
Accelerated Repurposing and Drug Development of Pulmonary Hypertension Therapies for COVID-19 Treatment Using an AI-Integrated Biosimulation Platform

 , , ,
, , ,
Abstract
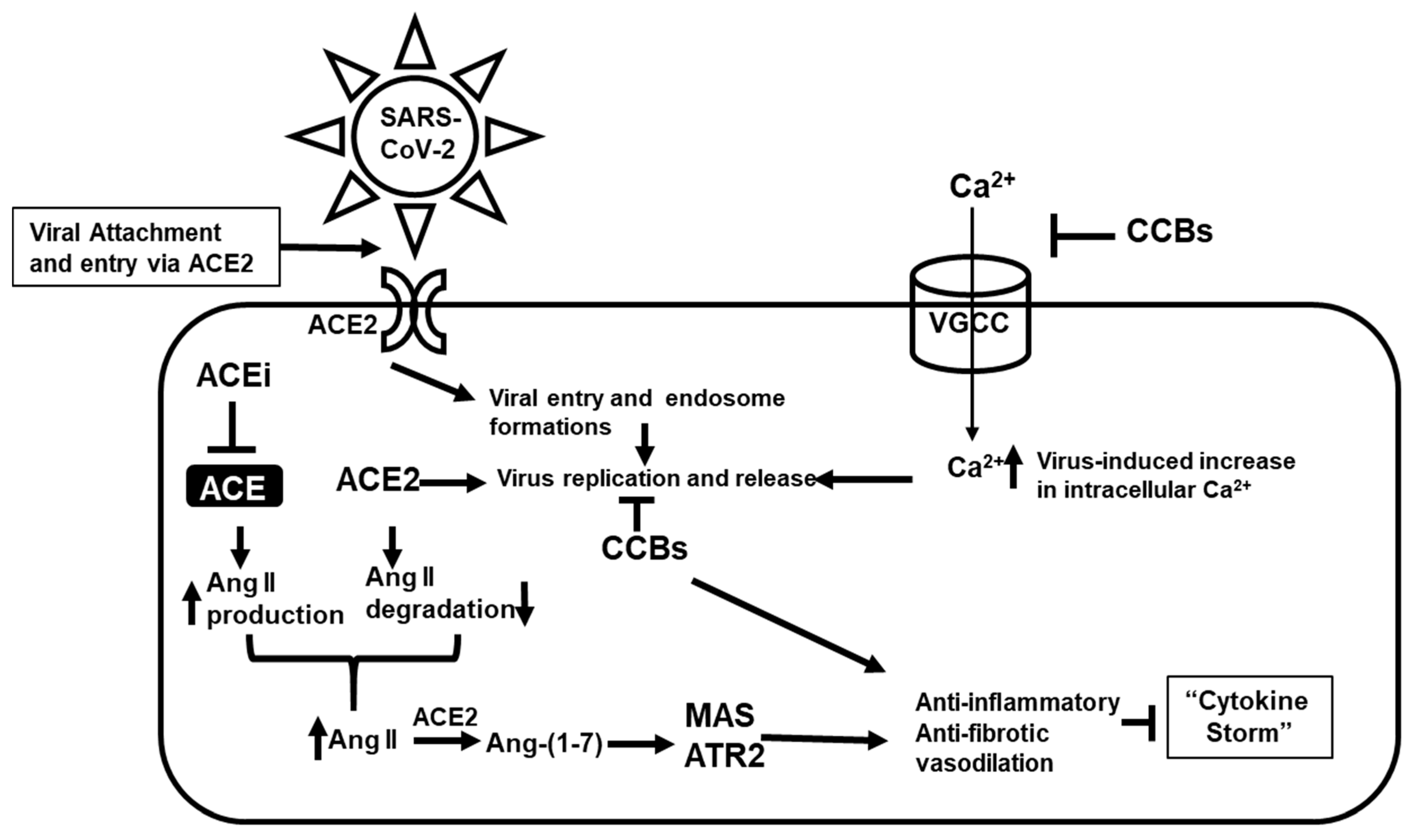
:1. Introduction
2. Results
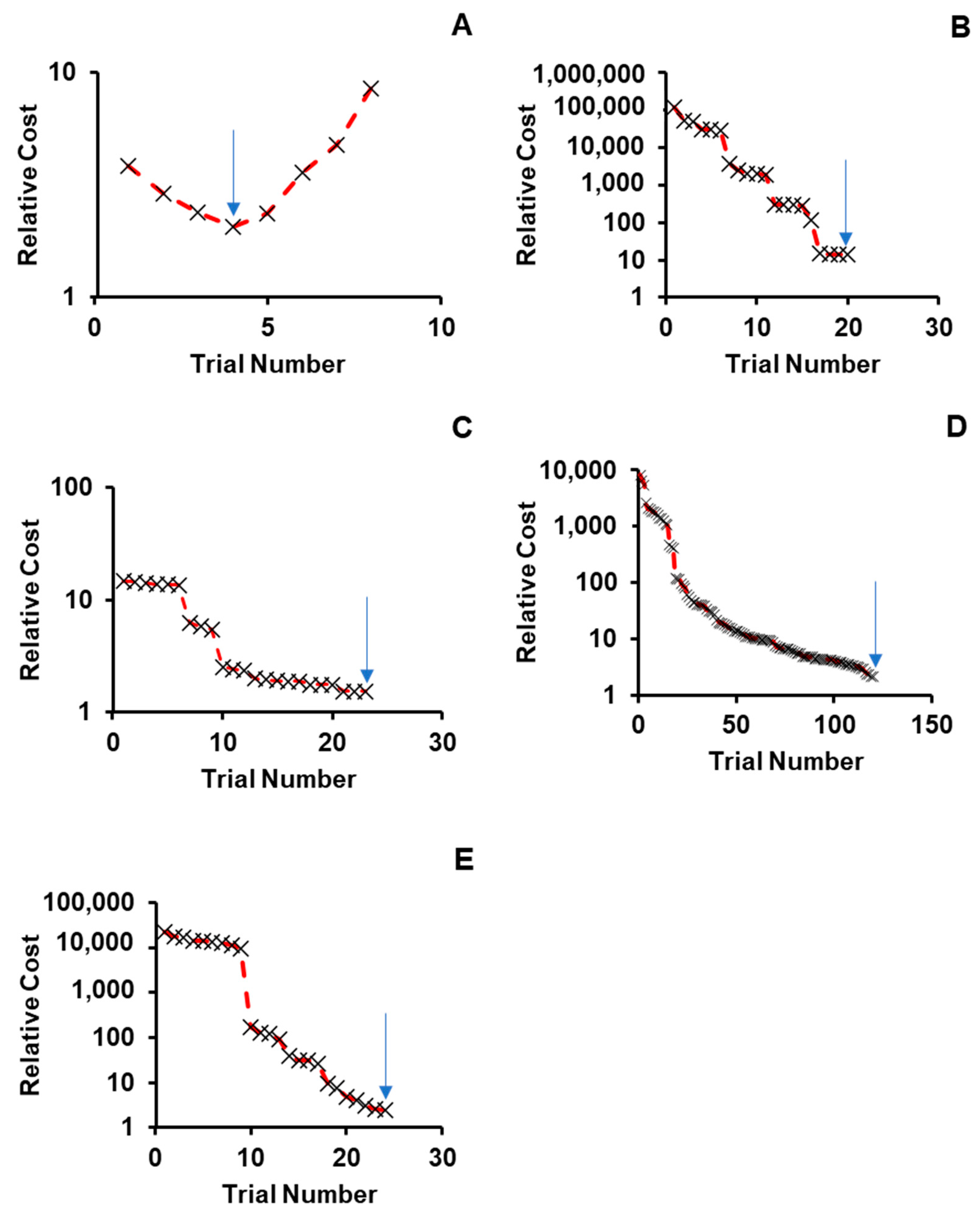
2.1. Sensitivity and Convergence Testing
2.2. Simulation Accuracy
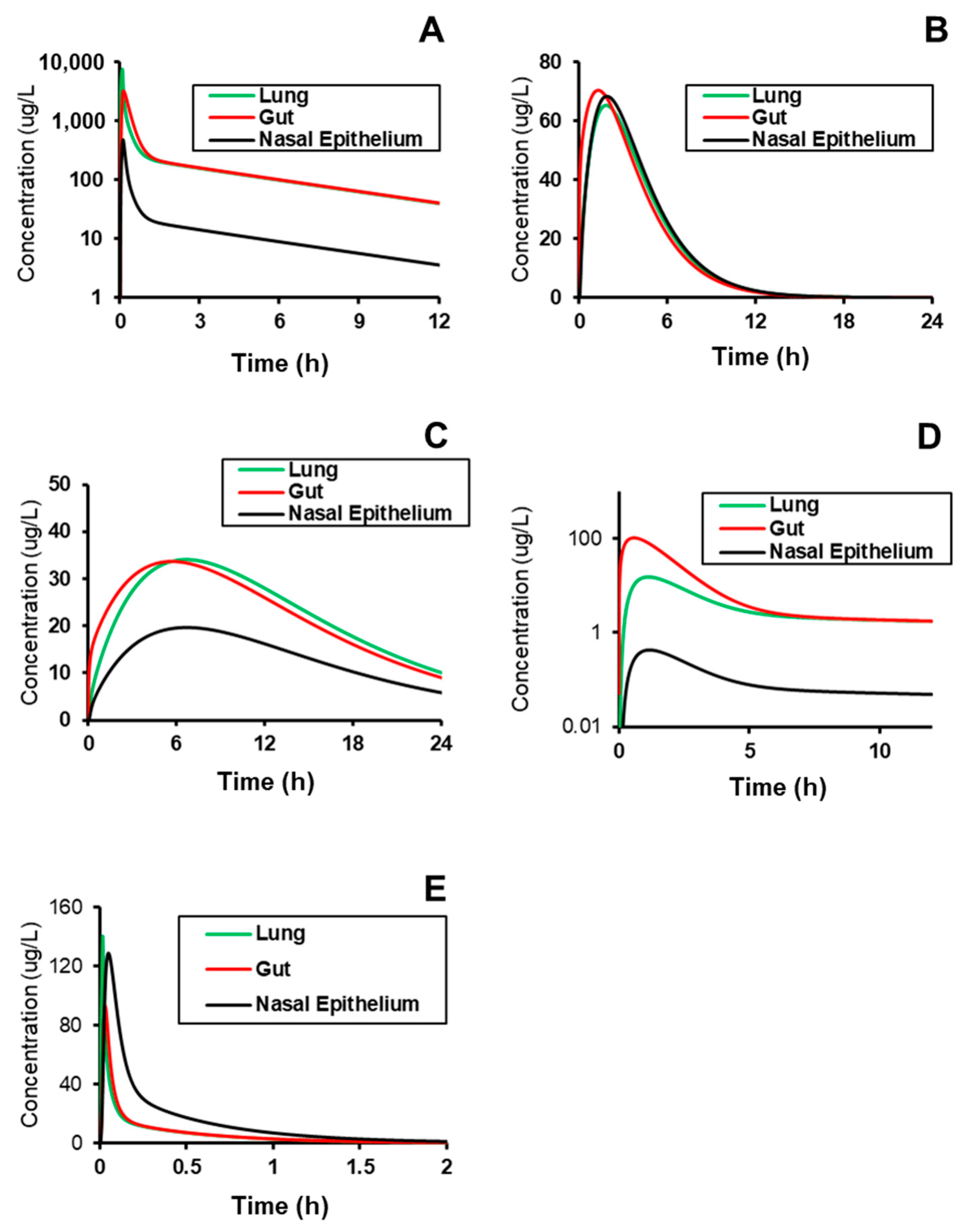
2.3. Simulating Distribution to Gut, Lung, and Nasal Epithelium
3. Discussion
4. Materials and Methods
4.1. Overview of BIOiSIM Platform
4.2. Modeling Compound Disposition
4.3. Rapid Repurposing Workflow
4.3.1. Test Dataset
4.3.2. Statistics and Tools
4.3.3. Subjects
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nicola, M.; O’Neill, N.; Sohrabi, C.; Khan, M.; Agha, M.; Agha, R. Evidence based management guideline for the COVID-19 pandemic—Review article. Int. J. Surg. 2020, 77, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Coronavirus Disease (COVID-19) Dashboard; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Rehman, M.F.U.; Fariha, C.; Anwar, A.; Shahzad, N.; Ahmad, M.; Mukhtar, S.; Ul Haque, F.M. Novel coronavirus disease (COVID-19) pandemic: A recent mini review. Comput. Struct. Biotechnol. J. 2021, 19, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Why do COVID death rates seem to be falling? Nature 2020, 587, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Mosser, P.C. Central bank responses to COVID-19. Bus. Econ. 2020, 55, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.M. The Covid-19 Vaccine-Development Multiverse. N. Engl. J. Med. 2020, 383, 1986–1988. [Google Scholar] [CrossRef] [PubMed]
- Lebrasseur, A.; Fortin-Bédard, N.; Lettre, J.; Bussières, E.-L.; Best, K.; Boucher, N.; Hotton, M.; Beaulieu-Bonneau, S.; Mercier, C.; Lamontagne, M.-E.; et al. Impact of COVID-19 on people with physical disabilities: A rapid review. Disabil. Health J. 2020, 14, 101014. [Google Scholar] [CrossRef]
- McMasters, M.; Blair, B.M.; Lazarus, H.M.; Alonso, C.D. Casting a wider protective net: Anti-infective vaccine strategies for patients with hematologic malignancy and blood and marrow transplantation. Blood Rev. 2020, 100779. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, J.; Li, H.; Luo, Z.; Ye, J.; Xu, Y.; Wang, Z.; Ye, D.; Liu, J.; Li, D.; et al. Recent progress of antiviral therapy for coronavirus disease 2019. Eur. J. Pharmacol. 2020, 890, 173646. [Google Scholar] [CrossRef]
