
Alteration in Nasopharyngeal Microbiota Profile in Aged Patients with COVID-19

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Methods
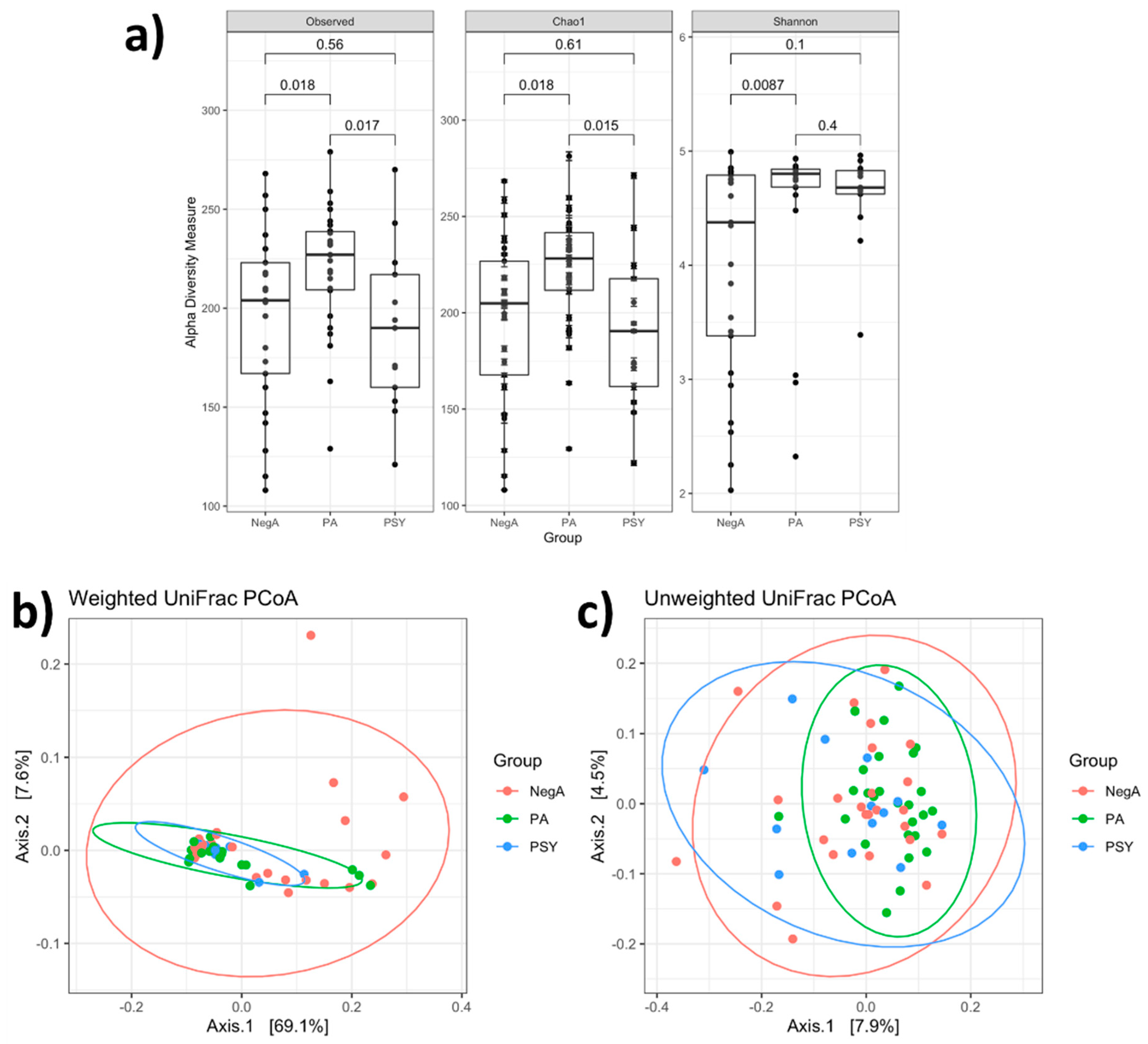
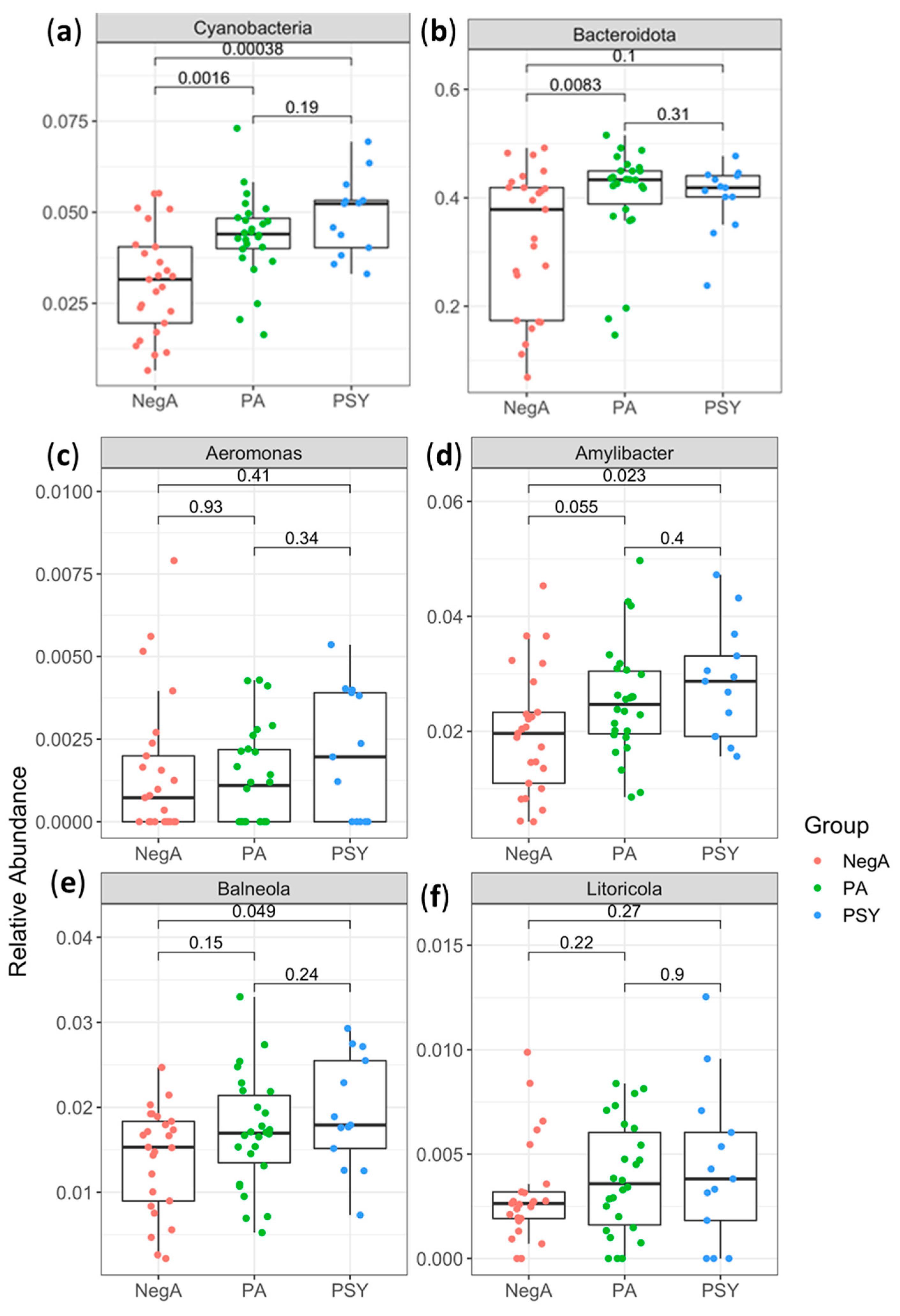
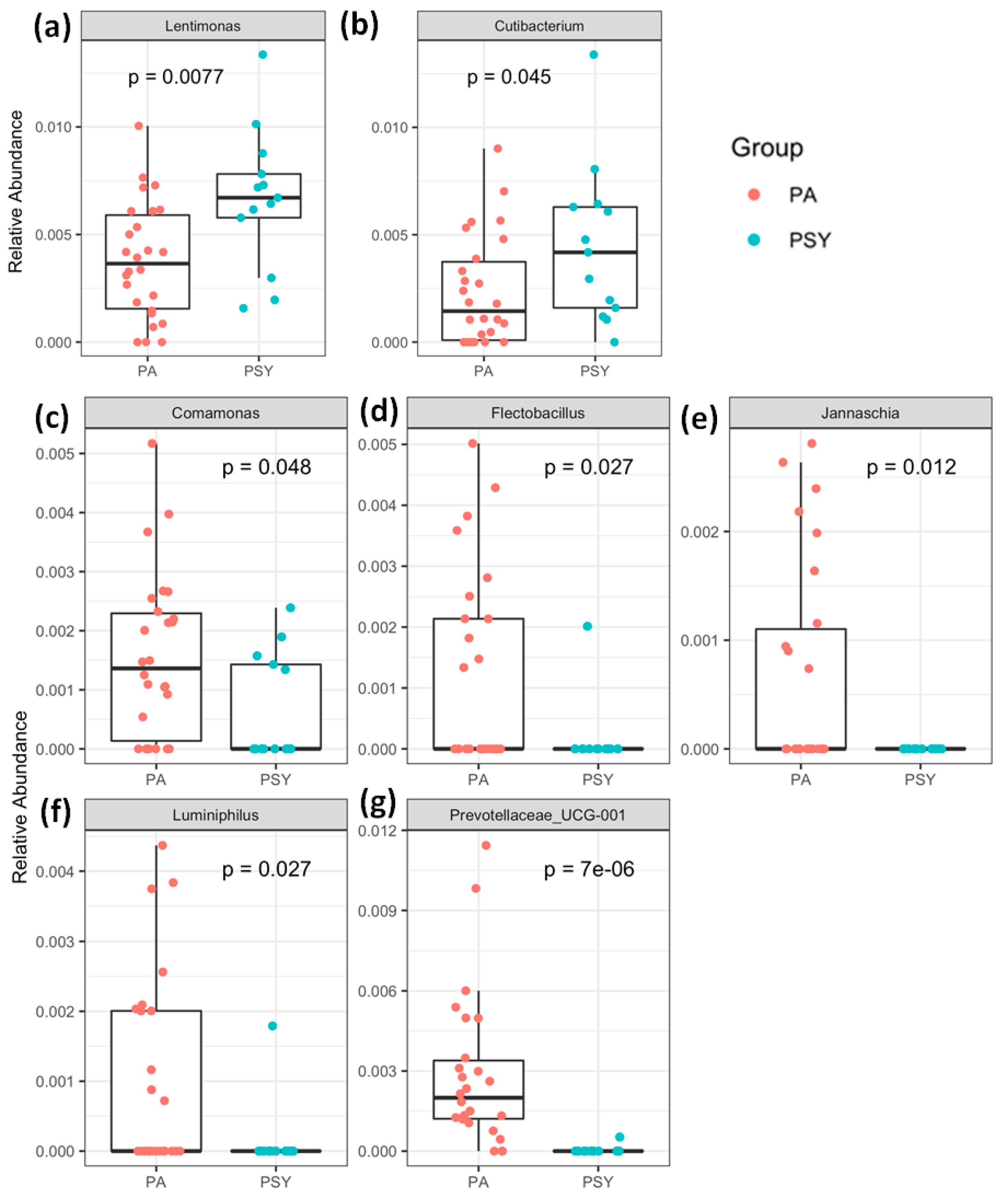
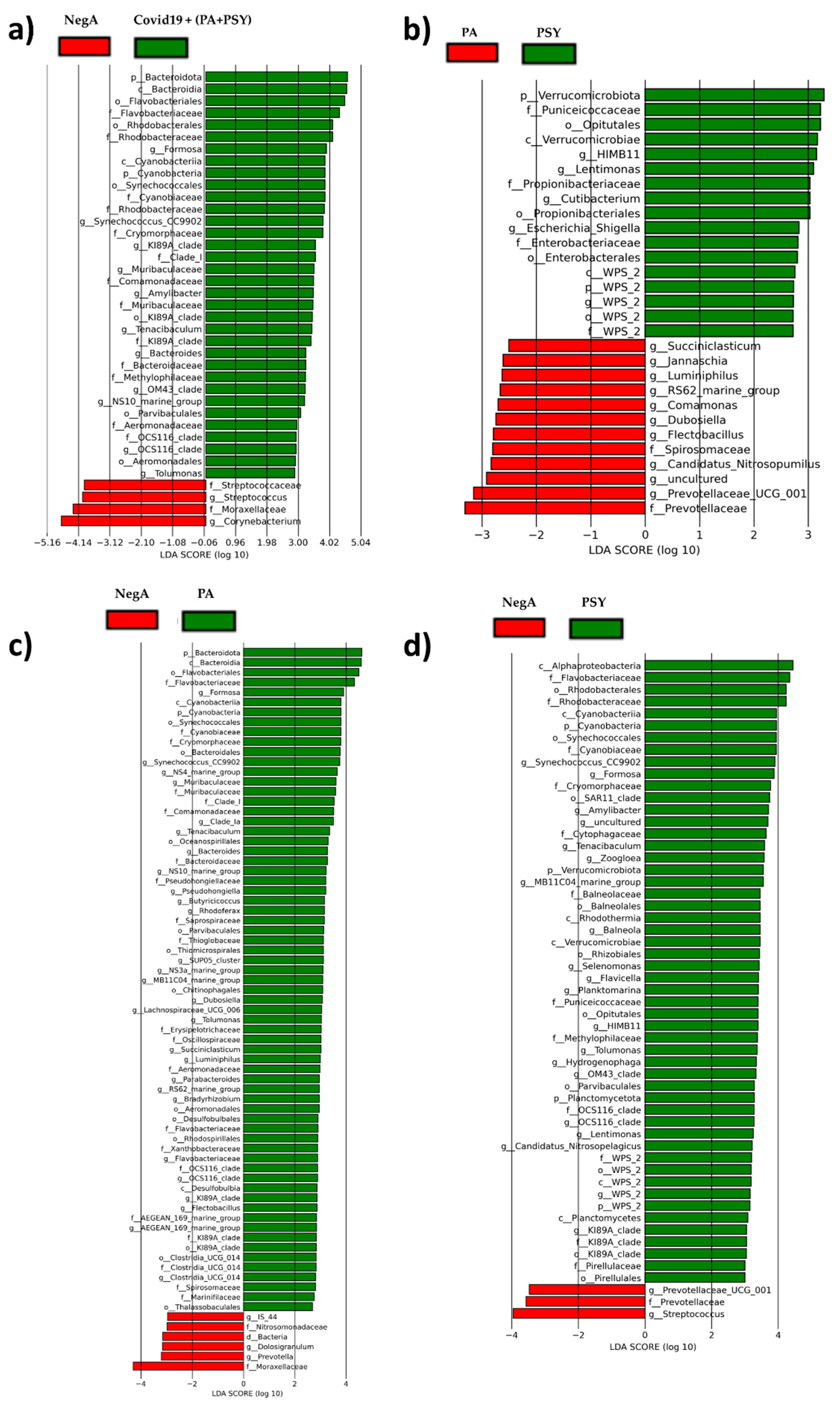
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Lefebvre, A.; Fiet, C.; Belpois-Duchamp, C.; Tiv, M.; Astruc, K.; Glélé, L.A. Case fatality rates of Ebola virus diseases: A meta-analysis of World Health Organization data. Médecine Et Mal. Infect. 2014, 44, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. Coronavirus: COVID-19 Has Killed More People than SARS and MERS Combined, Despite Lower Case Fatality Rate; British Medical Journal Publishing Group: London, UK, 2020. [Google Scholar]
