
Systemic Inflammation and COVID-19 Mortality in Patients with Major Noncommunicable Diseases: Chronic Coronary Syndromes, Diabetes and Obesity

Abstract
:1. Introduction
2. COVID-19, an Inflammatory Disease
3. COVID-19 Mortality and Chronic Inflammatory Status
4. Cardiovascular Dimension of COVID-19
5. CVD and COVID-19 in Children
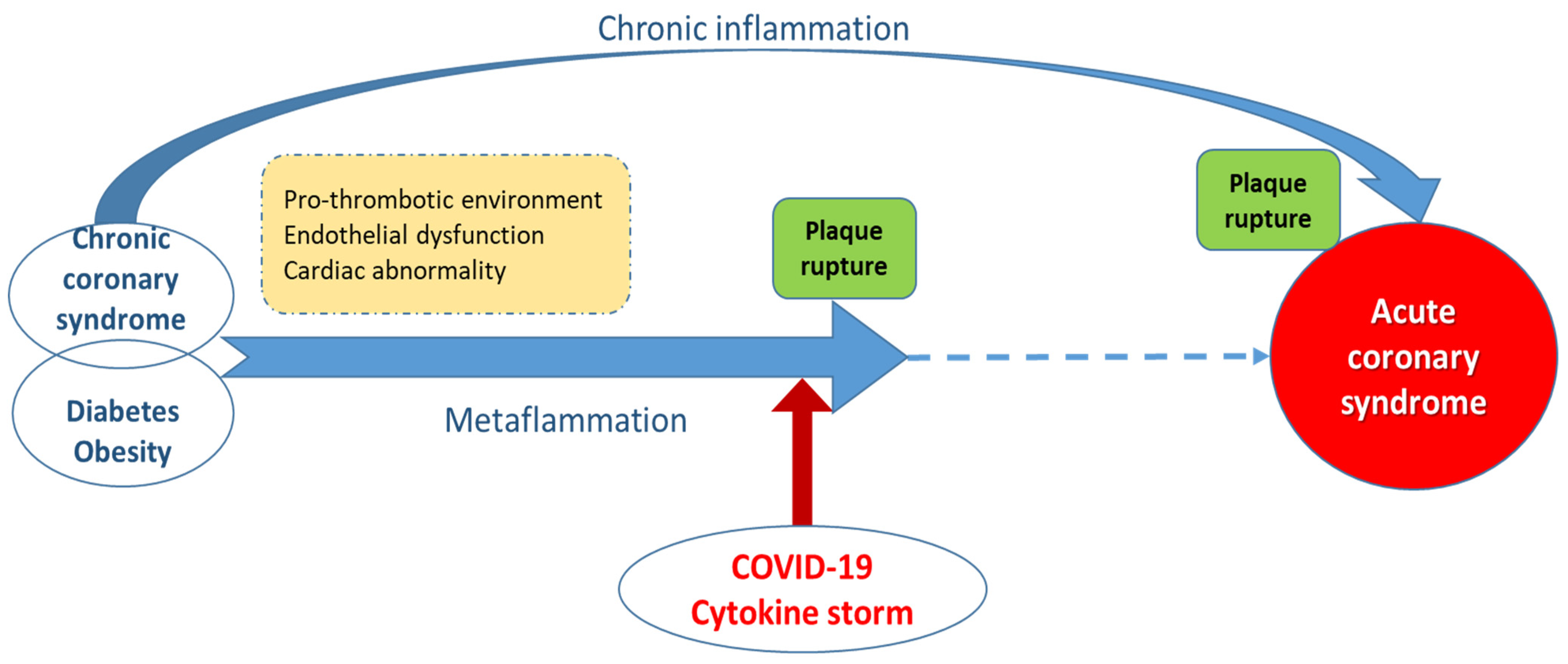
6. Inflammation, Acute Coronary Syndromes and COVID-19
7. Biomarkers Associated with Worse Prognosis in COVID-19 Patients Suffering Acute Coronary Syndromes
8. Statins and COVID-19 Disease Progression
9. Chronic Inflammation in Obesity and Type 2 Diabetes
10. Metabolic Dimension of COVID-19
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hendren, N.S.; Drazner, M.H.; Bozkurt, B.; Cooper, L.T., Jr. Description and Proposed Management of the Acute COVID-19 Cardiovascular Syndrome. Circulation 2020, 141, 1903–1914. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F.; et al. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 1666–1687. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Coronavirus Resource Center, Johns Hopkins University. 13 April 2020. Available online: https://coronavirus.jhu.edu/ (accessed on 1 April 2020).