- Baum, A.; Ajithdoss, D.; Copin, R.; Zhou, A.; Lanza, K.; Negron, N.; Ni, M.; Wei, Y.; Mohammadi, K.; Musser, B.; et al. REGN-COV2 antibodies prevent and treat SARS-CoV-2 infection in rhesus macaques and hamsters. Science 2020, 370, 1110–1115. [Google Scholar] [CrossRef]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with Covid-19. N. Engl. J. Med. 2021, 384, 238–251. [Google Scholar] [CrossRef]
- DeFrancesco, L. COVID-19 antibodies on trial. Nat. Biotechnol. 2020, 38, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xie, B.; Hashimoto, K. Current status of potential therapeutic candidates for the COVID-19 crisis. Brain Behav. Immun. 2020, 87, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Badagliacca, R.; Sciomer, S.; Petrosillo, N. Endothelin receptor antagonists for pulmonary arterial hypertension and COVID-19: Friend or foe? J. Heart Lung Transplant. 2020, 39, 729–730. [Google Scholar] [CrossRef]
- Chen, X.; Cao, R.; Zhong, W. Host Calcium Channels and Pumps in Viral Infections. Cells 2019, 9, 94. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Babaei, F.; Mirzababaei, M.; Nassiri-Asl, M.; Hosseinzadeh, H. Review of registered clinical trials for the treatment of COVID-19. Drug Dev. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Yang, M.; Tang, Q.L.; Hu, X.Y.; Willcox, M.L.; Liu, J.P. Characteristics of registered clinical trials on traditional Chinese medicine for coronavirus disease 2019 (COVID-19): A scoping review. Eur. J. Integr. Med. 2020, 41, 101251. [Google Scholar] [CrossRef]
- Bakovic, A.; Risner, K.; Bhalla, N.; Alem, F.; Chang, T.L.; Weston, W.; Harness, J.A.; Narayanan, A. Brilacidin, a COVID-19 Drug Candidate, Exhibits Potent In Vitro Antiviral Activity Against SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Omrani, A.S.; Pathan, S.A.; Thomas, S.A.; Harris, T.R.E.; Coyle, P.V.; Thomas, C.E.; Qureshi, I.; Bhutta, Z.A.; Mawlawi, N.A.; Kahlout, R.A.; et al. Randomized double-blinded placebo-controlled trial of hydroxychloroquine with or without azithromycin for virologic cure of non-severe Covid-19. EClinicalMedicine 2020, 29. [Google Scholar] [CrossRef]
