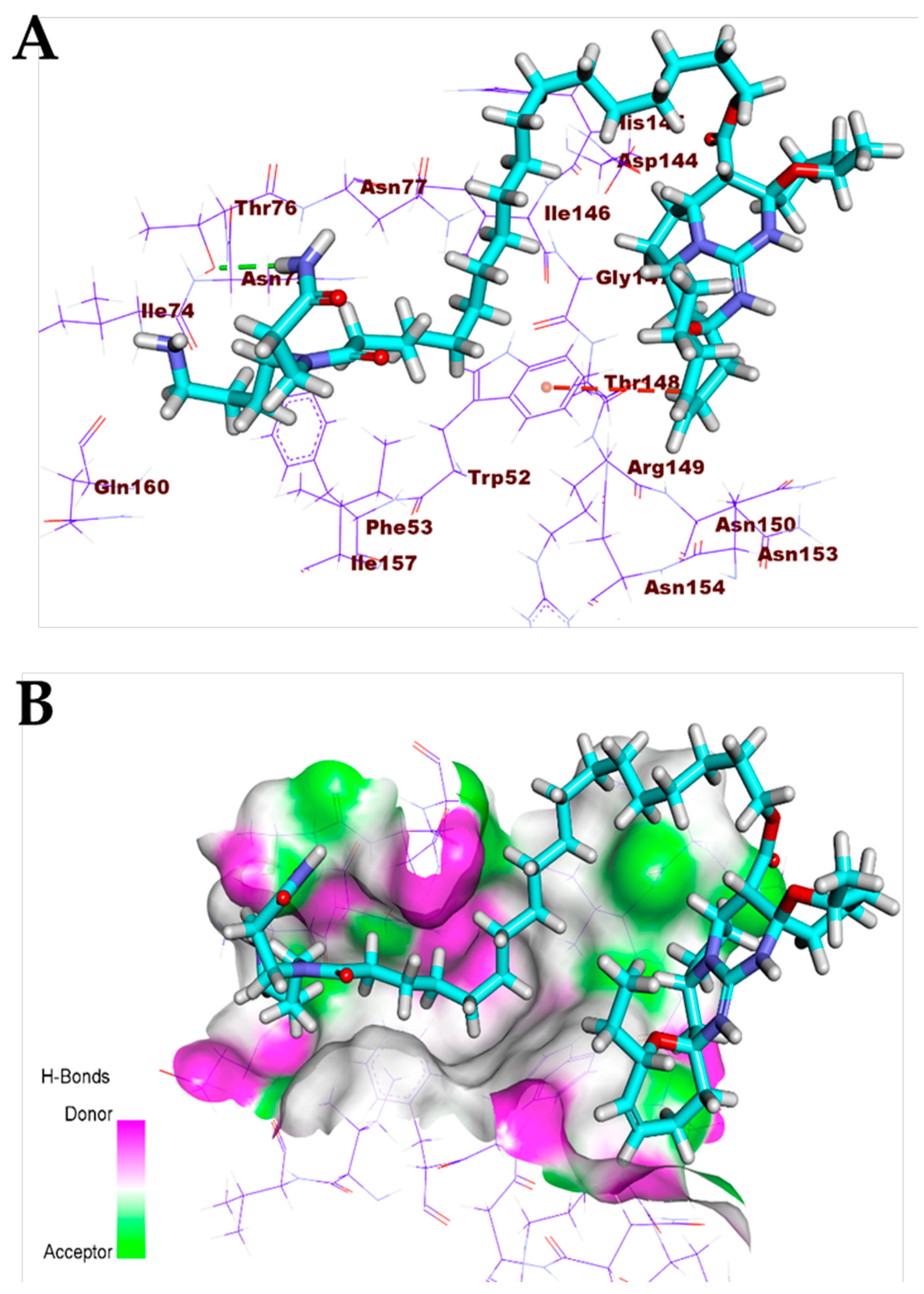
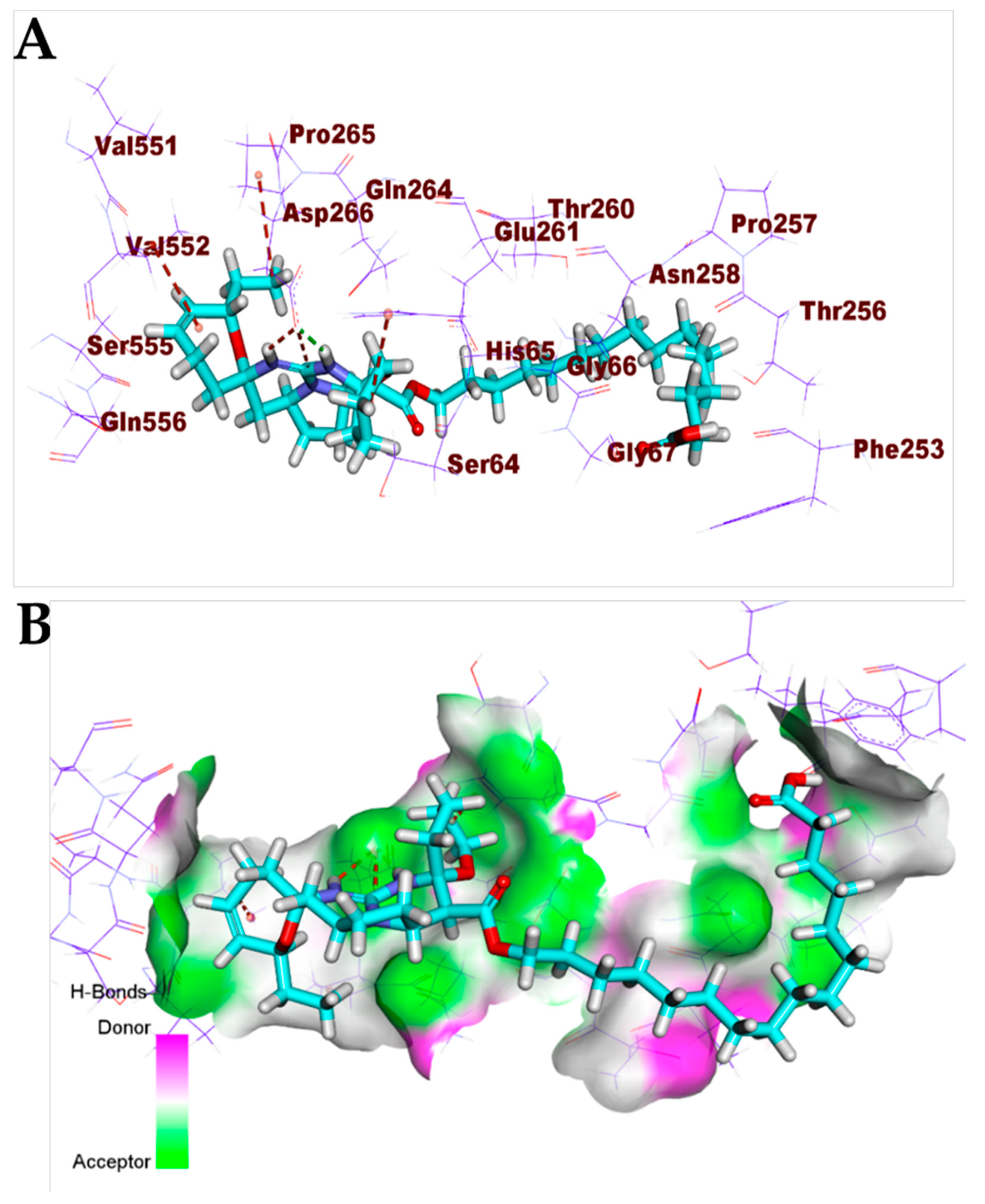
3.1. Validation of the Docking Processes
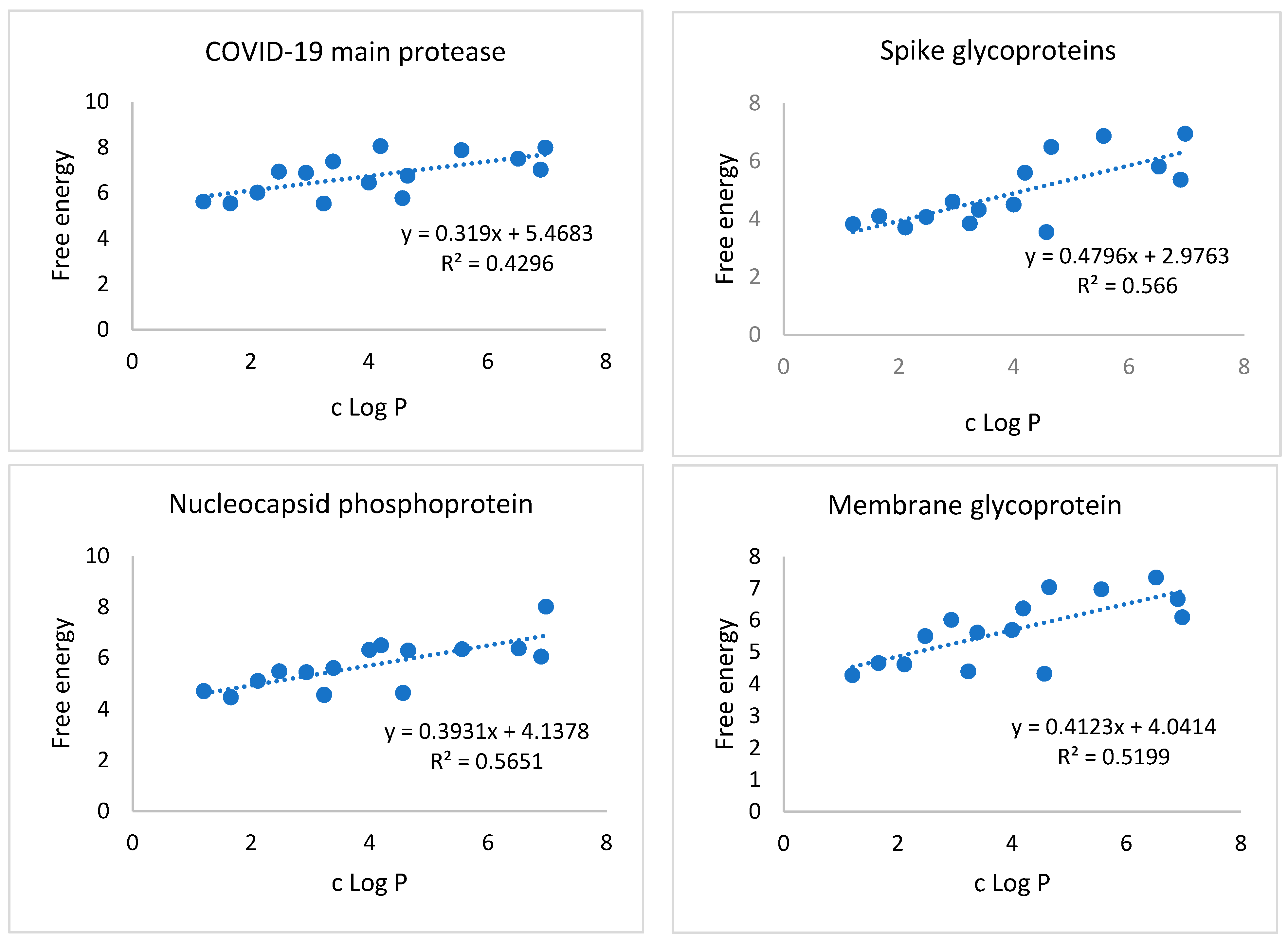
3.2. Correlation of c Log P with Free energy of Binding against SARS-CoV-2 Target Proteins
3.3. Structure-Activity Relationship (SAR)
3.4. In Silico ADMET Analysis
3.5. Toxicity Studies
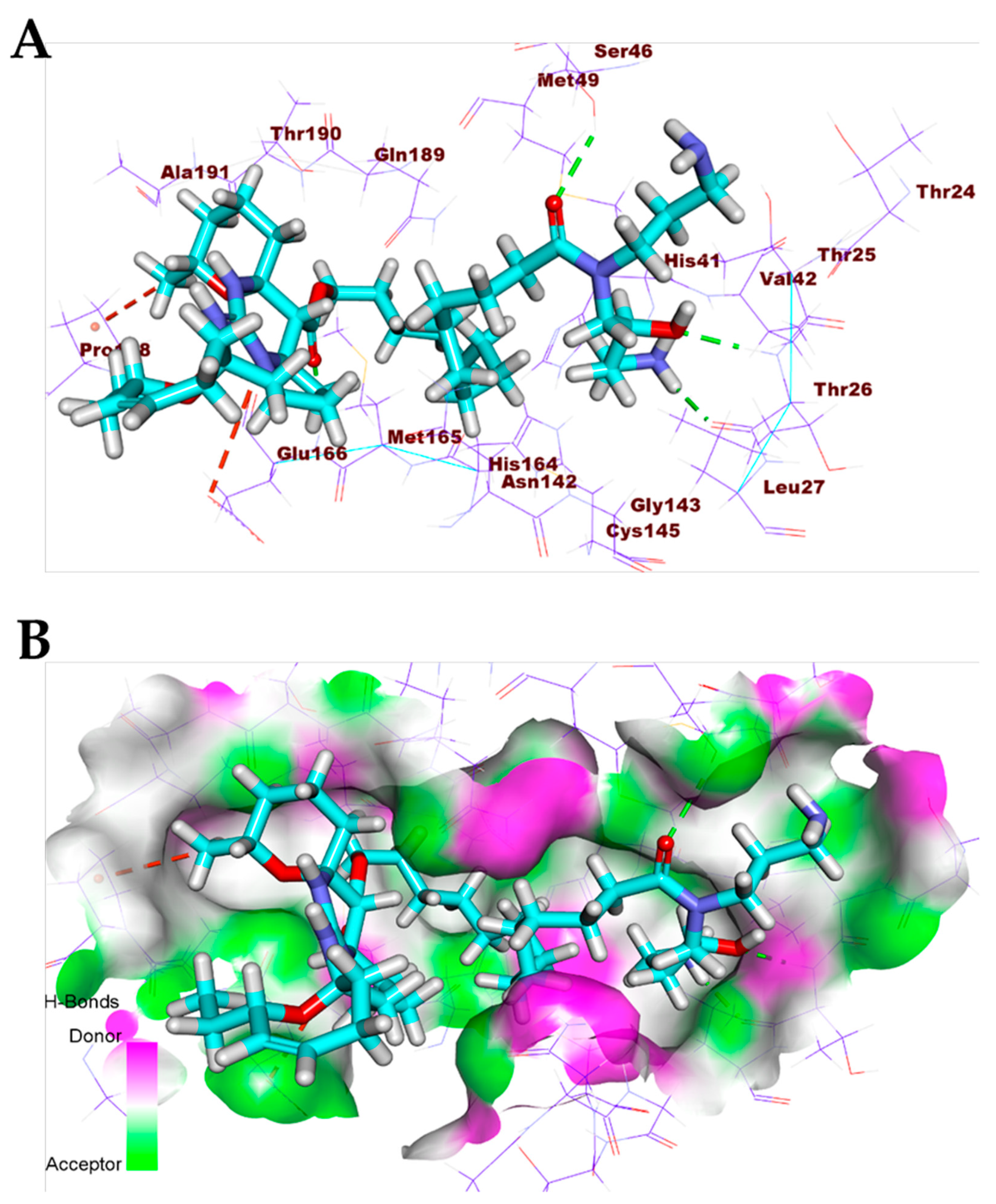
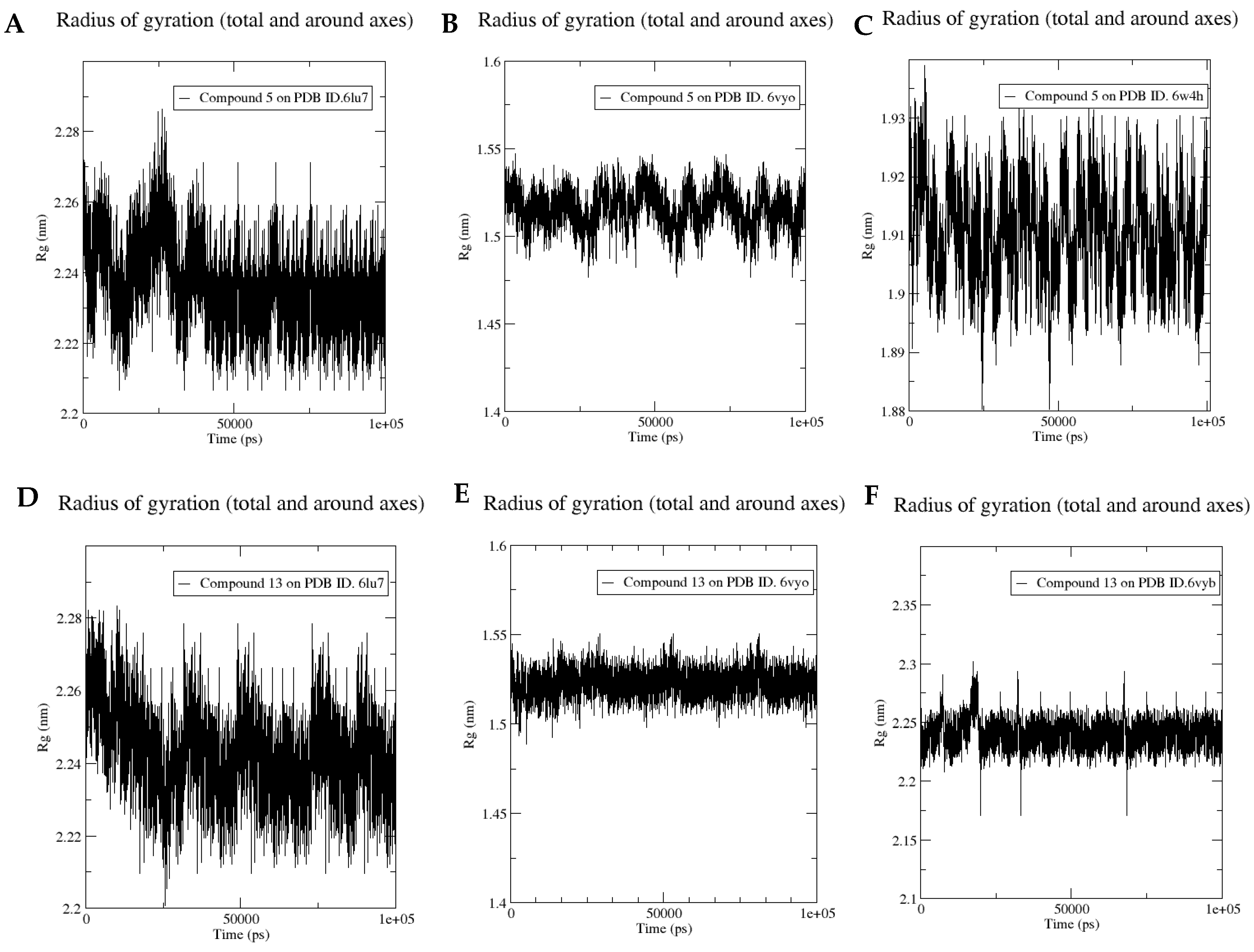
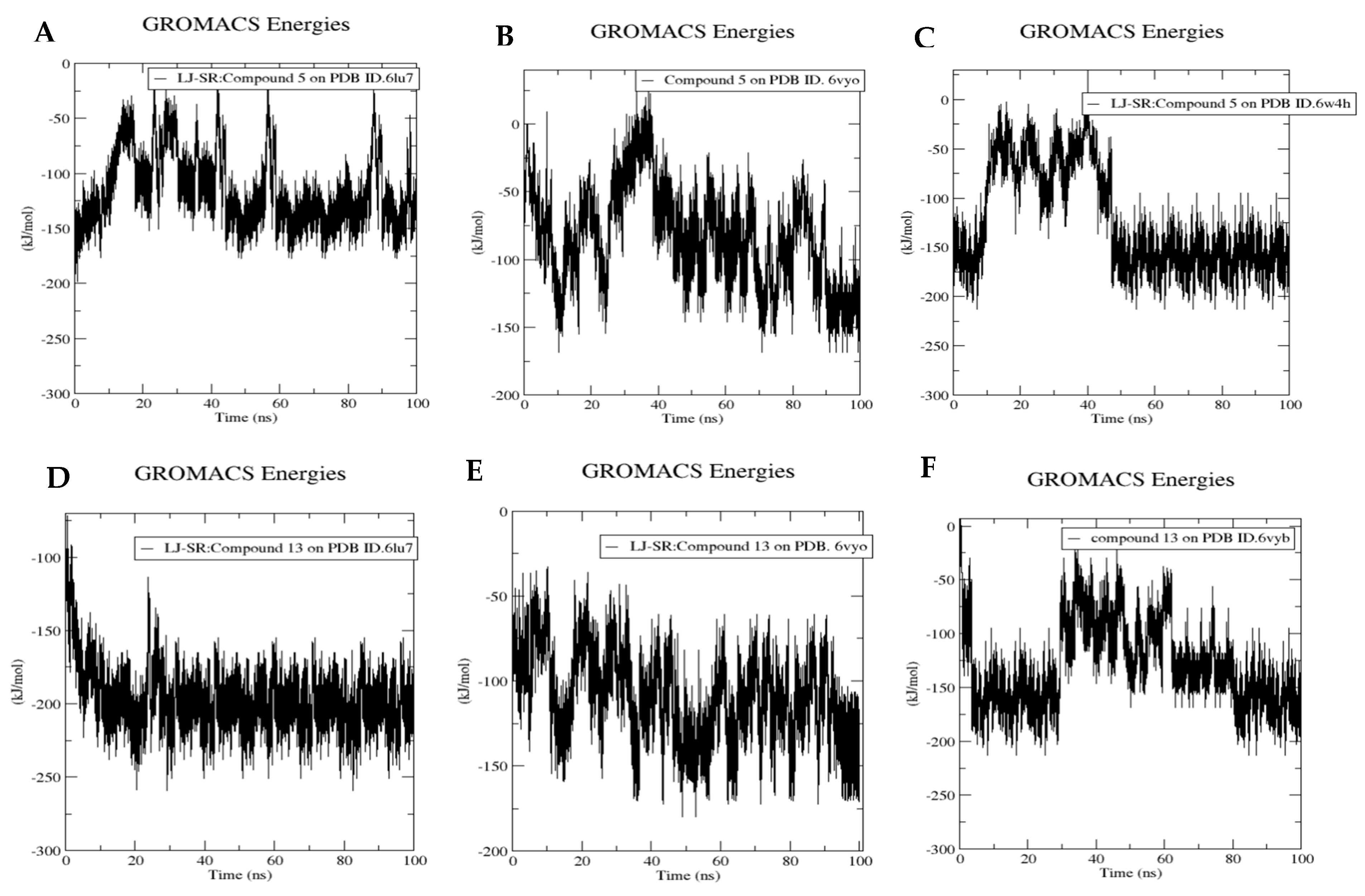
3.6. Molecular Dynamics Simulation for Compounds 5 and 13
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- El-Aziz, T.M.A.; Stockand, J.D. Recent progress and challenges in drug development against COVID-19 coronavirus (SARS-CoV-2)—An update on the status. Infect. Genet. Evol. 2020, 83, 104327. [Google Scholar] [CrossRef] [PubMed]
- Steffens, I. A hundred days into the coronavirus disease (COVID-19) pandemic. Euro Surveill. 2020, 25, 2000550. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 2005, 69, 635–664. [Google Scholar] [CrossRef] [Green Version]
- Bradburne, A.F.; Tyrrell, D.A. The propagation of “coronaviruses” in tissue-culture. Arch Gesamte Virusforsch. 1969, 28, 133–150. [Google Scholar] [CrossRef] [Green Version]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Molecular Evolution of Human Coronavirus Genomes. Trends Microbiol. 2017, 25, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Dong, W.; Milewska, A.; Golda, A.; Qi, Y.; Zhu, Q.K.; Marasco, W.A.; Baric, R.S.; Sims, A.C.; Pyrc, K.; et al. Human Coronavirus HKU1 Spike Protein Uses O-Acetylated Sialic Acid as an Attachment Receptor Determinant and Employs Hemagglutinin-Esterase Protein as a Receptor-Destroying Enzyme. J. Virol. 2015, 89, 7202–7213. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.Y.; Li, J.L.; Yang, X.L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed]
- de Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Ithete, N.L.; Stoffberg, S.; Corman, V.M.; Cottontail, V.M.; Richards, L.R.; Schoeman, M.C.; Drosten, C.; Drexler, J.F.; Preiser, W. Close relative of human Middle East respiratory syndrome coronavirus in bat, South Africa. Emerg. Infect. Dis. 2013, 19, 1697–1699. [Google Scholar] [CrossRef] [PubMed]
- Patiño-Galindo, J.Á.; Filip, I.; AlQuraishi, M.; Rabadan, R. Recombination and convergent evolution led to the emergence of 2019 Wuhan coronavirus. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.L.; Wang, Y.M.; Wu, Z.Q.; Xiang, Z.C.; Guo, L.; Xu, T.; Jiang, Y.Z.; Xiong, Y.; Li, Y.J.; Li, X.W.; et al. Identification of a novel coronavirus causing severe pneumonia in human: A descriptive study. Chin. Med J. 2020, 133, 1015–1024. [Google Scholar] [CrossRef]
- Rossmann, M.G. Structure of viruses: A short history. Q. Rev. Biophys. 2013, 46, 133. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Viruses. In Molecular Cell Biology, 4th ed.; WH Freeman: New York, NY, USA, 2000. [Google Scholar]