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, C.; Leone, O.; Rizzo, S.; De Gaspari, M.; van der Wal, A.C.; Aubry, M.C.; Bois, M.C.; Lin, P.T.; Maleszewski, J.J.; Stone, J.R. Pathological features of COVID-19-associated myocardial injury: A multicentre cardiovascular pathology study. Eur. Heart J. 2020, 41, 3827–3835. [Google Scholar] [CrossRef] [PubMed]
- Sheth, A.R.; Grewal, U.S.; Patel, H.P.; Thakkar, S.; Garikipati, S.; Gaddam, J.; Bawa, D. Possible mechanisms responsible for acute coronary events in COVID-19. Med. Hypotheses 2020, 143, 110125. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS- CoV-infected mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Law, H.K.; Cheung, C.Y.; Ng, H.Y.; Sia, S.F.; Chan, Y.O.; Luk, W.; Nicholls, J.M.; Peiris, J.S.; Lau, Y.L. Chemokine up-regulation in SARS-coronavirus- infected, monocyte-derived human dendritic cells. Blood 2005, 106, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Lau, C.C.Y.; Chan, K.H.; Li, C.P.Y.; Chen, H.; Jin, D.Y.; Chan, J.F.W.; Woo, P.C.Y.; Yuen, K.Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J. Gen. Virol. 2013, 94 Pt 12, 2679–2690. [Google Scholar] [CrossRef]
- Jain, U. Effect of COVID-19 on the Organs. Cureus 2020, 12, e9540. [Google Scholar] [CrossRef]
- Ramcharan, T.; Nolan, O.; Lai, C.Y.; Prabhu, N.; Krishnamurthy, R.; Richter, A.G.; Jyothish, D.; Kanthimathinathan, H.K.; Welch, S.B.; Hackett, S.; et al. Paediatric Inflammatory Multisystem Syndrome: Temporally Associated with SARS-CoV-2 (PIMS-TS): Cardiac Features, Management and Short-Term Outcomes at a UK Tertiary Paediatric Hospital. Pediatr. Cardiol. 2020, 41, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Gkoutzourelas, A.; Bogdanos, D.P.; Sakkas, L.I. Kawasaki Disease and COVID-19. Mediterr. J. Rheumatol. 2020, 31 (Suppl. 2), 268–274. [Google Scholar] [CrossRef]
- Wang, H.; Ma, S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am. J. Emerg. Med. 2008, 26, 711–715. [Google Scholar] [CrossRef]
- Frydrych, L.M.; Bian, G.; O’Lone, D.E.; Ward, P.A.; Delano, M.J. Obesity and type 2 diabetes mellitus drive immune dysfunction, infection development, and sepsis mortality. J. Leukoc. Biol. 2018, 104, 525–534. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.L.; Yuen, K.S.; Castaño-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Sattar, N. Inflammation and endothelial dysfunction: Intimate companions in the pathogenesis of vascular disease? Clin. Sci. 2004, 106, 443–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Korakas, E.; Ikonomidis, I.; Kousathana, F.; Balampanis, K.; Kountouri, A.; Raptis, A.; Palaiodimou, L.; Kokkinos, A.; Lambadiari, V. Obesity and COVID-19: Immune and metabolic derangement as a possible link to adverse clinical outcomes. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E105–E109. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Bae, J.H.; Kwon, H.S.; Nauck, M.A. COVID-19 and diabetes mellitus: From pathophysiology to clinical management. Nat. Rev. Endocrinol. 2021, 17, 11–30. [Google Scholar] [CrossRef]
- Huang, I.; Lim, M.A.; Pranata, R. Diabetes mellitus is associated with increased mortality and severity of disease in COVID-19 pneumonia—A systematic review, meta-analysis, and meta-regression. Diabetes Metab. Syndr. 2020, 14, 395–403. [Google Scholar] [CrossRef]
- Kumar, A.; Arora, A.; Sharma, P.; Anikhindi, S.A.; Bansal, N.; Singla, V.; Khare, S.; Srivastava, A. Is diabetes mellitus associated with mortality and severity of COVID-19? A meta-analysis. Diabetes Metab. Syndr. 2020, 14, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Shao, M.; Guo, Q.; Shi, J.; Zhao, Y.; Xiaokereti, J.; Tang, B. Diabetes Mellitus is Associated with Severe Infection and Mortality in Patients with COVID-19: A Systematic Review and Meta-analysis. Arch. Med. Res. 2020, 51, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Bello-Chavolla, O.Y.; Bahena-López, J.P.; Antonio-Villa, N.E.; Vargas-Vázquez, A.; González-Díaz, A.; Márquez-Salinas, A.; Fermín-Martínez, C.A.; Naveja, J.J.; Aguilar-Salinas, C.A. Predicting Mortality Due to SARS-CoV-2: A Mechanistic Score Relating Obesity and Diabetes to COVID-19 Outcomes in Mexico. J. Clin. Endocrinol. Metab. 2020, 105, dgaa346. [Google Scholar] [CrossRef]
- Huang, Y.; Lu, Y.; Huang, Y.M.; Wang, M.; Ling, W.; Sui, Y.; Zhao, H.L. Obesity in patients with COVID-19: A systematic review and meta-analysis. Metabolism 2020, 113, 154378. [Google Scholar] [CrossRef]