- Breining, P.; Frolund, A.L.; Hojen, J.F.; Gunst, J.D.; Staerke, N.B.; Saedder, E.; Cases-Thomas, M.; Little, P.; Nielsen, L.P.; Sogaard, O.S.; et al. Camostat mesylate against SARS-CoV-2 and COVID-19-Rationale, dosing and safety. Basic Clin. Pharmacol. Toxicol. 2020, 128, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, W.; Yoneda, T.; Koba, H.; Ueda, T.; Tsuji, N.; Ogawa, H.; Asakura, H. Potential mechanisms of nafamostat therapy for severe COVID-19 pneumonia with disseminated intravascular coagulation. Int. J. Infect. Dis. 2021, 102, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Dabbous, H.M.; Abd-Elsalam, S.; El-Sayed, M.H.; Sherief, A.F.; Ebeid, F.F.S.; El Ghafar, M.S.A.; Soliman, S.; Elbahnasawy, M.; Badawi, R.; Tageldin, M.A. Efficacy of favipiravir in COVID-19 treatment: A multi-center randomized study. Arch. Virol. 2021, 166, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035. [Google Scholar] [CrossRef] [PubMed]
- Kai, H.; Kai, M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors—Lessons from available evidence and insights into COVID-19. Hypertens. Res. 2020, 43, 648–654. [Google Scholar] [CrossRef]
- Baudin, B. New aspects on angiotensin-converting enzyme: From gene to disease. Clin. Chem. Lab. Med. 2002, 40, 256–265. [Google Scholar] [CrossRef]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angio-tensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Verma, A. COVID-19 and Angiotensin-Converting Enzyme Inhibitors and Angiotensin Receptor Blockers: What Is the Evidence? JAMA 2020, 323, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Donate-Macian, P.; Jungfleisch, J.; Perez-Vilaro, G.; Rubio-Moscardo, F.; Peralvarez-Marin, A.; Diez, J.; Valverde, M.A. The TRPV4 channel links calcium influx to DDX3X activity and viral infectivity. Nat. Commun. 2018, 9, 2307. [Google Scholar] [CrossRef]
- Ueda, M.; Daidoji, T.; Du, A.; Yang, C.S.; Ibrahim, M.S.; Ikuta, K.; Nakaya, T. Highly pathogenic H5N1 avian influenza virus induces extracellular Ca2+ influx, leading to apoptosis in avian cells. J. Virol. 2010, 84, 3068–3078. [Google Scholar] [CrossRef] [Green Version]
- Nathan, L.; Lai, A.L.; Millet, J.K.; Straus, M.R.; Freed, J.H.; Whittaker, G.R.; Daniel, S. Calcium Ions Directly Interact with the Ebola Virus Fusion Peptide To Promote Structure–Function Changes That Enhance Infection. ACS Infect. Dis. 2020, 6, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, L.K.; Li, S.F.; Zhang, S.F.; Wan, W.W.; Zhang, Y.L.; Xin, Q.L.; Dai, K.; Hu, Y.Y.; Wang, Z.B.; et al. Calcium channel blockers reduce severe fever with thrombocytopenia syndrome virus (SFTSV) related fatality. Cell Res. 2019, 29, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Lavanya, M.; Cuevas, C.D.; Thomas, M.; Cherry, S.; Ross, S.R. siRNA screen for genes that affect Junin virus entry uncovers voltage-gated calcium channels as a therapeutic target. Sci. Transl. Med. 2013, 5, 204ra131. [Google Scholar] [CrossRef] [Green Version]