- Pornillos, O.; Garrus, J.E.; Sundquist, W.I. Mechanisms of enveloped RNA virus budding. Trends Cell Biol. 2002, 12, 569–579. [Google Scholar] [CrossRef]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 19, 1–7. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Prajapat, M.; Sarma, P.; Shekhar, N.; Avti, P.; Sinha, S.; Kaur, H.; Kumar, S.; Bhattacharyya, A.; Kumar, H.; Bansal, S. Drug targets for corona virus: A systematic review. Indian J. Pharmacol. 2020, 52, 56. [Google Scholar]
- Bouvet, M.; Lugari, A.; Posthuma, C.C.; Zevenhoven, J.C.; Bernard, S.; Betzi, S.; Imbert, I.; Canard, B.; Guillemot, J.-C.; Lécine, P. Coronavirus Nsp10, a critical co-factor for activation of multiple replicative enzymes. J. Biol. Chem. 2014, 289, 25783–25796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, W.; Burke, D.C. Contributions to the study of marine products. XXXIX. The nucleosides of sponges. III.1 Spongothymidine and spongouridine2. J. Org. Chem. 1955, 20, 1501–1507. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, A.M.S.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharm. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef]
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmann, K.-H. Drugs from the Oceans: Marine Natural Products as Leads for Drug Discovery. Chim. Int. J. Chem. 2017, 71, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, C. Marine Natural Products in Medicinal Chemistry. ACS Med. Chem. Lett. 2018, 9, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F. Have marine natural product drug discovery efforts been productive and how can we improve their efficiency? Expert Opin. Drug Discov. 2019, 14, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Donia, M.; Hamann, M.T. Marine natural products and their potential applications as anti-infective agents. Lancet Infect. Dis. 2003, 3, 338–348. [Google Scholar] [CrossRef]
- Williams, D.E.; Andersen, R.J. Biologically active marine natural products and their molecular targets discovered using a chemical genetics approach. Nat. Prod. Rep. 2020, 37, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Bardón, M.; Pérez-Pertejo, Y.; Ordóñez, C.; Sepúlveda-Crespo, D.; Carballeira, N.M.; Tekwani, B.L.; Murugesan, S.; Martinez-Valladares, M.; García-Estrada, C.; Reguera, R.M.; et al. Screening Marine Natural Products for New Drug Leads against Trypanosomatids and Malaria. Mar. Drugs 2020, 18, 187. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Ali, A.; Wang, Q.; Irfan, M.; Khan, A.; Zeb, M.T.; Zhang, Y.-J.; Chinnasamy, S.; Wei, D.-Q. Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2—A molecular dynamic study. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2020, 37, 175–223. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara-Bell, J.; Lu, Y. Marine compounds and their antiviral activities. Antivir. Res. 2010, 86, 231–240. [Google Scholar] [CrossRef]