- Fulzele, S.; Sahay, B.; Yusufu, I.; Lee, T.J.; Sharma, A.; Kolhe, R.; Isales, C.M. COVID-19 Virulence in Aged Patients Might Be Impacted by the Host Cellular MicroRNAs Abundance/Profile. Aging Dis. 2020, 11, 509–522. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, J.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J. Allergy Clin. Immunol. 2020, 146, 110–118. [Google Scholar] [CrossRef]
- McCauley, K.; Durack, J.; Valladares, R.; Fadrosh, D.W.; Lin, D.L.; Calatroni, A.; LeBeau, P.K.; Tran, H.T.; Fujimura, K.E.; LaMere, B.; et al. Distinct nasal airway bacterial microbiotas differentially relate to exacerbation in pediatric patients with asthma. J. Allergy Clin. Immunol. 2019, 144, 1187–1197. [Google Scholar] [CrossRef]
- Korten, I.; Ramsey, K.; Mika, M.; Usemann, J.; Frey, U.; Hilty, M.; Latzin, P. Nasal microbiota and respiratory tract infections: The role of viral detection. Am. J. Respir. Crit. Care Med. 2019, 199, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Lehtinen, M.J.; Hibberd, A.A.; Männikkö, S.; Yeung, N.; Kauko, T.; Forssten, S.; Lehtoranta, L.; Lahtinen, S.J.; Stahl, B.; Lyra, A.; et al. Nasal microbiota clusters associate with inflammatory response, viral load, and symptom severity in experimental rhinovirus challenge. Sci. Rep. 2018, 8, 11411. [Google Scholar] [CrossRef] [Green Version]
- De Boeck, I.; van den Broek, M.F.; Allonsius, C.N.; Spacova, I.; Wittouck, S.; Martens, K.; Wuyts, S.; Cauwenberghs, E.; Jokicevic, K.; Vandenheuvel, D.; et al. Lactobacilli have a niche in the human nose. Cell Rep. 2020, 31, 107674. [Google Scholar] [CrossRef] [PubMed]
- Rullo, J.; Far, P.M.; Quinn, M.; Sharma, N.; Bae, S.; Irrcher, I.; Sharma, S. Local oral and nasal microbiota diversity in age-related macular degeneration. Sci. Rep. 2020, 10, 3862. [Google Scholar] [CrossRef] [Green Version]