- Yang, J.; Ma, Z.; Lei, Y. A meta-analysis of the association between obesity and COVID-19. Epidemiol. Infect. 2020, 149, e11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Gang, X.; He, G.; Li, Z.; Lv, Y.; Han, Q.; Wang, G. Obesity Increases the Severity and Mortality of Influenza and COVID-19: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2020, 11, 595109. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guan, B.; Su, T.; Liu, W.; Chen, M.; Bin Waleed, K.; Guan, X.; Gary, T.; Zhu, Z. Impact of cardiovascular disease and cardiac injury on in-hospital mortality in patients with COVID-19: A systematic review and meta-analysis. Heart 2020, 106, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Ssentongo, P.; Ssentongo, A.E.; Heilbrunn, E.S.; Ba, D.M.; Chinchilli, V.M. Association of cardiovascular disease and 10 other pre-existing comorbidities with COVID-19 mortality: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0238215. [Google Scholar] [CrossRef] [PubMed]
- Inciardi, R.M.; Lupi, L.; Zaccone, G.; Italia, L.; Raffo, M.; Tomasoni, D.; Cani, D.S.; Cerini, M.; Farina, D.; Gavazzi, E.; et al. Cardiac Involvement in a Patient With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 819–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babapoor-Farrokhran, S.; Gill, D.; Walker, J.; Rasekhi, R.T.; Bozorgnia, B.; Amanullah, A. Myocardial injury and COVID-19: Possible mechanisms. Life Sci. 2020, 253, 117723. [Google Scholar] [CrossRef]
- Ranard, L.S.; Fried, J.A.; Abdalla, M.; Anstey, D.E.; Givens, R.C.; Kumaraiah, D.; Kodali, S.K.; Takeda, K.; Karmpaliotis, D.; Rabbani, L.E.; et al. Approach to Acute Cardiovascular Complications in COVID-19 Infection. Circ. Heart Fail. 2020, 13, e007220. [Google Scholar] [CrossRef]
- Kwenandar, F.; Japar, K.V.; Damay, V.; Hariyanto, T.I.; Tanaka, M.; Lugito, N.P.H.; Kurniawan, A. Coronavirus disease 2019 and cardiovascular system: A narrative review. Int. J. Cardiol. Heart Vasc. 2020, 29, 100557. [Google Scholar] [CrossRef]
- Inciardi, R.M.; Adamo, M.; Lupi, L.; Cani, D.S.; Di Pasquale, M.; Tomasoni, D.; Italia, L.; Zaccone, G.; Tedino, C.; Fabbricatore, D.; et al. Characteristics and outcomes of patients hospitalized for COVID-19 and cardiac disease in Northern Italy. Eur. Heart J. 2020, 41, 1821–1829, Erratum in 2020, 41, 4591. [Google Scholar] [CrossRef]
- Wolfler, A.; Manarino, S.; Giacomet, V.; Camporesi, A.; Zuccotti, G. Acute myocardial injury: A novel clinical pattern in children with COVID-19. Lancet Child. Adolesc. Health 2020, 4, e26–e27. [Google Scholar] [CrossRef]
- Toubiana, J.; Poirault, C.; Corsia, A.; Bajolle, F.; Fourgeaud, J.; Angoulvant, F.; Debray, A.; Basmaci, R.; Salvador, E.; Biscardi, S.; et al. Kawasaki-like multisystem inflammatory syndrome in children during the covid-19 pandemic in Paris, France: Prospective observational study. BMJ 2020, 369, m2094. [Google Scholar] [CrossRef]
- Valverde, I.; Singh, Y.; Sanchez-de-Toledo, J.; Theocharis, P.; Chikermane, A.; Di Filippo, S.; Kuciñska, B.; Mannarino, S.; Tamariz-Martel, A.; Gutierrez-Larraya, F.; et al. Acute Cardiovascular Manifestations in 286 Children With Multisystem Inflammatory Syndrome Associated With COVID-19 Infection in Europe. Circulation 2021, 143, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Amirfakhryan, H. Kawasaki-like disease in children with COVID-19: A hypothesis. Med. Hypotheses 2020, 143, 110117. [Google Scholar] [CrossRef]
- Eberli, F.R.; Kurz, D. Cardiovascular aspects of COVID-19. Swiss Med. Wkly. 2020, 150, w20417. [Google Scholar]
- Whittaker, E.; Bamford, A.; Kenny, J.; Kaforou, M.; Jones, C.E.; Shah, P.; Ramnarayan, P.; Fraisse, A.; Miller, O.; Davies, P.; et al. Clinical Characteristics of 58 Children With a Pediatric Inflammatory Multisystem Syndrome Temporally Associated With SARS-CoV-2. JAMA 2020, 324, 259–269. [Google Scholar] [CrossRef]
- Feldstein, L.R.; Rose, E.B.; Horwitz, S.M.; Collins, J.P.; Newhams, M.M.; Son, M.B.F.; Newburger, J.W.; Kleinman, L.C.; Heidemann, S.M.; Martin, A.A.; et al. Multisystem Inflammatory Syndrome in U.S. Children and Adolescents. N. Engl. J. Med. 2020, 383, 334–346. [Google Scholar] [PubMed]
- Carlin, R.F.; Fischer, A.M.; Pitkowsky, Z.; Abel, D.; Sewell, T.B.; Landau, E.G.; Caddle, S.; Robbins-Milne, L.; Boneparth, A.; Milner, J.D.; et al. Discriminating Multisystem Inflammatory Syndrome in Children Requiring Treatment from Common Febrile Conditions in Outpatient Settings. J. Pediatr. 2021, 229, 26–32.e2. [Google Scholar] [CrossRef]