- Gogineni, V.; Schinazi, R.F.; Hamann, M.T. Role of Marine Natural Products in the Genesis of Antiviral Agents. Chem. Rev. 2015, 115, 9655–9706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Demerdash, A.; Atanasov, A.G.; Bishayee, A.; Abdel-Mogib, M.; Hooper, J.N.A.; Al-Mourabit, A. Batzella, Crambe and Monanchora: Highly Prolific Marine Sponge Genera Yielding Compounds with Potential Applications for Cancer and Other Therapeutic Areas. Nutrients 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sfecci, E.; Lacour, T.; Amade, P.; Mehiri, M. Polycyclic Guanidine Alkaloids from Poecilosclerida Marine Sponges. Mar. Drugs 2016, 14, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Demerdash, A.; Tammam, M.A.; Atanasov, A.G.; Hooper, J.N.A.; Al-Mourabit, A.; Kijjoa, A. Chemistry and Biological Activities of the Marine Sponges of the Genera Mycale (Arenochalina), Biemna and Clathria. Mar. Drugs 2018, 16, 214. [Google Scholar] [CrossRef] [Green Version]
- Kashman, Y.; Hirsh, S.; McConnell, O.J.; Ohtani, I.; Kusumi, T.; Kakisawa, H. Ptilomycalin A: A novel polycyclic guanidine alkaloid of marine origin. J. Am. Chem. Soc. 1989, 111, 8925–8926. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Ermolenko, L.; Gros, E.; Retailleau, P.; Thanh, B.N.; Anne, G.-B.; Al-Mourabit, A. Short-Cut Bio-Inspired Synthesis of Tricyclic Guanidinic Motifs of Crambescidins and Batzelladines Marine Alkaloids. Eur. J. Org. Chem. 2020. [Google Scholar] [CrossRef]
- Jares-Erijman, E.A.; Sakai, R.; Rinehart, K.L. Crambescidins: New antiviral and cytotoxic compounds from the sponge Crambe crambe. J. Org. Chem. 1991, 56, 5712–5715. [Google Scholar] [CrossRef]
- Rubiolo, J.A.; López-Alonso, H.; Roel, M.; Vieytes, M.R.; Thomas, O.; Ternon, E.; Vega, F.V.; Botana, L.M. Mechanism of cytotoxic action of crambescidin-816 on human liver-derived tumour cells. Br. J. Pharm. 2014, 171, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- El-Demerdash, A.; Moriou, C.; Martin, M.-T.; Rodrigues-Stien, A.d.S.; Petek, S.; Demoy-Schneider, M.; Hall, K.; Hooper, J.N.A.; Debitus, C.; Al-Mourabit, A. Cytotoxic Guanidine Alkaloids from a French Polynesian Monanchora n. sp. Sponge. J. Nat. Prod. 2016, 79, 1929–1937. [Google Scholar] [CrossRef]
- El-Demerdash, A.; Moriou, C.; Martin, M.-T.; Petek, S.; Debitus, C.; Al-Mourabit, A. Unguiculins A-C: Cytotoxic bis-guanidine alkaloids from the French Polynesian sponge, Monanchora n. sp. Nat. Prod. Res. 2018, 32, 1512–1517. [Google Scholar] [CrossRef]
- Mendez, A.G.; Juncal, A.B.; Silva, S.B.L.; Thomas, O.P.; Martín Vázquez, V.; Alfonso, A.; Vieytes, M.R.; Vale, C.; Botana, L.M. The Marine Guanidine Alkaloid Crambescidin 816 Induces Calcium Influx and Cytotoxicity in Primary Cultures of Cortical Neurons through Glutamate Receptors. ACS Chem. Neurosci. 2017, 8, 1609–1617. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, S.; Sorolla, A.; Fromont, J.; Blancafort, P.; Flematti, G.R. Crambescidin 800, Isolated from the Marine Sponge Monanchora viridis, Induces Cell Cycle Arrest and Apoptosis in Triple-Negative Breast Cancer Cells. Mar. Drugs 2018, 16, 53. [Google Scholar] [CrossRef] [Green Version]
- Shubina, L.K.; Makarieva, T.N.; von Amsberg, G.; Denisenko, V.A.; Popov, R.S.; Dyshlovoy, S.A. Monanchoxymycalin C with anticancer properties, new analogue of crambescidin 800 from the marine sponge Monanchora pulchra. Nat. Prod. Res. 2019, 33, 1415–1422. [Google Scholar] [CrossRef]
