
Abstract
Purpose
Trials of tocilizumab in patients with severe COVID-19 pneumonia have demonstrated mixed results, and the role of tocilizumab in combination with other treatments is uncertain. Here we evaluated whether tocilizumab plus remdesivir provides greater benefit than remdesivir alone in patients with severe COVID-19 pneumonia.
Methods
This randomized, double-blind, placebo-controlled, multicenter trial included patients hospitalized with severe COVID-19 pneumonia requiring > 6 L/min supplemental oxygen. Patients were randomly assigned (2:1 ratio) to receive tocilizumab 8 mg/kg or placebo intravenously plus ≤ 10 days of remdesivir. The primary outcome was time from randomization to hospital discharge or “ready for discharge” (defined as category 1, assessed by the investigator on a 7-category ordinal scale of clinical status) to day 28. Patients were followed for 60 days.
Results
Among 649 enrolled patients, 434 were randomly assigned to tocilizumab plus remdesivir and 215 to placebo plus remdesivir. 566 patients (88.2%) received corticosteroids during the trial to day 28. Median time from randomization to hospital discharge or “ready for discharge” was 14 (95% CI 12–15) days with tocilizumab plus remdesivir and 14 (95% CI 11–16) days with placebo plus remdesivir [log-rank P = 0.74; Cox proportional hazards ratio 0.97 (95% CI 0.78–1.19)]. Serious adverse events occurred in 128 (29.8%) tocilizumab plus remdesivir and 72 (33.8%) placebo plus remdesivir patients; 78 (18.2%) and 42 (19.7%) patients, respectively, died by day 28.
Conclusions
Tocilizumab plus remdesivir did not shorten time to hospital discharge or “ready for discharge” to day 28 compared with placebo plus remdesivir in patients with severe COVID-19 pneumonia.
Similar content being viewed by others

In this randomized controlled trial of patients with severe COVID-19 pneumonia, the median time from randomization to hospital discharge or “ready for discharge” was 14 days with tocilizumab plus remdesivir and 14 days with placebo plus remdesivir. Although large platform trials showed a survival benefit of tocilizumab in patients with severe COVID-19 and declining respiratory status, this trial did not confirm treatment benefit of tocilizumab in combination with remdesivir. |
Introduction
Coronavirus disease 2019 (COVID-19) has rapidly developed into a global health threat since emerging in China in late 2019 [1]. Most patients experience mild disease and recover with symptomatic treatment and supportive care. However, a subset experience more severe illness necessitating hospitalization [2]. Among patients who seek medical care, approximately 20% experience complications that may progress to acute respiratory distress syndrome (ARDS), multiorgan failure, and death [3].
Tocilizumab is a humanized monoclonal anti–interleukin-6 receptor-alpha antibody. Interleukin-6 levels are often increased in patients with acute lung injury and ARDS, including those with severe COVID-19 pneumonia [4, 5]. Tocilizumab improved survival, reduced the need for mechanical ventilation, and shortened the length of hospital stay compared with standard care in 2 large open-label platform trials (REMAP-CAP and RECOVERY) [6, 7]. EMPACTA, a randomized, double-blind, placebo-controlled trial of patients hospitalized with COVID-19 pneumonia but not receiving ventilatory support, showed reduced likelihood of progression to mechanical ventilation or death with tocilizumab but no survival benefit [8]. Other randomized controlled trials have not shown survival benefit with tocilizumab [9,10,11,12], although potential clinical benefits in reduced need for mechanical ventilation, duration of intensive care unit (ICU) stay, and length of hospital stay were observed in COVACTA and CORIMUNO-TOCI 1 [9, 11]. A prospective meta-analysis of 27 randomized trials involving more than 10,000 patients hospitalized for COVID-19 demonstrated that interleukin-6 antagonists were associated with lower 28-day all-cause mortality [13]. Rapidly evolving standards of care during the COVID-19 pandemic have resulted in further uncertainty about the use of tocilizumab in combination with other treatments, including corticosteroids and remdesivir [14].
Remdesivir is a selective inhibitor of the viral RNA-dependent RNA polymerase that delays RNA chain termination during replication of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) [15] and decreases the efficiency of viral nucleotide incorporation [16]. A 10-day course of remdesivir was superior to standard care alone in reducing time to recovery in a double-blind, randomized, placebo-controlled trial of patients hospitalized with severe COVID-19 [17], and a 5-day course demonstrated clinical benefit in moderate COVID-19 [18]. We hypothesized that combining remdesivir and tocilizumab could be more effective than adding remdesivir alone to standard care, which could include corticosteroids.
Methods
Trial design and patients
REMDACTA, a randomized, double-blind, placebo-controlled, multicenter, phase 3 trial, evaluated the efficacy and safety of tocilizumab plus remdesivir versus placebo plus remdesivir in patients aged 12 years and older hospitalized with severe COVID-19 pneumonia between June 2020 and March 2021. Patients were required to have a positive SARS-CoV-2 polymerase chain reaction test result within 7 days of randomization, pneumonia confirmed by chest x-ray or computed tomography, and hypoxemia requiring > 6 L/min supplemental oxygen. The protocol was amended in September 2020 to allow enrollment of patients who received ≤ 2 doses of remdesivir before randomization. Patients were excluded if the estimated glomerular filtration rate was < 30 mL/min (including patients undergoing hemodialysis or hemofiltration) or alanine aminotransferase or aspartate aminotransferase levels were > 5 × the upper limit of normal within 24 h of screening. Patients with suspected active bacterial, fungal, viral, or other infection except COVID-19 were excluded. Systemic corticosteroids for treatment of COVID-19 pneumonia were permitted. Treatment with convalescent plasma, chloroquine or hydroxychloroquine, antivirals, biologics, and Janus kinase inhibitors during the trial was prohibited.
Informed consent was obtained from the patient or legally authorized representative. The trial was conducted in accordance with the ICH E6 guidance for Good Clinical Practice and the principles of the Declaration of Helsinki or local laws and regulations, whichever afforded greater protection. The trial was approved by the institutional review board or ethics committee at each site. The sponsor, F. Hoffmann-La Roche Ltd., designed the trial and performed analyses; a contract research organization paid by the sponsor monitored the trial under the direction and supervision of the sponsor.