- Sharma, M.; Gorstein, S.; Aldrich, M.L.; Hsu, D.T.; Choueiter, N.F. Reversible Myocardial Injury Associated With SARS-CoV-2 in an Infant. JACC Case Rep. 2020, 2, 2348–2352. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Evans, C.; Kanthimathinathan, H.K.; Lillie, J.; Brierley, J.; Waters, G.; Johnson, M.; Griffiths, B.; du Pré, P.; Mohammad, Z.; et al. Intensive care admissions of children with paediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2 (PIMS-TS) in the UK: A multicentre observational study. Lancet Child. Adolesc. Health 2020, 4, 669–677, Erratum in 2020, 4, 669–677. [Google Scholar] [CrossRef]
- Oleszak, F.; Maryniak, A.; Botti, E.; Abrahim, C.; Salifu, M.O.; Youssef, M.; Henglein, V.L.; McFarlane, S.I. Myocarditis Associated With COVID-19. Am. J. Med. Case Rep. 2020, 8, 498–502. [Google Scholar] [CrossRef]
- Licu, R.A.; Blîndu, E.; Opincariu, D.; Benedek, T. Vulnerable Plaques Producing an Acute Coronary Syndrome Exhibit a Different CT Phenotype than Those That Remain Silent. J. Cardiovasc. Emergencies 2020, 6, 26–34. [Google Scholar] [CrossRef]
- Lazou, A.; Ikonomidis, I.; Bartekova, M.; Benedek, T.; Makavos, G.; Palioura, D.; Cabrera Fuentes, H.; Andreadou, I. Chronic inflammatory diseases, myocardial function and cardioprotection. Br. J. Pharmacol. 2020, 177, 5357–5374. [Google Scholar] [CrossRef] [PubMed]
- Benedek, T. COVID-19 Pandemic and Cardiovascular Challenges. J. Cardiovasc. Emergencies 2020, 6, 5–6. [Google Scholar] [CrossRef]
- Benedek, T.; Rodean, I.; Ratiu, M.; Rat, N.; Yero Eremie, L.; Biris, C.; Lazar, L.; Pacurar, M.; Benedek, I. Periodontal Disease, Infammation and Atherosclerosis Progression in Patients with Acute Coronary Syndromes—the ATHERODENT Study. J. Cardiovasc. Emergencies 2018, 4, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Rus, V.A.; Chitu, M.; Cernea, S.; Benedek, I.; Hodas, R.; Zavate, R.; Nyulas, T.; Hintea, M.; Benedek, T. Altered nutritional status, inflammation and systemic vulnerability in patients with acute myocardial infarction undergoing percutaneous coronary revascularisation: A prospective study in a level 3 cardiac critical care unit. Nutr. Diet. 2020, 77, 212–222. [Google Scholar] [CrossRef]
- Malavazos, A.E.; Goldberger, J.J.; Iacxobellis, G. Does epicardial fat contribute to COVID-19 myocardial inflammation? Eur. Heart J. 2020, 41, 2333. [Google Scholar] [CrossRef]
- Rus, A.; Rat, N.; Chitu, M.; Benedek, I. The Characteristics, Manifestations and Cardiopulmonary Imaging (CT/MRI) of COVID-19 in COVID-19 Patients. J. Interdiscip. Med. 2020. [Google Scholar] [CrossRef]
- Libenciuc, C.; Licu, R.A.; Hodas, R.; Chitu, M.; Benedek, I. Myocardial Injury and Myocarditis in SARS-CoV-2 Patients. J. Interdiscip. Med. 2020, 5, 101–104. [Google Scholar] [CrossRef]
- Biasucci, L.M.; Leo, M.; De Maria, G.L. Local and systemic mechanisms of plaque rupture. Angiology 2008, 59 (Suppl. 2), 73S–76S. [Google Scholar] [CrossRef]
- Mauriello, A.; Sangiorgi, G.; Fratoni, S.; Palmieri, G.; Bonanno, E.; Anemona, L.; Schwartz, R.S.; Spagnoli, L.G. Diffuse and active inflammation occurs in both vulnerable and stable plaques of the entire coronary tree: A histopathologic study of patients dying of acute myocardial infarction. J. Am. Coll. Cardiol. 2005, 45, 1585–1593. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, M.; Gasperetti, A.; Mancone, M. Redefining the prognostic value of high-sensitivity troponin in COVID-19 patients: The importance of concomitant coronary artery disease. J. Clin. Med. 2020, 9, 3263. [Google Scholar] [CrossRef]
- Galván-Román, J.M.; Rodríguez-García, S.C.; Roy-Vallejo, E.; Marcos-Jiménez, A.; Sánchez-Alonso, S.; Fernández-Díaz, C.; Alcaraz-Serna, A.; Mateu-Albero, T.; Rodríguez-Cortes, P.; Sánchez-Cerrillo, I.; et al. IL-6 serum levels predict severity and response to tocilizumab in COVID-19: An observational study. J. Allergy Clin. Immunol. 2021, 147, 72–80.e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, L.; Xu, M.; Wu, J.; Luo, D.; Zhu, Y.; Li, B.; Song, X.; Zhou, X. Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J. Clin. Virol. 2020, 127, 104370. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, T.; Han, M.; Li, X.; Wu, D.; Xu, Y.; Zhu, Y.; Liu, Y.; Wang, X.; Wang, L. Diagnostic utility of clinical laboratory data determinations for patients with the severe COVID-19. J. Med. Virol. 2020, 92, 791–796. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.M.; de Oliveira, M.H.S.; Benoit, S.; Plebani, M.; Lippi, G. Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): A meta-analysis. Clin. Chem. Lab. Med. 2020, 58, 1021–1028. [Google Scholar] [CrossRef] [Green Version]