- Palagiano, E.; De Marino, S.; Minale, L.; Riccio, R.; Zollo, F.; Iorizzi, M.; Carré, J.B.; Debitus, C.; Lucarain, L.; Provost, J. Ptilomycalin A, crambescidin 800 and related new highly cytotoxic guanidine alkaloids from the starfishes Fromia monilis and Celerina heffernani. Tetrahedron 1995, 51, 3675–3682. [Google Scholar] [CrossRef]
- Amade, P.; Charroin, C.; Baby, C.; Vacelet, J. Antimicrobial activities of marine sponges from the Mediterranean Sea. Mar. Biol. 1987, 94, 271–275. [Google Scholar] [CrossRef]
- Sun, X.; Sun, S.; Ference, C.; Zhu, W.; Zhou, N.; Zhang, Y.; Zhou, K. A potent antimicrobial compound isolated from Clathria cervicornis. Bioorg. Med. Chem. Lett. 2015, 25, 67–69. [Google Scholar] [CrossRef]
- Rubiolo, J.A.; Ternon, E.; López-Alonso, H.; Thomas, O.P.; Vega, F.V.; Vieytes, M.R.; Botana, L.M. Crambescidin-816 Acts as a Fungicidal with More Potency than Crambescidin-800 and -830, Inducing Cell Cycle Arrest, Increased Cell Size and Apoptosis in Saccharomyces cerevisiae. Mar. Drugs 2013, 11, 4419–4434. [Google Scholar] [CrossRef] [PubMed]
- Kasmiati, K.; Yoshioka, Y.; Okamoto, T.; Ojika, M. New Crambescidin-Type Alkaloids from the Indonesian Marine Sponge Clathria bulbotoxa. Mar. Drugs 2018, 16, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, P.E.; Ferreira Queiroz, E.; Marcourt, L.; Wolfender, J.L.; Sanchez, A.S.; Illien, B.; Al Mourabit, A.; Gauvin-Bialecki, A. Isolation and identification of new secondary metabolites from the marine sponge Monanchora unguiculata. Planta Med. 2016, 82, P580. [Google Scholar] [CrossRef]
- Campos, P.-E.; Wolfender, J.-L.; Queiroz, E.F.; Marcourt, L.; Al-Mourabit, A.; Frederich, M.; Bordignon, A.; De Voogd, N.; Illien, B.; Gauvin-Bialecki, A. Unguiculin A and Ptilomycalins E–H, Antimalarial Guanidine Alkaloids from the Marine Sponge Monanchora unguiculata. J. Nat. Prod. 2017, 80, 1404–1410. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, J.E.H.; Nitcheu, J.; Mahmoudi, N.; Ibana, J.A.; Mangalindan, G.C.; Black, G.P.; Howard-Jones, A.G.; Moore, C.G.; Thomas, D.A.; Mazier, D.; et al. Antimalarial Activity of Crambescidin 800 and Synthetic Analogues against Liver and Blood Stage of Plasmodium sp. J. Antibiot. 2006, 59, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Takishima, S.; Ishiyama, A.; Iwatsuki, M.; Otoguro, K.; Yamada, H.; Omura, S.; Kobayashi, H.; van Soest, R.W.; Matsunaga, S. Merobatzelladines A and B, anti-infective tricyclic guanidines from a marine sponge Monanchora sp. Org. Lett. 2009, 11, 2655–2658. [Google Scholar] [CrossRef]
- Suna, H.; Aoki, S.; Setiawan, A.; Kobayashi, M. Crambescidin 800, a pentacyclic guanidine alkaloid, protects a mouse hippocampal cell line against glutamate-induced oxidative stress. J. Nat. Med. 2007, 61, 288–295. [Google Scholar] [CrossRef]
- Nakao, Y.; Fusetani, N. Enzyme Inhibitors from Marine Invertebrates. J. Nat. Prod. 2007, 70, 689–710. [Google Scholar] [CrossRef]
- Mai, S.; Nagulapalli, V.; Patil, A.; Truneh, A.; Westley, J. Marine Compounds as HIV Inhibitors. U.S. Patent Application WO9301193 (A1), 21 January 1993. [Google Scholar]