- Omar, S.; Clarke, R.; Abdullah, H.; Brady, C.; Corry, J.; Winter, H.; Touzelet, O.; Power, U.F.; Lundy, F.; McGarvey, L.P.A.; et al. Respiratory virus infection up-regulates TRPV1, TRPA1 and ASICS3 receptors on airway cells. PLoS ONE 2017, 12, e0171681. [Google Scholar] [CrossRef] [Green Version]
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca2+ in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaron, J.R.; Gangaraju, S.; Rao, M.Y.; Kong, X.; Zhang, L.; Su, F.; Tian, Y.; Glenn, H.L.; Meldrum, D.R. K+ regulates Ca2+ to drive inflammasome signaling: Dynamic visualization of ion flux in live cells. Cell Death Dis. 2015, 6, e1954. [Google Scholar] [CrossRef] [Green Version]
- D’Elia, J.A.; Weinrauch, L.A. Calcium Ion Channels: Roles in Infection and Sepsis Mechanisms of Calcium Channel Blocker Benefits in Immunocompromised Patients at Risk for Infection. Int. J. Mol. Sci. 2018, 19, 2465. [Google Scholar] [CrossRef] [Green Version]
- Silva, I.V.G.; de Figueiredo, R.C.; Rios, D.R.A. Effect of Different Classes of Antihypertensive Drugs on Endothelial Function and Inflammation. Int. J. Mol. Sci. 2019, 20, 3458. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Han, S.J.; Kim, D.J.; Jang, H.C.; Lim, S.; Choi, S.H.; Kim, Y.H.; Shin, D.H.; Kim, S.H.; Kim, T.H.; et al. Effects of valsartan and amlodipine on oxidative stress in type 2 diabetic patients with hypertension: A randomized, multicenter study. Korean J. Intern. Med. 2017, 32, 497–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Lu, X.; Qie, S.; Xi, J. Thoughts on detecting tissue distribution of potential COVID-19 receptors. Future Virol. 2020, 15, 489–496. [Google Scholar] [CrossRef]
- Lombardo, F.; Berellini, G.; Obach, R.S. Trend Analysis of a Database of Intravenous Pharmacokinetic Parameters in Humans for 1352 Drug Compounds. Drug Metab. Dispos. 2018, 46, 1466–1477. [Google Scholar] [CrossRef] [Green Version]
- Benet, L.Z.; Broccatelli, F.; Oprea, T.I. BDDCS applied to over 900 drugs. AAPS J. 2011, 13, 519–547. [Google Scholar] [CrossRef] [Green Version]
- Krahenbuhl, S.; Grass, P.; Surve, A.; Kutz, K.; Reichen, J. Pharmacokinetics and haemodynamic effects of a single oral dose of the novel ACE inhibitor spirapril in patients with chronic liver disease. Eur. J. Clin. Pharmacol. 1993, 45, 247–253. [Google Scholar] [CrossRef]