- Coomes, E.A.; Haghbayan, H. Interleukin-6 in Covid-19: A systematic review and meta-analysis. Rev. Med Virol. 2020, 30, 1–9. [Google Scholar] [CrossRef]
- Zeng, F.; Huang, Y.; Guo, Y.; Yin, M.; Chen, X.; Xiao, L.; Deng, G. Association of inflammatory markers with the severity of COVID-19: A meta-analysis. Int. J. Infect. Dis. 2020, 96, 467–474. [Google Scholar] [CrossRef]
- Akbari, H.; Tabrizi, R.; Lankarani, K.B.; Aria, H.; Vakili, S.; Asadian, F.; Noroozi, S.; Keshavarz, P.; Faramarz, S. The role of cytokine profile and lymphocyte subsets in the severity of coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. Life Sci. 2020, 258, 118167. [Google Scholar] [CrossRef]
- Mo, P.; Xing, Y.; Xiao, Y.; Deng, L.; Zhao, Q.; Wang, H.; Xiong, Y.; Cheng, Z.; Gao, S.; Liang, K.; et al. Clinical characteristics of refractory COVID-19 pneumonia in Wuhan, China. Clin. Infect. Dis. 2020, ciaa270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.J.; Dong, X.; Cao, Y.Y.; Yuan, Y.D.; Yang, Y.B.; Yan, Y.Q.; Akdis, C.A.; Gao, Y.D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020, 75, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Cordeanu, E.M.; Duthil, N.; Severac, F.; Lambach, H.; Tousch, J.; Jambert, L.; Mirea, C.; Delatte, A.; Younes, W.; Frantz, A.S.; et al. Prognostic Value of Troponin Elevation in COVID-19 hospitalized patients. J. Clin. Med. 2020, 9, 4078. [Google Scholar] [CrossRef] [PubMed]
- Barman, H.A.; Atici, A.; Sahin, I.; Alici, G.; Tekin, E.A.; Baycan, Ö.F.; Ozturk, F.; Oflar, E.; Tugrul, S.; Yavuz, M.B.; et al. Prognostic significance of cardiac injury in COVID-19 patients wuth and without coronary artery disease. Coron. Artery Dis. 2020. [Google Scholar] [CrossRef]
- Peterson, E.; Lo, K.B.; DeJoy, R.; Salacup, G.; Pelayo, J.; Bhargav, R.; Gul, F.; Albano, J.; Azmaiparashvili, Z.; Amanullah, A.; et al. The relationship between coronary artery disease and clinical outcomes in COVID-19: A single-center retrospective analysis. Coron. Artery Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Grzegorowska, O.; Lorkowski, J. Possible correlations between atherosclerosis, acute coronary syndromes and COVID-19. J. Clin. Med. 2020, 9, 3746. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Chen, C.; Huihui, L.; Hang, W.; Wang, D.W. Cardiac injuries in coronavirus disease 2019 (COVID-19). J. Mol. Cell Cardiol. 2020, 145, 25–29. [Google Scholar]
- Han, H.; Xie, L.; Liu, R.; Yang, J.; Liu, F.; Wu, K.; Chen, L.; Hou, W.; Feng, Y.; Zhu, C. Analysis of heart injury laboratory parameters in 273 COVID-19 patients in one hospital in Wuhan, China. J. Med. Virol. 2020, 92, 819–823. [Google Scholar] [CrossRef]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic of Thromboembolic Disease: Implications for prevention, antithrombotic Therapy and Follow-Up. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M. Covid-19: The Rollercoaster of fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and their interactions with endothelial cells, platelets and erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168. [Google Scholar] [CrossRef]
- Rodriguez-Diez, R.R.; Tejera-Muñoz, A.; Marquez-Exposito, L.; Rayego-Mateos, S.; Santos Sanchez, L.; Marchant, V.; Tejedor Santamaria, L.; Ramos, A.M.; Ortiz, A.; Egido, J.; et al. Statins: Could and old friend help in the fight against COVID-19? Br. J. Pharmacol. 2020, 177, 4873–4886. [Google Scholar] [CrossRef] [PubMed]
- Subir, R.; Jagat, M.; Kalyan, G. Pros and cons for use of statins in people with coronavirus disease-19. Diabetes Metab. Syndr. 2020, 14, 1225–1229. [Google Scholar] [CrossRef]
- De Spiegeleer, A.; Bronselaer, A.; Teo, J.T.; Byttebier, G.; De Tré, G.; Belmans, L.; Dobson, R.; Wynendaele, E.; Van De Wiele, C.; Vandaele, F.; et al. The effects of ARBs, ACEis, and Statins on Clinical Outcomes of COVID-19 Infection Among Nursing Home Residents. JAMDA 2020, 21, 909–914. [Google Scholar] [CrossRef] [PubMed]
- Mittachione, G.; Schiavone, M.; Curnis, A.; Arca, M.; Antinori, S.; Gasperetti, A.; Mascioli, G.; Severino, P.; Sabato, F.; Caracciolo, M.M.; et al. Impact of prior statin use on clinical outcomes in COVID-19 patients: Data from tertiary referral hospitals during COVID-19 pandemic in Italy. J. Clin. Lipidol. 2021, 15, 68–78. [Google Scholar] [CrossRef]