- Patil, A.D.; Kumar, N.V.; Kokke, W.C.; Bean, M.F.; Freyer, A.J.; Brosse, C.D.; Mai, S.; Truneh, A.; Carte, B. Novel alkaloids from the sponge Batzella sp.: Inhibitors of HIV gp120-human CD4 binding. J. Org. Chem. 1995, 60, 1182–1188. [Google Scholar] [CrossRef]
- Bewley, C.A.; Ray, S.; Cohen, F.; Collins, S.K.; Overman, L.E. Inhibition of HIV-1 Envelope-Mediated Fusion by Synthetic Batzelladine Analogues. J. Nat. Prod. 2004, 67, 1319–1324. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Jares-Erijman, E.A. Crambescidins: New Antiviral and Cytotoxic Compounds from the Sponge Crambe crambe. U.S. Patent No. 5,756,734, 26 May 1998. [Google Scholar]
- Rinehart, K.L.; Shi, J.-G.; Sun, F. Crambescidin Compounds. U.S. Patent No. 6,028,077, 22 February 2000. [Google Scholar]
- Chang, L.; Whittaker, N.F.; Bewley, C.A. Crambescidin 826 and Dehydrocrambine A: New Polycyclic Guanidine Alkaloids from the Marine Sponge Monanchora sp. that Inhibit HIV-1 Fusion. J. Nat. Prod. 2003, 66, 1490–1494. [Google Scholar] [CrossRef]
- Gustafson, K.R.; Oku, N.; Milanowski, D.J. Antiviral Marine Natural Products. Curr. Med. Chem. Anti-Infect. Agents 2004, 3, 233–249. [Google Scholar] [CrossRef]
- Patil, A.D.; Freyer, A.J.; Taylor, P.B.; Carté, B.; Zuber, G.; Johnson, R.K.; Faulkner, D.J. Batzelladines F−I, Novel Alkaloids from the Sponge Batzella sp.: Inducers of p56lck-CD4 Dissociation. J. Org. Chem. 1997, 62, 1814–1819. [Google Scholar] [CrossRef]
- Patil, A.D.; Freyer, A.J.; Offen, P.; Bean, M.F.; Johnson, R.K. Three New Tricyclic Guanidine Alkaloids from the Sponge Batzella sp. J. Nat. Prod. 1997, 60, 704–707. [Google Scholar] [CrossRef]
- Olszewski, A.; Sato, K.; Aron, Z.D.; Cohen, F.; Harris, A.; McDougall, B.R.; Robinson, W.E.; Overman, L.E.; Weiss, G.A. Guanidine alkaloid analogs as inhibitors of HIV-1 Nef interactions with p53, actin, and p56lck. Proc. Natl. Acad. Sci. USA 2004, 101, 14079–14084. [Google Scholar] [CrossRef] [Green Version]
- Hua, H.-M.; Peng, J.; Dunbar, D.C.; Schinazi, R.F.; de Castro Andrews, A.G.; Cuevas, C.; Garcia-Fernandez, L.F.; Kelly, M.; Hamann, M.T. Batzelladine alkaloids from the Caribbean sponge Monanchora unguifera and the significant activities against HIV-1 and AIDS opportunistic infectious pathogens. Tetrahedron 2007, 63, 11179–11188. [Google Scholar] [CrossRef]
- Harvey, A.L. Presynaptic effects of toxins. Int. Rev. Neurobiol. 1990, 32, 201–239. [Google Scholar]
- El-Gamal, K.M.; El-Morsy, A.M.; Saad, A.M.; Eissa, I.H.; Alswah, M. Synthesis, docking, QSAR, ADMET and antimicrobial evaluation of new quinoline-3-carbonitrile derivatives as potential DNA-gyrase inhibitors. J. Mol. Struct. 2018, 1166, 15–33. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; Li, W.; Li, H.; Yang, L.; Wang, J.; Mahdy, H.A.; Mehany, A.; Jaiash, D.A.; Santali, E.Y. Screening of Some Sulfonamide and Sulfonylurea Derivatives as Anti-Alzheimer’s Agents Targeting BACE1 and PPARγ. J. Chem. 2020, 2020, 1631243. [Google Scholar] [CrossRef]