Eligible patients were randomly assigned in a 2:1 ratio using an interactive web-based response system and a permuted block method to receive blinded treatment with tocilizumab plus remdesivir or placebo plus remdesivir. Randomization was stratified by geographic region (North America, Europe, other) and by a 2-level factor based on clinical status at screening (ordinal scale categories 4–5, category 6) on a 7-category ordinal scale (additional details are in the Online Resource). Remdesivir was administered intravenously, followed by a single intravenous dose of tocilizumab 8 mg/kg (maximum, 800 mg) or placebo on day 1. Patients with sustained fever or clinically significant worsening of signs and symptoms of COVID-19 (e.g., increased supplemental oxygen requirement) could receive a second infusion of blinded tocilizumab or placebo within 8 to 24 h of the first infusion. Patients were monitored through day 60, and the primary end point was assessed at day 28. Additional details and the complete trial protocol are in the Online Resource. The trial is registered on ClinicalTrials.gov (https://clinicaltrials.gov/ct2/show/NCT04409262).
Outcomes
The primary outcome was time from randomization to hospital discharge or “ready for discharge,” defined as category 1, assessed by the investigator on the 7-category ordinal scale to day 28. Patients achieved the outcome at the time of discharge or when they achieved category 1 on the 7-category ordinal scale, whichever occurred first, provided they had no further ordinal scale assessments greater than category 1 on or before day 28, they were not readmitted to the hospital on or before day 28, and they did not die on or before day 28.
The primary outcome was initially defined as clinical status assessed by the investigator using a 7-category ordinal scale of clinical status on day 28, but this was changed to time from randomization to hospital discharge or “ready for discharge” to day 28 in response to evolving external data, including results from COVACTA and EMPACTA, which suggested that time to discharge was a more sensitive outcome for trials in this patient population [8, 9]. The amendment was finalized in September 2020, and the original primary outcome was retained as a secondary outcome. Ordinal scale categories are as follows: 1, discharged or “ready for discharge” (as evidenced by normal body temperature and respiratory rate, and stable oxygen saturation on ambient air or ≤ 2 L/min supplemental oxygen); 2, non–ICU hospital ward, not requiring supplemental oxygen; 3, non–ICU hospital ward, requiring supplemental oxygen; 4, ICU or non–ICU hospital ward, requiring noninvasive ventilation or high-flow oxygen; 5, ICU, requiring intubation and mechanical ventilation; 6, ICU, requiring extracorporeal membrane oxygenation or mechanical ventilation and additional organ support; 7, death. Key secondary efficacy outcomes assessed to day 28 were time from randomization to mechanical ventilation or death, clinical status on the ordinal scale at day 14, and time to death. Mortality was assessed at day 28 and day 60. Adverse events were recorded according to the Medical Dictionary for Regulatory Activities, version 23.1.
Statistical analysis
Assuming a median time to hospital discharge or “ready for discharge” of 11 days in the placebo plus remdesivir arm [17], a sample size of 650 patients accruing approximately 520 events was calculated to provide 80% power to detect a hazard ratio of 1.3 or an approximate 2.5-day reduction in median time to hospital discharge or “ready for discharge” for tocilizumab plus remdesivir.
Efficacy was assessed in the modified intention-to-treat population, defined as all randomly assigned patients who received any amount of tocilizumab or placebo grouped according to randomly assigned treatment arm. Safety was assessed in the safety-evaluable population, defined as all patients who received any amount of study medication (remdesivir, tocilizumab/placebo) according to treatment received.
The primary outcome was compared between the treatment arms using a log-rank test stratified by region (North America, Europe, other) and baseline ordinal scale category (4–5, 6). Treatment arms were compared using a Cox proportional hazards model adjusted for stratification factors. Patients who died on or before day 28 were censored at day 28, and patients who withdrew or were lost to follow-up before achieving the primary outcome (not followed by death) were censored at their last recorded ordinal scale assessment. The primary end point was tested at a 2-sided 5% significance level. Kaplan–Meier plots, medians, 95% CIs, and P values from the stratified log-rank test were calculated for time-to-event primary and secondary efficacy outcomes, and distributions were compared using the Cox model. Ordinal data were analyzed using a proportional odds model comparing treatment arms and accounting for stratification factors at randomization. Effect sizes, P values, and 95% CIs were calculated based on odds ratios for treatment effect from the proportional odds model. The Cochran–Mantel–Haenszel test adjusted by the stratification factors at randomization was used to analyze mortality at day 28 and day 60. Additional details are in the Online Resource.
Results
Patients
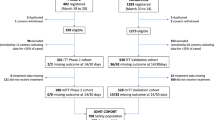
Overall, 709 patients were screened and 649 were enrolled from Brazil [n = 154 (23.7%)], Russia [n = 49 (7.6%)], Spain [n = 14 (2.2%)], and the United States [n = 432 (66.6%)] between June 2020 and January 2021 at 53 trial sites; 434 were randomly assigned to the tocilizumab plus remdesivir arm and 215 to the placebo plus remdesivir arm (Fig. 1). Four patients in the tocilizumab plus remdesivir arm and five patients in the placebo plus remdesivir arm did not receive tocilizumab or placebo and were excluded from the modified intention-to-treat population (additional details on the analysis populations are in the Online Resource). Approximately three-quarters of patients completed the trial to day 28: 336 (77.4%) in the tocilizumab plus remdesivir arm and 160 (74.4%) in the placebo plus remdesivir arm. Death was the most common reason for not completing the trial; 78 patients (18%) in the tocilizumab plus remdesivir arm and 42 (19.5%) in the placebo plus remdesivir arm died on or before day 28. Among the remaining patients, in the tocilizumab plus remdesivir arm, 9 (2.1%) were lost to follow-up, 7 (1.6%) withdrew consent, 2 (0.5%) discontinued because of adverse events, 1 (0.2%) discontinued following a protocol deviation, and 1 (0.2%) discontinued for other reasons that were not specified; in the placebo plus remdesivir arm, 7 patients (3.3%) were lost to follow-up, 3 (1.4%) withdrew consent, and 3 (1.4%) discontinued following protocol deviations (Fig. 1).