- Yang, B.; Wu, C.; Ji, B.; Wu, M.; He, Z.; Shang, L.; Sun, J. Virtual population pharmacokinetic using physiologically based pharmacokinetic model for evaluating bioequivalence of oral lacidipine formulations in dogs. Asian J. Pharm. Sci. 2017, 12, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.R.; Bryson, H.M. Lacidipine. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic po-tential in the treatment of hypertension. Drugs 1994, 48, 274–296. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Nagar, S.; Korzekwa, K. A physiologically based pharmacokinetic model to predict the pharmacokinetics of highly protein-bound drugs and the impact of errors in plasma protein binding. Biopharm. Drug Dispos. 2016, 37, 123–141. [Google Scholar] [CrossRef]
- Eichelbaum, M.; Mikus, G.; Vogelgesang, B. Pharmacokinetics of (+)-, (-)- and (+/-)-verapamil after intravenous administration. Br. J. Clin. Pharmacol. 1984, 17, 453–458. [Google Scholar] [CrossRef] [Green Version]
- Beermann, B. Pharmacokinetics of lisinopril. Am. J. Med. 1988, 85, 25–30. [Google Scholar] [CrossRef]
- Albashir, A.A.D. Renin-Angiotensin-Aldosterone System (RAAS) Inhibitors and Coronavirus Disease 2019 (COVID-19). South Med. J. 2021, 114, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Da Ros, L.; Squassante, L.; Milleri, S. Dose linearity of lacidipine pharmacokinetics after single and repeated oral doses in healthy volunteers. Clin. Pharmacokinet. 2003, 42, 99–106. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.-L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Sanga, V.; Barton, M. Potential harmful effects of discontinuing ACE-inhibitors and ARBs in COVID-19 patients. eLife 2020, 9, e57278. [Google Scholar] [CrossRef] [Green Version]
- Maharao, N.; Antontsev, V.; Hou, H.; Walsh, J.; Varshney, J. Scalable in silico Simulation of Transdermal Drug Permeability: Application of BIOiSIM Platform. Drug Des. Devel. Ther. 2020, 14, 2307–2317. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Leahy, D.; Rowland, M. Physiologically based pharmacokinetic modeling 1: Predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94, 1259–1276. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Physiologically based pharmacokinetic modelling 2: Predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J. Pharm. Sci. 2006, 95, 1238–1257. [Google Scholar] [CrossRef]