- Ibrahim, M.K.; Eissa, I.H.; Alesawy, M.S.; Metwaly, A.M.; Radwan, M.M.; ElSohly, M.A. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of quinazolin-4 (3H)-one derivatives as potential PPARγ and SUR agonists. Bioorganic Med. Chem. 2017, 25, 4723–4744. [Google Scholar] [CrossRef]
- Elmetwally, S.A.; Saied, K.F.; Eissa, I.H.; Elkaeed, E.B. Design, synthesis and anticancer evaluation of thieno [2,3-d] pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers. Bioorganic Chem. 2019, 88, 102944. [Google Scholar] [CrossRef]
- Mahdy, H.A.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.; El-Gamal, K.M.; El-Sharkawy, A.; Elhendawy, M.A.; Radwan, M.M.; Elsohly, M.A. Design, synthesis, molecular modeling, in vivo studies and anticancer evaluation of quinazolin-4 (3H)-one derivatives as potential VEGFR-2 inhibitors and apoptosis inducers. Bioorganic Chem. 2020, 94, 103422. [Google Scholar] [CrossRef]
- El-Zahabi, M.A.; Elbendary, E.R.; Bamanie, F.H.; Radwan, M.F.; Ghareib, S.A.; Eissa, I.H. Design, synthesis, molecular modeling and anti-hyperglycemic evaluation of phthalimide-sulfonylurea hybrids as PPARγ and SUR agonists. Bioorganic Chem. 2019, 91, 103115. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Eissa, I.H.; Belal, A.; El-Sayed, A.A. Design, eco-friendly synthesis, molecular modeling and anticancer evaluation of thiazol-5 (4 H)-ones as potential tubulin polymerization inhibitors targeting the colchicine binding site. RSC Adv. 2020, 10, 2791–2811. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Mannhold, R.; Kubinyi, H.; Folkers, G. Pharmacokinetics and Metabolism in Drug Design; John Wiley & Sons: Hoboken, NJ, USA, 2012; Volume 51. [Google Scholar]
- Klopman, G.; Stefan, L.R.; Saiakhov, R.D. ADME evaluation: 2. A computer model for the prediction of intestinal absorption in humans. Eur. J. Pharm. Sci. 2002, 17, 253–263. [Google Scholar] [CrossRef]
- Roy, P.P.; Roy, K. QSAR studies of CYP2D6 inhibitor aryloxypropanolamines using 2D and 3D descriptors. Chem. Biol. Drug Des. 2009, 73, 442–455. [Google Scholar] [CrossRef]
- Ghafourian, T.; Amin, Z. QSAR models for the prediction of plasma protein binding. Bioimpacts 2013, 3, 21. [Google Scholar] [PubMed]
- Xia, X.; Maliski, E.G.; Gallant, P.; Rogers, D. Classification of kinase inhibitors using a Bayesian model. J. Med. Chem. 2004, 47, 4463–4470. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA. QSAR, ADMET and Predictive Toxicology. Available online: https://www.3dsbiovia.com/products/collaborative-science/biovia-discovery-studio/qsar-admet-and-predictive-toxicology.html (accessed on 17 May 2020).
- Venkatapathy, R.; Wang, N.C.Y.; Martin, T.M.; Harten, P.F.; Young, D. Structure–Activity Relationships for Carcinogenic Potential. Gen. Appl. Syst. Toxicol. 2009. [Google Scholar] [CrossRef]
- Goodrnan, G.; Wilson, R. Comparison of the dependence of the TD50 on maximum tolerated dose for mutagens and nonmutagens. Risk Anal. 1992, 12, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Council, N.R. Correlation Between Carcinogenic Potency and the Maximum Tolerated Dose: Implications for Risk Assessment. In Issues in Risk Assessment; National Academies Press (US): Cambridge, MA, USA, 1993. [Google Scholar]