- Cernea, S.; Dobreanu, M. Diabetes and beta cell function: From mechanisms to evaluation and clinical implications. Biochem. Med. 2013, 23, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Prattichizzo, F.; De Nigris, V.; Spiga, R.; Mancuso, E.; La Sala, L.; Antonicelli, R.; Testa, R.; Procopio, A.D.; Olivieri, F.; Ceriello, A. Inflammageing and metaflammation: The yin and yang of type 2 diabetes. Ageing Res. Rev. 2018, 41, 1–17. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Bastard, J.P.; Maachi, M.; Van Nhieu, J.T.; Jardel, C.; Bruckert, E.; Grimaldi, A.; Robert, J.J.; Capeau, J.; Hainque, B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J. Clin. Endocrinol. Metab. 2002, 87, 2084–2089. [Google Scholar] [CrossRef]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Chylikova, J.; Dvorackova, J.; Tauber, Z.; Kamarad, V. M1/M2 macrophage polarization in human obese adipose tissue. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech. Repub. 2018, 162, 79–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Zhao, Q.; Yang, T.; Ding, W.; Zhao, Y. Cellular metabolism and macrophage functional polarization. Int. Rev. Immunol. 2015, 34, 82–100. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Galván-Peña, S.; O’Neill, L.A. Metabolic reprograming in macrophage polarization. Front. Immunol. 2014, 5, 420. [Google Scholar]
- Soto-Heredero, G.; Gómez de Las Heras, M.M.; Gabandé-Rodríguez, E.; Oller, J.; Mittelbrunn, M. Glycolysis—A key player in the inflammatory response. FEBS J. 2020, 287, 3350–3369. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.R.; Milner, J.J.; Makowski, L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol. Rev. 2012, 249, 218–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiem, K.; Keating, S.T.; Netea, M.G.; Riksen, N.P.; Tack, C.J.; van Diepen, J.; Stienstra, R. Hyperglycemic Memory of Innate Immune Cells Promotes In Vitro Proinflammatory Responses of Human Monocytes and Murine Macrophages. J. Immunol. 2021, 206, 807–813. [Google Scholar] [CrossRef]
- Lachmandas, E.; Vrieling, F.; Wilson, L.G.; Joosten, S.A.; Netea, M.G.; Ottenhoff, T.H.; van Crevel, R. The effect of hyperglycaemia on in vitro cytokine production and macrophage infection with Mycobacterium tuberculosis. PLoS ONE 2015, 10, e0117941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dror, E.; Dalmas, E.; Meier, D.T.; Wueest, S.; Thévenet, J.; Thienel, C.; Timper, K.; Nordmann, T.M.; Traub, S.; Schulze, F.; et al. Postprandial macrophage-derived IL-1β stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 2017, 18, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Diepen, J.A.; Thiem, K.; Stienstra, R.; Riksen, N.P.; Tack, C.J.; Netea, M.G. Diabetes propels the risk for cardiovascular disease: Sweet monocytes becoming aggressive? Cell Mol. Life Sci. 2016, 73, 4675–4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbadi, A.; Loftis, J.; Wang, A.; Yu, M.; Wang, Y.; Shakya, S.; Li, X.; Maytin, E.; Hascall, V. Heparin inhibits proinflammatory and promotes anti-inflammatory macrophage polarization under hyperglycemic stress. J. Biol. Chem. 2020, 295, 4849–4857. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef]
- Guzik, T.J.; Cosentino, F. Epigenetics and Immunometabolism in Diabetes and Aging. Antioxid. Redox Signal. 2018, 29, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Brodsky, S.V.; Goligorsky, D.M.; Hampel, D.J.; Li, H.; Gross, S.S.; Goligorsky, M.S. Glycated collagen I induces premature senescence-like phenotypic changes in endothelial cells. Circ. Res. 2002, 90, 1290–1298. [Google Scholar] [CrossRef] [Green Version]
- Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.; Roche, H.M. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 56, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles-Messance, H.; Mitchelson, K.A.J.; De Marco Castro, E.; Sheedy, F.J.; Roche, H.M. Regulating metabolic inflammation by nutritional modulation. J. Allergy Clin. Immunol. 2020, 146, 706–720. [Google Scholar] [CrossRef]
- Tilg, H.; Zmora, N.; Adolph, T.E.; Elinav, E. The intestinal microbiota fuelling metabolic inflammation. Nat. Rev. Immunol. 2020, 20, 40–54. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1β by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2012, 425, 121–126. [Google Scholar] [CrossRef]