The safety population was defined as all patients who received any amount of study medication (remdesivir, tocilizumab/placebo) with patients grouped according to treatment received. aIncludes 1 patient who was randomly assigned to the tocilizumab plus remdesivir arm but received placebo plus remdesivir. bDoes not include 1 patient who was randomly assigned to the tocilizumab plus remdesivir arm but received placebo plus remdesivir; this patient was included in the placebo plus remdesivir arm of the safety population. cIncludes the patient who was randomly assigned to the tocilizumab plus remdesivir arm but received placebo plus remdesivir and 2 patients (1 from each treatment arm) who received remdesivir but not tocilizumab or placebo. ALT alanine aminotransferase, AST aspartate aminotransferase, eGFR estimated glomerular filtration rate, PCR, polymerase chain reaction, ULN upper limit of normal
A second dose of tocilizumab or placebo was administered to 85 patients (19.8%) in the tocilizumab plus remdesivir arm and 48 (22.7%) in the placebo plus remdesivir arm (safety population) 8 to 24 h after the first tocilizumab/placebo dose for sustained fever or clinically significant worsening of signs or symptoms. Among all randomly assigned patients, 266 (41%) discontinued remdesivir early, 171 (39.4%) in the tocilizumab plus remdesivir arm, and 95 (44.2%) in the placebo plus remdesivir arm. The most common reason for discontinuing remdesivir was discharge from the study hospital before completing 10 days of treatment [tocilizumab plus remdesivir, 99 patients (22.8%); placebo plus remdesivir, 55 patients (25.6%)].
Demographics and disease characteristics at baseline were generally well balanced between treatment arms (Table 1). Most patients were at ordinal scale category 4 at baseline [tocilizumab plus remdesivir, 336 (78.1%); placebo plus remdesivir, 175 (83.3%)]. Similar proportions of patients received remdesivir before randomization (tocilizumab plus remdesivir, 19.3%; placebo plus remdesivir, 19%). Most patients received systemic corticosteroids at baseline [tocilizumab plus remdesivir, 357/429 (83.2%); placebo plus remdesivir 184/213 (86.4%); safety population] or during the trial to day 28 [tocilizumab plus remdesivir, 378/429 (88.1%); placebo plus remdesivir 188/213 (88.3%); safety population].
Primary outcome
Median time from randomization to hospital discharge or “ready for discharge” to day 28 was 14 (95% CI 12–15) days in the tocilizumab plus remdesivir arm and 14 (95% CI 11–16) days in the placebo plus remdesivir arm [log-rank P = 0.74; Cox proportional hazards ratio 0.97 (95% CI 0.78–1.19)] (Fig. 2a, Table 2). The proportion of patients discharged or “ready for discharge” to day 28 was 66% in the tocilizumab plus remdesivir arm and 67.1% in the placebo plus remdesivir arm. There were no significant differences in time to hospital discharge or “ready for discharge” between treatment arms among subgroups of patients according to demographics, corticosteroid use at baseline, remdesivir use before randomization, or mechanical ventilation at baseline. Time to hospital discharge or “ready for discharge” did not differ according to baseline ordinal scale category except among patients in ordinal category 5 at baseline (39 in the tocilizumab plus remdesivir group, 9 in the placebo plus remdesivir group) for whom tocilizumab was associated with longer time to discharge [hazard ratio 0.36 (95% CI 0.14–0.91)]; however, the small number of patients in this subgroup limits interpretability (Online Resource Fig. S1). No difference was observed between treatment arms for the original primary outcome: clinical status at day 28 on the 7-category ordinal scale (Online Resource Fig. S2).

Time to (a) hospital discharge or “ready for discharge” (primary outcome), (b) mechanical ventilation or death, (c) death. Data are shown as (a) 1 minus the Kaplan–Meier curve, (b) Kaplan–Meier curve for time to mechanical ventilation or death, and (c) Kaplan–Meier curve for time to death (modified intention-to-treat population). Panel a shows time to hospital discharge or “ready for discharge” defined as days from randomization to hospital discharge or “ready for discharge” not followed by ordinal scale category > 1, hospital readmission, or death. Patients who discontinued or were lost to follow-up for any reason before hospital discharge or “ready for discharge” criteria were met were censored at their last recorded ordinal scale assessment. Panel b shows time to mechanical ventilation or death defined as the time from randomization to the first occurrence of death or mechanical ventilation. For patients already receiving mechanical ventilation at baseline, only death was counted as an event. One patient had a missing baseline mechanical ventilation record; therefore, the baseline ordinal scale category (category 3: non-ICU hospital ward or “ready for hospital ward” requiring supplemental oxygen) was used to impute baseline mechanical ventilation status as not receiving mechanical ventilation. Patients who withdrew or were lost to follow-up before discharge (not followed by death) were censored at their last assessment of vital signs. Patients who withdrew or who were lost to follow-up on or after the day of discharge (not followed by death or hospital readmission) were censored at day 28. Panel c shows time to death defined as the time from randomization to death. Patients who withdrew or were lost to follow-up before discharge (not followed by death) were censored at the last known date they were alive. Patients who withdrew or were lost to follow-up on or after the day of discharge (not followed by death or hospital readmission) were censored at day 28. ICU intensive care unit
Secondary outcomes
Mechanical ventilation or death to day 28 occurred in 123 patients (28.6%) in the tocilizumab plus remdesivir arm and 61 patients (29%) in the placebo plus remdesivir arm. Median time to mechanical ventilation or death to day 28 was nonevaluable in both treatment arms because it occurred in less than half the patients [log-rank P = 0.90; hazard ratio 0.98 (95% CI 0.72–1.34)] (Fig. 2b, Table 2). Mean clinical status on the 7-category ordinal scale at day 14 was 2.8 (95% CI 2.6–3) in the tocilizumab plus remdesivir arm and 2.9 (95% CI 2.6–3.2) in the placebo plus remdesivir arm [difference –0.07 (–0.4, 0.3); P = 0.72] (Table 2) [because the assumption of proportional odds was not met (Online Resource Table S1), the prespecified difference in means analysis is presented]. Proportions of patients in each ordinal scale category at day 14 were similar between treatment arms (Table 2, Online Resource Fig. S3). Seventy-eight patients (18.1%) in the tocilizumab plus remdesivir arm and 41 (19.5%) in the placebo plus remdesivir arm died by day 28 [weighted difference –1.3% (95% CI –7.8%, 5.2%); P = 0.69]. Median time to death was nonevaluable in both arms [log-rank P = 0.79; hazard ratio 0.95 (95% CI, 0.65–1.39)] (Fig. 2c, Table 2). No significant difference was observed in time to death to day 28 between treatment arms among subgroups of patients based on demographics, corticosteroid use at baseline, remdesivir use before randomization, or mechanical ventilation at baseline (Online Resource Fig. S1). By day 60, 97 patients (22.6%) in the tocilizumab plus remdesivir arm and 54 (25.7%) in the placebo plus remdesivir arm had died [weighted difference –3% (95% CI –10.1%, 4%); P = 0.39].