- Schmitt, W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol. In Vitro 2008, 22, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.; Sarangapani, R.; Gentry, R.; Clewell, H.; Covington, T.; Frederick, C.B. Application of a hybrid CFD-PBPK nasal dosimetry model in an inhalation risk assessment: An example with acrylic acid. Toxicol. Sci. 2000, 57, 312–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boger, E.; Evans, N.; Chappell, M.; Lundqvist, A.; Ewing, P.; Wigenborg, A.; Friden, M. Systems Pharmacology Approach for Prediction of Pulmonary and Systemic Pharmacokinetics and Receptor Occupancy of Inhaled Drugs. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Frederick, C.B.; Bush, M.L.; Lomax, L.G.; Black, K.A.; Finch, L.; Kimbell, J.S.; Morgan, K.T.; Subramaniam, R.P.; Morris, J.B.; Ultman, J.S. Application of a hybrid computational fluid dynamics and physiologically based inhalation model for interspecies dosimetry extrapolation of acidic vapors in the upper airways. Toxicol. Appl. Pharmacol. 1998, 152, 211–231. [Google Scholar] [CrossRef]
- Jia, L.; Wong, H. In vitro and in vivo assessment of cellular permeability and pharmacodynamics of S-nitrosylated captopril, a nitric oxide donor. Br. J. Pharmacol. 2001, 134, 1697–1704. [Google Scholar] [CrossRef]
- O’Hagan, S.; Kell, D.B. The apparent permeabilities of Caco-2 cells to marketed drugs: Magnitude, and independence from both biophysical properties and endogenite similarities. PeerJ 2015, 3, e1405. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Kou, L.; Ma, P.; Gao, L.; Li, B.; Li, R.; Luo, C.; Shentu, J.; He, Z.; Sun, J. Interspecies prediction of oral pharmacokinetics of different lacidipine formulations from dogs to human: Physiologically based pharmacokinetic modelling combined with bi-orelevant dissolution. RSC Adv. 2015, 5, 19844–19852. [Google Scholar] [CrossRef]
- Integrated Physiology Database—PK-Sim®; Bayer Technology Services GmbH: Leverkusen, Germany, 2012; Available online: http://www.systems-biology.com/products/pk-sim.html (accessed on 10 October 2020).
- US Environmental Protection Agency Office of Health and Environmental Assessment. Physiological Parameter Values for PBPK Models: A Report Prepared by the International Life Sciences Institute and Risk Sciences Institute; International Life Sciences Institute and Risk Sciences Institute: Washington, DC, USA, 1994.