- Gonella Diaza, R.; Manganelli, S.; Esposito, A.; Roncaglioni, A.; Manganaro, A.; Benfenati, E. Comparison of in silico tools for evaluating rat oral acute toxicity. Sar. Qsar. Environ. Res. 2015, 26, 1–27. [Google Scholar] [CrossRef]
- Pizzo, F.; Benfenati, E. In silico models for repeated-dose toxicity (RDT): Prediction of the no observed adverse effect level (NOAEL) and lowest observed adverse effect level (LOAEL) for drugs. In Silico Methods for Predicting Drug Toxicity; Springer: Berlin/Heidelberg, Germany, 2016; pp. 163–176. [Google Scholar]
- Venkatapathy, R.; Moudgal, C.J.; Bruce, R.M. Assessment of the oral rat chronic lowest observed adverse effect level model in TOPKAT, a QSAR software package for toxicity prediction. J. Chem. Inf. Comput. Sci. 2004, 44, 1623–1629. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | COVID-19 Main Protease | Spike Glycoproteins | Nucleocapsid Phosphoprotein | Membrane Glycoprotein | nsp10 | c Log P |
|---|---|---|---|---|---|---|
| Monanchoradin A (1) | −5.62 | −3.83 | −4.70 | −4.27 | −6.12 | 1.204 |
| Monanchoradin B (2) | −5.54 | −4.10 | −4.46 | −4.65 | −5.73 | 1.66 |
| Monanchoradin C (3) | −6.01 | −3.71 | −5.10 | −4.61 | −6.08 | 2.116 |
| Dehydrocrambescin A2 418 (4) | −6.45 | −4.50 | −6.31 | −5.69 | −7.19 | 3.998 |
| Crambescidin 786 (5) | −8.05 | −5.60 | −6.49 | −6.37 | −9.06 | 4.195 |
| Crambescidin 814 (6) | −7.87 | −6.87 | −6.34 | −6.97 | −7.50 | 5.563 |
| Norcrambescidic acid (7) | −7.50 | −5.81 | −6.37 | −7.34 | −7.35 | 6.52 |
| Monalidine A (8) | −5.77 | −3.55 | −4.63 | −4.32 | −5.63 | 4.566 |
| (−)-crambescin A2 392 (9) | −6.93 | −4.07 | −5.47 | −5.50 | −6.61 | 2.479 |
| (−)-crambescin A2 406 (10) | −6.88 | −4.60 | −5.44 | −6.01 | −10.54 | 2.936 |
| (−)-crambescin A2 420 (11) | −7.38 | −4.32 | −5.60 | −5.61 | −6.53 | 3.392 |
| Crambescidin 800 (12) | −6.75 | −6.49 | −6.29 | −7.04 | −7.22 | 4.651 |
| Crambescidin 826 (13) | −7.99 | −6.95 | −8.01 | −6.09 | −8.39 | 6.979 |
| Crambescidic acid (14) | −7.02 | −5.36 | −6.05 | −6.66 | −7.38 | 6.898 |
| Crambescidin 359 (15) | −5.53 | −3.85 | −4.55 | −4.39 | −4.72 | 3.235 |
| Co-crystallized ligand (PRD_002214) | −8.18 | - | - | - | - | - |
| Co-crystallized ligand (NAG) | - | −3.56 | - | - | - | - |
| Co-crystallized ligand (MES) | - | - | −3.80 | - | - | - |
| Co-crystallized ligand (NAG) | - | - | - | −3.63 | - | - |
| Co-crystallized ligand (SAM) | - | - | - | - | −5.77 | - |
| Compounds | BBB Level a | Solubility Level b | Absorption Level c | CYP2D6 Prediction d | Hepatotoxicity Prediction e | PPB Prediction f |
|---|---|---|---|---|---|---|
| Monanchoradin A (1) | 3 | 4 | 0 | FALSE | FALSE | FALSE |
| Monanchoradin B (2) | 3 | 3 | 0 | FALSE | FALSE | FALSE |
| Monanchoradin C (3) | 3 | 3 | 0 | FALSE | FALSE | FALSE |
| Dehydrocrambescin A2 418 (4) | 4 | 3 | 2 | FALSE | FALSE | FALSE |
| Crambescidin 786 (5) | 4 | 4 | 3 | FALSE | FALSE | FALSE |
| Crambescidin 814 (6) | 4 | 4 | 3 | FALSE | FALSE | FALSE |