Safety to day 28
In the safety population, the number of patients who experienced ≥ 1 adverse event was 320 of 429 (74.6%) in the tocilizumab plus remdesivir arm and 147 of 213 (69%) in the placebo plus remdesivir arm. Serious adverse events were reported in 128 patients (29.8%) in the tocilizumab plus remdesivir arm and 72 (33.8%) in the placebo plus remdesivir arm (Table 3, Online Resource Table S2). Forty-six patients (10.7%) in the tocilizumab plus remdesivir arm and 28 (13.1%) in the placebo plus remdesivir arm discontinued treatment because of an adverse event. Adverse events of special interest were balanced between both treatment arms, including serious infections [86 (20%) and 53 (24.9%)] and serious bleeding events [11 (2.6%) and 7 (3.3%)]. The most common serious infections (reported in > 1% of patients in each treatment arm) were progression of underlying COVID-19 pneumonia (required to be reported as a serious adverse event if COVID-19 resulted in death), septic shock, pneumonia, sepsis, and bacterial pneumonia.
Discussion
In this trial, there was no difference between tocilizumab plus remdesivir and placebo plus remdesivir in time from randomization to hospital discharge or “ready for discharge” to day 28. There were also no differences between treatment arms in the key secondary outcomes of time to mechanical ventilation or death to day 28, clinical status at day 14 assessed by the investigator on the 7-category ordinal scale, and time to death to day 28. No new safety signals were identified in this trial, and the safety profile was consistent with the known safety profiles of tocilizumab and remdesivir. Adverse events of special interest, including the incidence and types of serious infections, were balanced between the treatment arms and consistent with the known disease course of severe COVID-19.
Compared with other randomized placebo-controlled trials of tocilizumab in severe COVID-19, REMDACTA differed with respect to target patient population and background treatments, in part because of the continuing evolution of standard care. In EMPACTA, which demonstrated a treatment benefit on time to mechanical ventilation or death for tocilizumab versus placebo, 64.2% of patients were receiving low-flow oxygen at enrollment, 54.6% received remdesivir, and > 80% received systemic corticosteroids. COVACTA recruited patients with a broader range of disease severity, and fewer patients received effective background therapy (< 10% of patients received remdesivir, < 50% received corticosteroids). These differences in patient selection and treatments may account for the different outcomes between the studies.
Based on results of the REMAP-CAP [6] and RECOVERY [7] open-label platform trials, tocilizumab in combination with dexamethasone is recommended by the National Institutes of Health COVID-19 Treatment Guidelines Panel in certain patients hospitalized with COVID-19 who exhibit rapid respiratory decompensation and, at a minimum, need for high-flow oxygen supplementation [19]. In RECOVERY, patients considered for random allocation to the tocilizumab arm were required to have clinical evidence of progressive COVID-19 after their first random allocation in the trial. In REMAP-CAP, patients were enrolled within 24 h of initiation of organ support in the ICU, defined as invasive or noninvasive mechanical ventilation (including high-flow nasal cannula with flow rate > 30 L/min of and fraction of inspired oxygen > 0.4) or intravenous infusion of any vasopressor or inotrope. REMDACTA enrolled patients who required > 6 L/min of supplemental oxygen irrespective of time since symptom onset time since or since diagnosis of COVID-19 and evidence of recent or rapid deterioration was not required. Together, these findings suggest that patients with declining respiratory status may be more likely to benefit from treatment with tocilizumab. Additional studies are needed to confirm this hypothesis.
The endorsement of interleukin-6 receptor blockade in patients with severe and critical COVID-19 by the World Health Organization and others will likely increase the adoption of tocilizumab as standard of care in these patients [14]. Although trials of remdesivir have not demonstrated a clear mortality benefit [20], remdesivir remains standard of care in many parts of the world based on trials that demonstrated shortened time to recovery and other clinical benefits [17, 18]. Only 27% of patients in RECOVERY and 33% of patients in REMAP-CAP received remdesivir. Thus, it is important to better understand the role of tocilizumab in combination with remdesivir and other treatments, including newer antivirals, neutralizing antibodies, and other immunomodulators. Additional studies or patient-level meta-analyses may be required to achieve this [21].
Strengths and weaknesses
To our knowledge, REMDACTA is the first multicenter, double-blind, randomized, placebo-controlled trial to investigate the inhibition of interleukin-6 signaling in patients with COVID-19, all of whom received remdesivir and most of whom also received systemic corticosteroids. However, REMDACTA was not powered to detect the relatively small but clinically meaningful mortality benefit demonstrated in larger platform studies. REMDACTA was designed and initiated relatively early in the pandemic, before results from other randomized controlled trials of tocilizumab were available. Thus, the primary outcome of the trial was changed from clinical status on the ordinal scale to time to discharge or “ready for discharge” when enrollment was approximately half-completed based on the results from other trials, which suggested that time to discharge or “ready for discharge” is a more sensitive and clinically meaningful outcome in patients with severe COVID-19 [8, 9]. REMDACTA eligibility criteria were also modified to allow enrollment of patients who had received up to 2 doses of remdesivir before randomization because remdesivir was increasingly administered as standard of care. These changes were implemented while patients were still enrolling in the trial, and the protocol and statistical analysis plan were finalized on February 22, 2021, before unblinding on March 1, 2021.