- Bekkering, S.; Quintin, J.; Joosten, L.A.; van der Meer, J.W.; Netea, M.G.; Riksen, N.P. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- Rajani, C.; Jia, W. Disruptions in gut microbial-host co-metabolism and the development of metabolic disorders. Clin. Sci. 2018, 132, 791–811. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Sagulenko, V.; Zamoshnikova, A.; Richards, A.A.; Cridland, J.A.; Irvine, K.M.; Stacey, K.J.; Sweet, M.J. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012, 217, 1325–1329. [Google Scholar] [CrossRef] [Green Version]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Levy, M.; Grosheva, I.; Zheng, D.; Soffer, E.; Blacher, E.; Braverman, S.; Tengeler, A.C.; Barak, O.; Elazar, M.; et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 2018, 359, 1376–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sell, H.; Habich, C.; Eckel, J. Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 2012, 8, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.L.; Savelieff, M.G.; Hayek, S.S.; Pennathur, S.; Kretzler, M.; Pop-Busui, R. COVID-19 and Diabetes: A Collision and Collusion of Two Diseases. Diabetes 2020, 69, 2549–2565. [Google Scholar] [CrossRef] [PubMed]
- Brufsky, A. Hyperglycemia, hydroxychloroquine, and the COVID-19 pandemic. J. Med. Virol. 2020, 92, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Bode, B.; Garrett, V.; Messler, J.; McFarland, R.; Crowe, J.; Booth, R.; Klonoff, D.C. Glycemic Characteristics and Clinical Outcomes of COVID-19 Patients Hospitalized in the United States. J. Diabetes Sci. Technol. 2020, 14, 813–821. [Google Scholar] [CrossRef]
- Suleyman, G.; Fadel, R.A.; Malette, K.M.; Hammond, C.; Abdulla, H.; Entz, A.; Demertzis, Z.; Hanna, Z.; Failla, A.; Dagher, C.; et al. Clinical Characteristics and Morbidity Associated with Coronavirus Disease 2019 in a Series of Patients in Metropolitan Detroit. JAMA Netw. Open 2020, 3, e2012270. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, C.M.; Jones, S.A.; Yang, J.; Rajagopalan, H.; O’Donnell, L.; Chernyak, Y.; Tobin, K.A.; Cerfolio, R.J.; Francois, F.; Horwitz, L.I. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: Prospective cohort study. BMJ 2020, 369, m1966. [Google Scholar] [CrossRef]
- Zhu, L.; She, Z.G.; Cheng, X.; Qin, J.J.; Zhang, X.J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metab. 2020, 31, 1068–1077.e3. [Google Scholar] [CrossRef]
- Chao, W.C.; Tseng, C.H.; Wu, C.L.; Shih, S.J.; Yi, C.Y.; Chan, M.C. Higher glycemic variability within the first day of ICU admission is associated with increased 30-day mortality in ICU patients with sepsis. Ann. Intensive Care 2020, 10, 17. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Biomarker | Reference | Study/Patients Characteristics | Study Findings |
|---|---|---|---|
| IL-6 | Galván-Román, J.M. et al. [64] | 146 patients; 66% males; median age: 63 years | Baseline levels >30 pg/mL predicts invasive mechanical ventilation requirement (OR: 7.1; p < 0.001) |
| Liu, F. et al. [65] | 140 patients; 35% males; age: 65.5 (54.3–73.0) years | IL-6 >32.1 pg/mL predicted severe complications (HR: 2.375; 95%CI: 1.058–5.329; p < 0.001) | |
| Gao, Y. et al. [66] | 43 adult patients; age 45.20 years (severe group) and 42.96 years (mild group); 26 males | Independent risk factor for the severity (OR: 17.304 (95%CI: 2.416, 123.933); p = 0.005) | |
| Qin, C. et al. [67] | 452 patients (286 with severe infection); median age: 58 years; 235 were males | Increased levels in severe vs. nonsevere cases (25.2 vs. 13.3 pg/mL; p < 0.001) | |
| Henry, B.M. et al. [68] | MA of 21 studies (n = 3377 patients) | Higher levels in severe vs. nonsevere disease (WMD: 1.70 (0.8, 2.6) pg/mL); significantly greater increases in nonsurvivors vs. survivors (WMD: 4.6 (3.4, 5.8] pg/mL) | |
| Coomes, E.A. et al. [69] | MA of 10 studies (n = 1798) | 2.9-fold higher levels in patients with complicated disease compared with patients with noncomplicated disease; Patients requiring ICU admission had higher values (random of means: 3.24 (95%CI: 2.54, 4.14]). | |
| Zeng, F. et al. [70] | MA of 16 studies (n = 3962) | Nonsevere group had lower levels (WMD: −21.32 ng/L (95%CI: −28.34, −14.31), p < 0.001) | |
| Akbari, H. et al. [71] | MA of 44 studies (n = 7865) | Higher levels in severe groups (WMD: 17.79 pg/mL (95%CI: 14.24, 21.33), p < 0.001) | |
| CRP | Liu, F. et al. [65] | 140 patients; 35% males; age: 65.5 (54.3–73.0) years | CRP >41.8 mg/L predicted severe complications (HR: 4.394 (95%CI: 1.924–10.033); p < 0.001) |
| Qin, C. et al. [67] | 452 patients (286 with severe infection); median age: 58 years; 235 were males | Increased levels in severe vs. nonsevere cases (57.9 vs. 33.2 mg/L; p < 0.001) | |