- ICRP. Basic Anatomical and Physiological Data for Use in Radiological Protection Reference Values. Ann. ICRP 2002, 32, 3–4. [Google Scholar]
- Maharao, N.; Antontsev, V.; Wright, M.; Varshney, J. Entering the era of computationally driven drug development. Drug Metab. Rev. 2020, 52, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Peters, S. Physiologically-Based Pharmacokinetic (PBPK) Modeling and Simulations: Principles, Methods, and Applications in the Pharmaceutical Industry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Campbell, J.L.; Andersen, M.E.; Clewell, H.J. A hybrid CFD-PBPK model for naphthalene in rat and human with IVIVE for nasal tissue metabolism and cross-species dosimetry. Inhal. Toxicol. 2014, 26, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, L.M.; Kirman, C.R.; Gargas, M.L.; Carson, M.L.; Tardiff, R.G. Development of a physiologically-based toxicokinetic model of acrylamide and glycidamide in rats and humans. Food Chem. Toxicol. 2010, 48, 668–685. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Development | Overview | Source |
|---|---|---|---|
| Remdesivir | Gilead, Approved by FDA | Antiviral, host factor-targeted. RNA-dependent/RNA polymerase-targeted | [20] |
| APN01 | APEIRON Biologics, Phase I | Pilot trial ongoing in China | [21] |
| Brilacidin | Innovation Pharmaceuticals, Phase II | Defensin mimetic drug candidate. Has shown antibacterial, anti-inflammatory, and immunomodulatory properties in several clinical trials | [22] |
| Hydroxychloroquine | Repurposed, Rejected | Host factor-targeted. Antimalarial drug that affects endosomal function and blocks autophagosome-lysosome fusion | [23] |
| Azithromycin | Repurposed | Host factor-targeted. Broad-spectrum antibiotic, blocks autophagosome clearance in human cells | [23] |
| Camostat | Repurposed | Host factor-targeted. TMPRSS2 inhibitor | [24] |
| Nafamostat | Repurposed | Host factor-targeted. TMPRSS2 inhibitor | [25] |
| Favipiravir | Repurposed, Approved in Russia, Japan | Host factor-targeted. RNA-dependent/RNA polymerase-targeted | [26] |
| Drug Name | Class | ka | LogP | pKa | Fu,p | B:P | Clearance (L/h/kg) | Kplung | Kpgut |
|---|---|---|---|---|---|---|---|---|---|
| Lisinopril | ACEi | 0.17 * | −1.115 [49] | 3.17 (acid), 10.21 (base) [49] | 0.99 [49] | 0.71 * | 0.072 [49] | 0.57 | 0.50 |
| Captopril | ACEi | N/A | 0.34 [50] | 4.01 (acid), −1.2 [49] | 0.73 [49] | 0.45 * | 0.72 [49] | 0.15 * | 0.15 * |
| Spirapril | ACEi | 0.53 [51] | 0 * | 3.62 (acid), 5.2 (base) [49] | 0.314678 * | 0.74 ** | 0.43 [51] | 0.21 * | 0.16 * |
| Lacidipine | CCB | 1.7843 * | 5.51 [52] | 19.47 (acid), −6.4 (base) [49] | 0.05 [53] | 0.70 * | 1.23 [49] | 11.72 * | 11.72 * |
| Verapamil | CCB | N/A | 3.795 [54] | 9.68 (base) [49] | 0.064 [55] | 0.88 [36,54] | 0.84 [49,54,55] | 3.69 * | 3.69 * |
| Drug Name | Formulation | Experimental Setup | Reference |
|---|---|---|---|
| Lisinopril | 20 mg, oral dose | 20 mg of Lisinopril was given orally for 10 consecutive days. 8 subjects in the study. | [56] |
| Captopril | 2.78 mg, 5.67 mg, 11.4 mg, IV dose | 1 mL of intravenous injection at three different dosage levels was administered to 7 healthy subjects. | [57] |
| Spirapril | 25 mg, oral dose | 25 mg spirapril p.o. prepared by dissolving 25 mg of lyophilized spirapril in 50 mL tap water was given to the subjects. 16 subjects. | [51] |
| Lacidipine | 2 mg, 4 mg, oral dose | Single dose of 2 mg and 4 mg of Lacidipine was administered. The study has a total of 24 subjects (12 male, 12 female) | [58] |
| Verapamil | 50 mg, IV dose | 5 subjects received 5 mg verapamil dissolved in 30 mL of saline infused over 5 min. | [55] |