Despite randomization, there were some slight imbalances in baseline characteristics between the treatment arms (e.g., more patients aged 65 and older, more patients requiring invasive mechanical ventilation, and fewer patients receiving corticosteroids in the tocilizumab plus remdesivir arm). These imbalances were unlikely to have been important individually but could have created a cumulative bias in favor of placebo plus remdesivir. A slightly higher proportion of patients in the placebo plus remdesivir arm than the tocilizumab plus remdesivir arm discontinued remdesivir before completing 10 days of treatment (44.2% vs 39.4%); however, most of these early discontinuations were the result of hospital discharge, consistent with remdesivir use in other trials [17] and in clinical practice.
Conclusion
In this randomized, double-blind, placebo-controlled trial, tocilizumab plus remdesivir did not shorten time to hospital discharge or “ready for discharge” to day 28 compared with placebo plus remdesivir in patients with severe COVID-19 pneumonia, most of whom received systemic corticosteroids. Serious infections were not more frequent with tocilizumab treatment, and no new safety signals were identified.
References
World Health Organization. Coronavirus disease (COVID-19) pandemic. https://www.who.int/emergencies/diseases/novel-coronavirus-2019. Accessed July 6, 2021
World Health Organization. Clinical management of severe acute respiratory infection (SARI) when COVID-19 disease is suspected: interim guidance 13 March 2020. https://www.who.int/docs/default-source/coronaviruse/clinical-management-of-novel-cov.pdf. Accessed July 6, 2021
World Health Organization. Coronavirus disease (COVID-2019) weekly epidemiological update and weekly operational update. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports. Accessed July 6, 2021
Bain W, Yang H, Shah FA, Suber T, Drohan C, Al-Yousif N et al (2021) COVID-19 versus non-COVID ARDS: comparison of demographics, physiologic parameters, inflammatory biomarkers and clinical outcomes. Ann Am Thorac Soc. https://doi.org/10.1513/AnnalsATS.202008-1026OC
McElvaney OJ, McEvoy NL, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM et al (2020) Characterization of the inflammatory response to severe COVID-19 illness. Am J Respir Crit Care Med 202:812–821. https://doi.org/10.1164/rccm.202005-1583OC
The REMAP-CAP Investigators (2021) Interleukin-6 receptor antagonists in critically ill patients with Covid-19. N Engl J Med 384:1491–1502. https://doi.org/10.1056/NEJMoa2100433
RECOVERY Collaborative Group (2021) Tocilizumab in patients admitted to hospital with COVID-19 (RECOVERY): a randomised, controlled, open-label, platform trial. Lancet 397:1637–1645
Salama C, Han J, Yau L, Reiss WG, Kramer B, Neidhart JD et al (2021) Tocilizumab in patients hospitalized with Covid-19 pneumonia. N Engl J Med 384:20–30. https://doi.org/10.1056/NEJMoa2030340
Rosas IO, Bräu N, Waters M, Go RC, Hunter BD, Bhagani S et al (2021) Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. N Engl J Med 384:1503–1516. https://doi.org/10.1056/NEJMoa2028700
Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS et al (2020) Efficacy of tocilizumab in patients hospitalized with Covid-19. N Engl J Med 383:2333–2344. https://doi.org/10.1056/NEJMoa2028836
Hermine O, Mariette X, Tharaux PL, Resche-Rigon M, Porcher R, Ravaud P (2021) Effect of tocilizumab vs usual care in adults hospitalized with COVID-19 and moderate or severe pneumonia: a randomized clinical trial. JAMA Intern Med 181:32–40. https://doi.org/10.1001/jamainternmed.2020.6820
Salvarani C, Dolci G, Massari M, Merlo DF, Cavuto S, Savoldi L et al (2021) Effect of tocilizumab vs standard care on clinical worsening in patients hospitalized with COVID-19 pneumonia: a randomized clinical trial. JAMA Intern Med 181:24–31. https://doi.org/10.1001/jamainternmed.2020.6615
The WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group (2021) Association between administration of IL-6 antagonists and mortality among patients hospitalized for COVID-19: a meta-analysis. JAMA. https://doi.org/10.1001/jama.2021.11330
World Health Organization (2020) A living WHO guideline on drugs for covid-19. BMJ 370:m3379. https://doi.org/10.1136/bmj.m3379
Gordon CJ, Tchesnokov EP, Woolner E, Perry JK, Feng JY, Porter DP et al (2020) Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J Biol Chem 295:6785–6797. https://doi.org/10.1074/jbc.RA120.013679
Tchesnokov EP, Gordon CJ, Woolner E, Kocinkova D, Pery JK, Feng JY et al (2020) Template-dependent inhibition of coronavirus RNA-dependent RNA polymerase by remdesivir reveals a second mechanism of action. J Biol Chem 295:16156–16165. https://doi.org/10.1074/jbc.AC120.015720
Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman S, Kalil AC et al (2020) Remdesivir for the treatment of Covid-19—final report. N Engl J Med 383:1813–1826. https://doi.org/10.1056/NEJMoa2007764
Spinner CD, Gottlieb RL, Criner GJ, Arribas López JR, Cattelan AM, Soriano Viladomiu A et al (2020) Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. JAMA 324:1048–1057. https://doi.org/10.1001/jama.2020.16349
National Institutes of Health. The COVID-19 Treatment Guidelines Panel’s statement on the use of tocilizumab for the treatment of COVID-19. March 5, 2021. https://files.covid19treatmentguidelines.nih.gov/guidelines/archive/updated-statement-on-tocilizu-03-05-2021.pdf. Accessed July 6, 2021
Pan H, Peto R, Henao-Restrepo AM, Preziosi MP, Sathiyamoorthy V, Karim QA et al (2021) Repurposed antiviral drugs for Covid-19—interim WHO solidarity trial results. N Engl J Med 384:497–511. https://doi.org/10.1056/NEJMoa2023184
Butler E, Warrer Munch M, Venkatesh B (2021) Time for tocilizumab in COVID-19? Intensive Care Med 47:692–694
Acknowledgements
The first draft of the manuscript was written with medical writing support provided by Sara Duggan, PhD, of ApotheCom; this support was funded by F. Hoffmann-La Roche Ltd.