| Mo, P. et al. [72] | 155 patients (45.2% refractory patients); age: 54 (42–66) years; 55.5% males | Increased levels in refractory cases (46 (22–106) mg/L vs. 23 (10–47) mg/L; p = 0.001) | |
| Zhang, J.J. et al. [73] | 140 patients; age: 57 (25–87) years; 50.7% males | Increased levels in severe vs. nonsevere cases (47.6 (20.6–87.1) mg/L vs. 28.7 (9.5–52.1) mg/L, p < 0.001) | |
| Zeng, F. et al. [70] | MA of 16 studies (n = 3962) | Nonsevere group had lower levels (WMD: −41.78 mg/L (95%CI: −52.43, −31.13), p < 0.001) | |
| Akbari, H. et al. [71] | MA of 44 studies (n = 7865) | Higher levels in severe groups (WMD: 41.07 mg/L (95%CI: 29.76, 52.38), p < 0.001) | |
| Procalcitonin | Liu, F. et al. [65] | 140 patients; 35% males; age: 65.5 (54.3–73.0) years | PCT >0.07 ng/mL predicts severe complications (HR: 4.908 (95%CI: 1.797, 13.402), p = 0.002) |
| Qin, C. et al. [67] | 452 patients (286 with severe infection); median age: 58 years; 235 were males | Increased levels in severe vs. nonsevere cases(0.1 vs. 0.05 ng/mL; p < 0.001) | |
| Zhang, J.J. et al. [73] | 140 patients; age: 57 (25–87) years; 50.7% males | Increased in severe vs. nonsevere cases (0.1 (0.06–0.3) ng/mL vs. 0.05 (0.03–0.1) ng/mL; p < 0.001) | |
| Zeng, F. et al. [70] | MA of 16 studies (n = 3962) | Nonsevere group had lower levels (WMD: −0.13 ng/mL (95%CI: −0.20, −0.05), p < 0.001) | |
| Akbari, H. et al. [71] | MA of 44 studies (n = 7865) | Higher levels in severe groups (WMD: 0.07 ng/mL (95%CI: 0.05, 0.09), p< 0.001) | |
| Ferritin | Qin, C. et al. [67] | 452 patients (286 with severe infection); median age: 58 years; 235 were males | Increased levels in severe vs. nonsevere cases (800.4 vs. 523.7 ng/mL; p < 0.001) |
| Henry, B.M. et al. [68] | MA of 21 studies (n = 3377 patients) | Discriminate between severe vs. nonsevere disease (WMD: 408.28 (311.12, 505.44) ng/mL); significantly greater increases in nonsurvivors vs. survivors (WMD: 760.2 (560.84, 959.53) ng/mL) | |
| Zeng, F. et al. [70] | MA of 16 studies (n = 3962) | Nonsevere group had lower levels (WMD: −398.80 mg/L (95%CI: −625.89, −171.71), p < 0.001) | |
| Akbari, H. et al. [71] | MA of 44 studies (n = 7865) | Higher levels in severe groups (WMD: 594.25 μg/L (95%CI: 438.10, 750.39), p < 0.001) | |
| ESR | Zeng, F. et al. [70] | MA of 16 studies (n = 3962) | Nonsevere group had lower levels (WMD: −8 mm/h (95%CI: −14, −2), p = 0.005) |
| Akbari, H. et al. [71] | MA of 44 studies (n = 7865) | Higher levels in severe groups (WMD: 23.39 mm/h, (95%CI: 16.51, 30.27), p < 0.001) | |
| TNF-α | Akbari, H. et al. [71] | MA of 44 studies (n = 7865) | Higher levels in severe groups (WMD: 0.24 pg/mL (95%CI: 0.01, 0.47), p < 0.001) |
| Biomarkers of Cardiovascular Involvement in COVID-19 Patients | Markers of Rapid Deterioration in Adult Patients with COVID-19 | Associated with the Cytokine Storm | Associated with Cardiac Damage and Coagulation and Predicts Complications | Markers of Plaque Instability and Increased Thrombotic Risk | Associated with Coagulation Activation and Thrombin Generation | Markers of Inflammation and Cardiac Involvement in Children |
|---|---|---|---|---|---|---|
| NT-proBNP/BNP | ✓ | ✓ | ||||
| Troponin | ✓ | ✓ | ✓ | |||
| D-dimer | ✓ | ✓ | ✓ | |||
| Procalcitonin | ✓ | |||||
| Ferritin | ✓ | |||||
| IL-6 | ✓ | ✓ | ✓ | ✓ | ||
| C-reactive protein | ✓ | ✓ | ||||
| ESR | ✓ | |||||
| Deterioration of lymphocyte counts | ✓ | |||||
| TNF-α | ✓ | ✓ | ||||
| IL-7 | ✓ | |||||
| IL-22 | ✓ | |||||
| CXCL10 | ✓ | |||||
| IL-1B | ✓ | |||||
| IL-8 | ✓ | |||||
| IL-1 | ✓ | |||||
| CK | ✓ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buicu, A.-L.; Cernea, S.; Benedek, I.; Buicu, C.-F.; Benedek, T. Systemic Inflammation and COVID-19 Mortality in Patients with Major Noncommunicable Diseases: Chronic Coronary Syndromes, Diabetes and Obesity. J. Clin. Med. 2021, 10, 1545. https://doi.org/10.3390/jcm10081545
Buicu A-L, Cernea S, Benedek I, Buicu C-F, Benedek T. Systemic Inflammation and COVID-19 Mortality in Patients with Major Noncommunicable Diseases: Chronic Coronary Syndromes, Diabetes and Obesity. Journal of Clinical Medicine. 2021; 10(8):1545. https://doi.org/10.3390/jcm10081545
Chicago/Turabian StyleBuicu, Andreea-Luciana, Simona Cernea, Imre Benedek, Corneliu-Florin Buicu, and Theodora Benedek. 2021. "Systemic Inflammation and COVID-19 Mortality in Patients with Major Noncommunicable Diseases: Chronic Coronary Syndromes, Diabetes and Obesity" Journal of Clinical Medicine 10, no. 8: 1545. https://doi.org/10.3390/jcm10081545