| Norcrambescidic acid (7) | 4 | 2 | 2 | FALSE | FALSE | FALSE |
| Monalidine A (8) | 1 | 2 | 0 | TRUE | TRUE | TRUE |
| (-)-crambescin A2 392 (9) | 4 | 4 | 1 | FALSE | FALSE | FALSE |
| (-)-crambescin A2 406 (10) | 4 | 3 | 1 | FALSE | FALSE | FALSE |
| (-)-crambescin A2 420 (11) | 4 | 3 | 2 | FALSE | FALSE | FALSE |
| Crambescidin 800 (12) | 4 | 4 | 3 | FALSE | FALSE | FALSE |
| Crambescidin 826 (13) | 4 | 4 | 3 | FALSE | FALSE | FALSE |
| Crambescidic acid (14) | 4 | 2 | 2 | FALSE | FALSE | FALSE |
| Crambescidin 359 (15) | 1 | 2 | 0 | FALSE | FALSE | FALSE |
| Daclatasvir | 4 | 3 | 3 | FALSE | TRUE | TRUE |
| Compounds | FDA Rodent Carcinogenicity | Carcinogenic Potency TD50 Mouse a | Rat Maximum Tolerated Dose (Feed) b | Rat Oral LD50 b | Rat Chronic LOAEL b,* |
|---|---|---|---|---|---|
| Monanchoradin A (1) | Non-Carcinogen | 51.0661 | 0.085 | 0.399 | 0.0168 |
| Monanchoradin B (2) | Non-Carcinogen | 52.712 | 0.091 | 0.457 | 0.0167 |
| Monanchoradin C (3) | Non-Carcinogen | 54.2866 | 0.098 | 0.509 | 0.0166 |
| Dehydrocrambescin A2 418 (4) | Non-Carcinogen | 19.5925 | 0.573 | 10.139 | 0.0450 |
| Crambescidin 786 (5) | Non-Carcinogen | 1.91771 | 0.063 | 10.559 | 0.0019 |
| Crambescidin 814 (6) | Non-Carcinogen | 1.91977 | 0.071 | 13.415 | 0.0017 |
| Norcrambescidic acid (7) | Non-Carcinogen | 5.77105 | 0.043 | 11.836 | 0.0013 |
| Monalidine A (8) | Non-Carcinogen | 32.2161 | 0.123 | 3.156 | 0.0448 |
| (-)-crambescin A2 392 (9) | Non-Carcinogen | 39.9613 | 0.310 | 2.634 | 0.0171 |
| (-)-crambescin A2 406 (10) | Non-Carcinogen | 40.6645 | 0.329 | 2.970 | 0.0168 |
| (-)-crambescin A2 420 (11) | Non-Carcinogen | 41.3406 | 0.350 | 3.269 | 0.0165 |
| Crambescidin 800 (12) | Non-Carcinogen | 1.91899 | 0.065 | 11.440 | 0.0018 |
| Crambescidin 826 (13) | Non-Carcinogen | 1.30045 | 0.042 | 14.200 | 0.0012 |
| Crambescidic acid (14) | Non-Carcinogen | 5.07065 | 0.040 | 8.153 | 0.0018 |
| Crambescidin 359 (15) | Non-Carcinogen | 0.779067 | 0.027 | 1.829 | 0.0021 |
| Daclatasvir | Non-Carcinogen | 0.970599 | 0.022 | 0.677 | 0.0063 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Demerdash, A.; Metwaly, A.M.; Hassan, A.; Abd El-Aziz, T.M.; Elkaeed, E.B.; Eissa, I.H.; Arafa, R.K.; Stockand, J.D. Comprehensive Virtual Screening of the Antiviral Potentialities of Marine Polycyclic Guanidine Alkaloids against SARS-CoV-2 (COVID-19). Biomolecules 2021, 11, 460. https://doi.org/10.3390/biom11030460
El-Demerdash A, Metwaly AM, Hassan A, Abd El-Aziz TM, Elkaeed EB, Eissa IH, Arafa RK, Stockand JD. Comprehensive Virtual Screening of the Antiviral Potentialities of Marine Polycyclic Guanidine Alkaloids against SARS-CoV-2 (COVID-19). Biomolecules. 2021; 11(3):460. https://doi.org/10.3390/biom11030460
Chicago/Turabian StyleEl-Demerdash, Amr, Ahmed M. Metwaly, Afnan Hassan, Tarek Mohamed Abd El-Aziz, Eslam B. Elkaeed, Ibrahim H. Eissa, Reem K. Arafa, and James D. Stockand. 2021. "Comprehensive Virtual Screening of the Antiviral Potentialities of Marine Polycyclic Guanidine Alkaloids against SARS-CoV-2 (COVID-19)" Biomolecules 11, no. 3: 460. https://doi.org/10.3390/biom11030460