REMDACTA Investigators: BRAZIL - CEMEC – Centro Multidisciplinar de Estudos Clínicos, São Bernardo Do Campo: Adilson Joaquim Westheimer Cavalcante, Loni Dorigo, Julia Minghini, Marina Miranda, Julia Ribeiro, Priscila Barboza; Hospital de Base, São José do Rio Preto: Suzana Margareth Lobo, Graziela Bernadin Luckemeyer, Paula Spinasse Borges, Fernando Henrique Scatena Garcia, Tamiris Moimaz, Luana Machado, Juliane Garcia, Ana Carolina Affonso, Keulle Candido, Gabriella Navarro, Gabriela Taparo, Danelli Frasatto, Maria Fernanda Santos; Hospital Nossa Senhora das Graças, Curitiba: Clovis Cunha, Fernanda Pedroso, Gustvo Giarovini, Maicon Pinto, Allan Silva, Eduardo Detgel, Murilo Henrique Guedes, Ana Paula Cunha, Açucena Maximo, Gisele Padilha, Kelly Ferreira; Instituto de Infectologia Emilio Ribas, São Paulo: Tâmara Newman Lobato Souza, Maria Silvia Biagioni Santos, Ana Luiza de Castro Conde Toscano, Bernardo Porto Maia, Gustavo Dittmar, Denise Peluso, Luciana Borges, Mario Gonzalez, Anna Karina Mostachio, Luiz Carlos Pereira Junior; Instituto de Pesquisa Clínica Evandro Chagas FIOCRUZ, Rio de Janeiro: Beatriz Grinsztejn, Sandra Wagner Cardoso, Isabel Mendes, Isabel Tavares, Rafael Fraga, Monique Silveira, Mariah Pires, Maria Pia Diniz Ribero, Rodrigo Escada, Larissa Melo Villela, Gabriela Robleda, Lucimar Santos Salgado, Jessica Felix; Instituto do Coração – HCFMUSP, São Paulo: Ludhmila Abrahão Hajjar, Fernando Oliveira, Fernanda Andrade, Thalita Gonzales, Brenno Rizerio Gomes, Katia Sansivieri, Alexandra Vieira; Santa Casa de Misericordia de Belo Horizonte, Belo Horizonte: Antonio Tarcísio de Faria Freire, Douglas Dias e Silva, Alexandre Barros, Claudia Oliveira, Thayse Kayser, Flavia Medeiros, Graziele Vieira, Daniela Pereira Mendes, Jaqueline Soares de Oliveira Gualberto, Viviane Santos, Joelma Souza. RUSSIAN FEDERATION - City Clinical Hospital #52, Moscow: Daria Fomina, Maryana Lysenko; City Pokrovskaya Hospital, St. Petersburg: Alexander Vishnevskiy, Eugeniy Morozov, Dmitriy Nikolaev, Anton Sizov, Maria Kiseleva, Mikhail Terskikh; Medsi Clinic, Moscow: Anastasiya Mochalova, Mirzakhmed Aliekserov, Armen Oganesyan, Viktoria Mikhailova; City Clinical Hospital No. 15, Moscow: Ivan Gordeev, Kseniya Polubatonova, Kseniya Komissarova, Vitaliy Firstov, Vartan Grigoryan, Ilya Kokorin, Nataliya Suvorova, Anna Kozlova, Sevinch Mamedguseyinova, Valentin Kokorin, Nina Lapochkina. SPAIN - Hospital General Universitario de Guadalajara, Guadalajara: Olga Mediano, Pilar Resano, Ana Ampuero, Sofia RomeroPeralta, Rosa Mediano, Laura Silgado Martinez, Esther Viejo; Bellvitge University Hospital, L’Hospitalet del Llobregat: Jordi Carratalà, Alexander Rombauts, Gabriela Abelenda Alonso, Elena Fabra; Hospital Universitario Fundación Jiménez Díaz-Universidad Autónoma de Madrid, Madrid, Spain: Miguel Górgolas Hernández-Mora, Aws Valeed Mohammed Al-Hayani, Alfonso Cabello Úbeda, Laura Prieto Perez, Beatriz Álvarez Álvarez, Irene Carrillo Acosta, Cristina Algar Arevalo, Inmaculada Burillo, Marta Fernández Pinilla; Hospital Universitario HM Torrelodones, Torrelodones: Pablo Alejandro Cardinal Fernandez, Eduardo Domínguez, Gema Aguila, Mercedes Villareal, Manuel Francisco Gil, Adrian Nieto; Hospital Universitario Principe de Asturias, Alcala de Henares: Jose Sanz Moreno, Maria Novella-Mena, Cristina Hernandez-Gutierrez. UNITED STATES of AMERICA - Advocate Christ Medical Center, Oak Lawn, IL: Kenneth Dodd, David Barounis, Michael TeKippe, Ronda Oram, Adam Treitman, Valerie Griffeth, Kathy Hesse, Colleen Valenti, Mary Murray, Nicole Hannigan, Muthana Sweis, Paul Maze, Christopher Doherty, Sherri Velez; Baylor St Luke’s Medical Center, Houston, TX: Ivan Rosas, Nicola Hanania, Kalpalatha Guntupalli, Fernando Poli de Frias, Laura Bertrand, Mustafa Atik, Hinali Zaveri; Baylor Scott & White Medical Center – College Station, College Station, TX: Robert L. Gottlieb, Timothy M. Byrd, Seth J. Sullivan, Marvin Dao, Samantha Beevers, Vanessa Hoelscher, Rachel Lovett; Baylor Scott & White Medical Center – Irving, Irving, TX: Robert L. Gottlieb, Steven G. Davis, Robert M. Engle, Tania Purkayastha, Jon Thammavong, Brittany Garcia, Melissa Ainslie, Alyssa Boudreau, Krizia Duque, Demi Lira, Dedra Preece, Noelia Ramirez, Felicity Vaughan, Mary Wallace, Decolia Washington, Amy C. Thomas, Susan Ferdosian, Chad Samuelson; Baylor Scott & White Medical Center – Plano, Plano, TX: Robert L. Gottlieb, J. Michael DiMaio, Aasim Afzal, Katrina Youngblood, Julie McCracken, Christine Brooks, Penni Dolton, Raven Harding, Anne LaRock, Pauline Natheri, Jacqueline Nguyen; Baylor Scott & White Medical Center – Temple, Temple, TX: Robert L. Gottlieb, John Midturi, Thomas Delmas, Shekhar Ghamande, Heath White, Ludmyla Ploskanych, Rohini Bagewadi, Wanda Fikes, Mackenzie Morquecho, Dayna Sawyer, Jessica Christian, Katelyn Brockway, John Thompson, Stefani Lehman, Martha Zayed; Baylor University Medical Center, Dallas, TX: Robert L. Gottlieb, Cedric W. Spak , Shelley A. Hall, Mark Hupert, Janet Jerrow, Katalin