| Compounds | Lisinopril | Captopril | Spirapril | Lacidipine | Verapamil | ||||
|---|---|---|---|---|---|---|---|---|---|
| Output metrics | ROA | Oral | IV | IV | IV | Oral | Oral | Oral | IV |
| Dose, mg | 20 | 2.78 | 5.67 | 11.4 | 25 | 2 | 4 | 5 | |
| AUC0-t, µg·h/L | Observed | N/A | N/A | N/A | N/A | N/A | 3.66 | 7.66 | N/A |
| Calculated | 752.00 | 42.97 | 93.93 | 215.69 | 991.83 | 3.12 | 6.80 | 703.03 | |
| Predicted | 823.50 | 49.16 | 92.55 | 212.12 | 977.60 | 3.27 | 8.80 | 494.26 | |
| AAFE | 1.10 | 1.14 | 1.01 | 1.02 | 1.01 | 1.05 | 1.29 | 1.42 | |
| AFE | 1.10 | 1.14 | 0.99 | 0.98 | 0.99 | 1.05 | 1.29 | 0.70 | |
| Cmax, µg/L | Observed | N/A | N/A | N/A | N/A | 430.00 | 1.24 | 3.09 | N/A |
| Calculated | 57.40 | 104.64 | 234.55 | 454.71 | 378.00 | 1.17 | 2.87 | 1176.82 | |
| Predicted | 53.93 | 105.81 | 183.46 | 497.25 | 196.17 | 1.00 | 2.01 | 1696.96 | |
| AAFE | 1.06 | 1.01 | 1.28 | 1.09 | 1.93 | 1.16 | 1.43 | 1.44 | |
| AFE | 0.94 | 1.01 | 0.78 | 1.09 | 0.52 | 0.86 | 0.70 | 1.44 | |
| Tmax, h | Observed | N/A | N/A | N/A | N/A | 0.90 | 1.13 | 1.25 | N/A |
| Calculated | 6.04 | 0.15 | 0.19 | 0.13 | 1.00 | 1.23 | 1.05 | 0.09 | |
| Predicted | 6.03 | 0.15 | 0.19 | 0.13 | 1.75 | 1.05 | 1.05 | 0.09 | |
| AAFE | 1.00 | 1.00 | 1.00 | 1.00 | 1.75 | 1.17 | 1.00 | 1.00 | |
| AFE | 1.00 | 1.00 | 1.00 | 1.00 | 1.75 | 0.86 | 1.00 | 1.00 | |
| Statistics | Chi-squared | 2803.13 * | 22.56 | 23.01 | 26.85 | 2.70 | 107.9 * | 182.15 * | 11.57 * |
| p-values | >0.50 | >0.50 | >0.50 | >0.50 | <0.001 | >0.50 | >0.50 | 0.36 | |
| Metrics | Tissue | Lisinopril, 20 mg, oral | Captopril, 2.78 mg, IV | Spirapril, 25 mg, oral | Lacidipine, 2 mg, oral | Verapamil, 50 mg, IV |
|---|---|---|---|---|---|---|
| AUC0–t, µg·h/L | Lung | 341.24 | 52.36 | 2315.64 | 13.86 | 344.12 |
| Gut | 354.03 | 196.74 | 2310.31 | 13.86 | 354.74 | |
| Nasal tissue | 196.91 | 1.48 | 207.74 | 31.63 | 360.48 | |
| Nasal epithelium | 196.34 | 1.48 | 207.58 | 31.63 | 360.48 | |
| Cmax, µg/L | Lung | 34.07 | 15.26 | 7514.65 | 140.24 | 65.26 |
| Gut | 33.68 | 103.16 | 3213.00 | 92.77 | 70.37 | |
| Nasal tissue | 19.67 | 0.43 | 657.99 | 256.36 | 68.36 | |
| Nasal epithelium | 19.67 | 0.43 | 478.23 | 128.78 | 68.35 | |
| Tmax, h | Lung | 6.65 | 1.13 | 0.08 | 0.02 | 1.84 |
| Gut | 5.68 | 0.57 | 0.13 | 0.03 | 1.30 | |
| Nasal tissue | 6.66 | 1.13 | 0.09 | 0.02 | 1.85 | |
| Nasal epithelium | 6.69 | 1.17 | 0.11 | 0.05 | 1.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakravarty, K.; Antontsev, V.G.; Khotimchenko, M.; Gupta, N.; Jagarapu, A.; Bundey, Y.; Hou, H.; Maharao, N.; Varshney, J. Accelerated Repurposing and Drug Development of Pulmonary Hypertension Therapies for COVID-19 Treatment Using an AI-Integrated Biosimulation Platform. Molecules 2021, 26, 1912. https://doi.org/10.3390/molecules26071912
Chakravarty K, Antontsev VG, Khotimchenko M, Gupta N, Jagarapu A, Bundey Y, Hou H, Maharao N, Varshney J. Accelerated Repurposing and Drug Development of Pulmonary Hypertension Therapies for COVID-19 Treatment Using an AI-Integrated Biosimulation Platform. Molecules. 2021; 26(7):1912. https://doi.org/10.3390/molecules26071912
Chicago/Turabian StyleChakravarty, Kaushik, Victor G. Antontsev, Maksim Khotimchenko, Nilesh Gupta, Aditya Jagarapu, Yogesh Bundey, Hypatia Hou, Neha Maharao, and Jyotika Varshney. 2021. "Accelerated Repurposing and Drug Development of Pulmonary Hypertension Therapies for COVID-19 Treatment Using an AI-Integrated Biosimulation Platform" Molecules 26, no. 7: 1912. https://doi.org/10.3390/molecules26071912