Martits-Chalangari, Erica Lusk, Dianna Robinson, Sarah Burris, Maris Adams, Rebecca Baker, Giselle Carino, Laura Clariday, Tanqunisha Coleman, Rania El Tomi, Nazia Iram, Felecia Jones, Catherine Kumwenda-Wilson, Kiara Leonard, Eva Patel, Shannon Perez, Raven Robert, Caren Swift, Emma Turner, Samantha Wang, Aaron Killian, Adriana Palacios, Edilia Solis; Baystate Medical Center, Springfield, MA: Daniel Skiest, Hans Schlecht, Abigail Orenstein, Armando Paez, Mark Tidswell, Durane Walker, Sherell Thornton-Thompson; Ben Taub General Hospital, Houston, TX: Ivan Rosas, Nicola Hanania, Kalpalatha Guntupalli, Fernando Poli de Frias, Mustafa Atik, Laura Bertrand, Benazir Khan, Juan Rodriguez, Rene Plascencia, Hinali Zaveri; Boston Medical Center, Boston, MA: Nina H. Lin, Archana Asundi, Tara Bouton, Samantha Roche, Yunhan Chen, Athina Schmidt, Jennifer Bombard; Henry Ford Medical Center, Detroit, MI: Mayur Ramesh, George Alangaden, Katrina Williams, Melissa Resk, Victoria Churchill; Hoag Hospital Newport Beach and Irvine, Irvine, CA: Philip Robinson, Rosie Blancas, Bahram Alavynejad, Neema Aghamohammadi, Mazda Aghamohammadi, Martin Fee, Nathan Gilmore, Farjad Sarafian, Usman Shah, Laura Heim, Laurie Hendricks, Marcus Breit, Rachel Romansik, Adriene Swietlikowski, Wendy Price, Atessa Kiani; Holy Cross Health, Fort Lauderdale, FL: Zdenka Segota, Stephen Renae, Margaret Juliana Gorensek, Carlos Octavio Guerra, Frank Peter Hull, Ricardo R. Reyes, E. Tracey Melhuish, Denise Pichardo, Eileen Georgi, Christine Lepurage; Houston Methodist Hospital, Houston, TX: Deepa Gotur, Katherine Perez, Cullen Hebert, Christopher Cortes, Siraya Jaijakul, Jiejian Lin, Raquel Bunge, Ashley Szczepanek, Sheila Moore, Sergio Ibarra Cortez, Rosbel Brito, , Christopher Sanchez, Andrea Green, Aaron Barnes, Kim Donlon, Connie Okon, Krystin Nieto; Intermountain LDS Hospital, Salt Lake City, UT: Bradley Hunter, Daanish Hoda, Andy Badke, Brandon Webb, Samuel Brown, Ithan Peltan, Lindsay Leither, Darija Runjaic Ward, Kelie Taylor, Molly Baxter, Jacki Anderson; Larkin Community Hospital, Hialeah, FL: Luis Mendez-Mullet, Tania Morejon, Lilybet Rodriguez, Pedram Rad, Amy Martinez; Larkin Community Hospital Palm Springs Campus, Hialeah, FL: Luis Mendez-Mullet, Tania Morejon, Amy Martinez; Lehigh Valley Health Network, Allentown, PA: Marcelo Gareca, Mark Knouse, Amy Slenker, Carla Rossi, Eric Young, Thomas Eames, Andrew Orzel, Matthew Karpowicz, Nicholas Trask; Liver Institute at Methodist Dallas, Dallas, TX: Parvez Mantry, Edward Dominguez, Shandra Silas, Melba Townsend, Jeny Rendon, Keyshania Smith; Medical University of South Carolina, Charleston, SC: Nandita Nadig, Susan Dorman, Alice Boylan, Timothy Whelan, Angela Millare, Amy Chamberlain, Abby Grady, Angel Darrow, April Rasberry, Max Lento; Novant Health (Presbyterian Medical Center, Forsyth Medical Center and Rowan Medical Center), Charlotte, NC; Winston-Salem, NC and Salisbury, NC: Alan Skarbnik, Michael Morgan, Heather Michael, Michael Blocker, James Dugan, Abayomi Agbebi, David Priest, William Harley, Pai-Ling Richards, Shannon Bowman, Latisha Morgan, Alanza Ferguson, Laurie Lowder, Tara Villalta, Heather Povinelli; Ochsner Clinic Foundation, New Orleans, LA: Julia Garcia-Diaz, Samuel King, Julie Castex, Derek Vonderhaar, Freda Dunhurst, Dana Comeaux, Meghan Rich, Hailey Anderson; OhioHealth Research Institute, Columbus, OH: Brian Zeno, Edward Cordasco, Kitra Hunter, Kevin Miller, Vicki Rezash, Emily Mulvany; Prime Healthcare Services – Saint Michael’s Medical Center, Newark, NJ: Jihad Slim, Maria Szabela, Herbert Galang, Justin Matthew; Providence Portland Medical Center, Portland, OR: Jason Wells, Tobias Pusch, David Hotchkin, Imran Mohamedy, Jeffrey Robinson, Nicholas Stucky, Amy Dechet, Ronald Dworkin, Roberto Patron, Wendi Drummond, Justin Jin, Jennifer Marfori, Cameron Cover, Chris Fountain, Kimberly Sutcliffe, Tara Foote, George Morris, Laurie Miller, Deirdre Cochran; Providence Regional Medical Center Everett, Everett, WA: George Diaz, Robert Choi, Daniel McClung, Albert Pacifico, Courtney Bohland, Vanessa Elan, Rebecca Watson, Dean Rocco; Providence St. John’s Health Center, Santa Monica, CA: Terese Hammond, Anmol Rangoola, Fabian Romero, Trevan Fischer, Ana Rocha, Hahn Ngyuen Thuy, Aleksandra Dubinchik, Marisol Savage, Courtney Johnson, Silvia Estrada; San Juan Oncology Associates, Farmington, NM: Jeffrey Neidhart, Sardar Zakariya Imam, Ankit Anand, Rosemarie Mestas, Jessica Barnes; St. Luke's Clinic – Rheumatology: Boise, Boise, ID: Karen Miller, Lynn Ann Hoppert, Derek Linderman, Sogol Nowbar, Lejla Godusevic, Adriana Datcu, Kelli Bruni, Jessica Grunke, William Dittrich, Brian Goltry, Dixie Durham; Thomas Jefferson University, Philadelphia, PA: Katherine Belden, Lily Ackermann, Michael Baram, Tricia Royer, Jessica Most, Dagan Coppock, Bryan Hess, Nana Aburjania, Katie Fitzgerald, Maura Wasilewski, Lyntovia Hylton, Linda Hosler, Diana Clarkson Naylor, Barbara Gallagher, Elizabeth Duddy, Minal Hatwar; University of Maryland, Baltimore, MD: Joseph Rabin, Leslie Sult, Lauren Offer; University of Miami, Miami, FL: David De La Zerda, Kunal Gawri, Annette Amoros, Jose Gonzales Zamora, Laiqua Khalid, Jovanna Bertran-Lopez, Tanya Quiroz, Emilia Faraj, Pamela Barletta, Patricia Rebolledo; Valleywise Health Medical Center, Phoenix, AZ: Michael White, Andres Alvarez, Raina Rahman, Christelle Kassis, Mary Mulrow, Amanda Frary; Velocity Clinical Research, San Diego, CA: J. Scott Overcash, Michael Waters, Hanh Chu, Kia Lee, Karla Zepeda, Rosalynn Landazuri, Shandel Odom, Cecilia Barbabosa, Allison Davis, Andrea Garcia, Kim Quillin, Kaitlyn Sandler, Angela Anorve, Jacob Pineda, Erica Sanchez, Dalia Tovar; West Virginia University Hospital, Morgantown, WV: Rahul Sangani, Sally Hodder, Rafia Zulfikar, Sarah Hadique, Nicole Kovacic, Elyse Krupp, Yanmin (Alice) Li, Tracy Yang, Cynthia Fisher-Duda, Louise Moore; Wyckoff Heights Medical Center, Brooklyn, NY: Parvez Mir, Zeyar Thet, Asif Khan, Judy Pham; Yale University School of Medicine, New Haven, CT: Onyema Ogbuagu, Thilinie Bandaranayake, Jessica Tuan, Laurie Andrews, Frances Santiago.
Funding
This trial was supported by F. Hoffmann-La Roche Ltd.
Author information
Authors and Affiliations
Contributions
IOR had full access to all the data in the trial and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors vouch for accuracy and adherence to the protocol during trial conduct. The manuscript was prepared by the authors with writing assistance funded by the sponsor. Concept and design: IOR, LT, JKO, OG, KT, HC, DB. Acquisition, analysis, or interpretation of data: All authors. Drafting of the manuscript: JKO. Critical revision of the manuscript for important intellectual content: All authors. Statistical analysis: EG and NL-K. All authors read and approved the final version of the manuscript
Corresponding author
Ethics declarations
Conflicts of interest
IOR Grant from Roche/Genentech related to the submitted work and grant from Genentech and personal fees from Genentech, Boehringer, and Bristol Myers Squibb outside the submitted work. GD: Grants from Gilead Sciences, Regeneron Inc., Roche, Boehringer Ingelheim, and Edasa Biotech outside the submitted work. Gilead Medical Affairs sentinel panel and Scientific Advisory Board for Safeology Inc. RLG: Personal fees from Eli Lilly, Gilead Sciences Inc., GlaxoSmithKline, Johnson and Johnson, and Roivant Sciences Inc. and nonfinancial support from Gilead Sciences Inc. outside the submitted work. SML: Investigator fees from Roche/Genentech during the conduct of the trial. PR: Nothing to disclose. BDH: Personal fees from Kite Pharma and Novartis outside the submitted work. AWC: Nothing to disclose. JSO: Institutional funding from Roche during conduct of the trial. NAH: Grants from GlaxoSmithKline, Sanofi, AstraZeneca, Genentech, Boehringer Ingelheim, Novartis, and Gossamer Bio; personal fees from GlaxoSmithKline, Sanofi, AstraZeneca, Genentech, Novartis, Regeneron, Teva, and Amgen outside the submitted work. AS: Personal fees from Genentech, AbbVie, Pharmacyclics, Janssen, Kite Pharma, Celgene, Verastem, BeiGene, Novartis, TG Therapeutics, Seattle Genetics, Morphosys, Jazz Pharmaceuticals, and Gilead Sciences and nonfinancial support from Bristol Myers Squibb outside the submitted work. JG-D: Nothing to disclose. IG: Nothing to disclose. JC: Grant provided to institution from Roche/Genentech during conduct of the trial. Grants and personal fees from Gilead Sciences Inc. outside the submitted work. OG: Employee of Roche and shareholder of Roche Holding AG. EG: Employee of Roche. NL-K: Employee of Roche/Genentech. LT: Employee of Roche/Genentech and has an unpublished patent pending, “Tocilizumab and Remdesivir Combination Therapy for COVID-19 Pneumonia.” KT: Former employee of Roche/Genentech and owns stock in Roche/Genentech. HC: Employee of Gilead Sciences Inc. DB: Former employee of and holds stock in Gilead Sciences Inc. JKO: Employee of Roche/Genentech.
Role of the funder/sponsor
F. Hoffmann-La Roche Ltd was involved in designing the trial, performing the analyses, and writing the report. A contract research organization paid by the sponsor monitored the trial under direction and supervision of the sponsor.
Data sharing statement
Qualified researchers may request access to individual patient level data upon publication through the clinical trial data request platform (https://vivli.org/). Further details on Roche's criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical trial documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Katie Tuckwell, Huyen Cao and Diana Brainard were affiliated with the above institutions at the time of the trial.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Rosas, I.O., Diaz, G., Gottlieb, R.L. et al. Tocilizumab and remdesivir in hospitalized patients with severe COVID-19 pneumonia: a randomized clinical trial. Intensive Care Med 47, 1258–1270 (2021). https://doi.org/10.1007/s00134-021-06507-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00134-021-06507-x