- Sahajpal, N.S.; Mondal, A.K.; Njau, A.; Ananth, S.; Jones, K.; Ahluwalia, P.K.; Ahluwalia, M.; Jilani, Y.; Chaubey, A.; Hegde, M.; et al. Proposal of RT-PCR-Based Mass Population Screening for Severe Acute Respiratory Syndrome Coronavirus 2 (Coronavirus Disease 2019). J. Mol. Diagn. 2020, 22, 1294–1299. [Google Scholar] [CrossRef]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. mSystems 2017, 2, e00127-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Salzano, F.A.; Marino, L.; Salzano, G.; Botta, R.M.; Cascone, G.; D’Agostino Fiorenza, U.; Selleri, C.; Casolaro, V. Microbiota Composition and the Integration of Exogenous and Endogenous Signals in Reactive Nasal Inflammation. J. Immunol. Res. 2018, 2018, 2724951. [Google Scholar] [CrossRef] [PubMed]
- El-Anwar, M.W.; Elzayat, S.; Fouad, Y.A. ENT manifestation in COVID-19 patients. Auris Nasus Larynx 2020, 47, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Finlay, B.B.; Finlay, J.M. The Whole-Body Microbiome: How to Harness Microbes—Inside and out—For Lifelong Health Hardcover, 1st ed.; The Experiment: New York, NY, USA, 2019; pp. 238–260. [Google Scholar]
- Lee, K.H.; Gordon, A.; Shedden, K.; Kuan, G.; Ng, S.; Balmaseda, A.; Foxman, B. The respiratory microbiome and susceptibility to influenza virus infection. PLoS ONE 2019, 14, e0207898. [Google Scholar] [CrossRef] [Green Version]
- De Maio, F.; Posteraro, B.; Ponziani, F.R.; Cattani, P.; Gasbarrini, A.; Sanguinetti, M. Nasopharyngeal Microbiota Profiling of SARS-CoV 2 Infected Patients. Biol. Proced. Online 2020, 22, 18. [Google Scholar] [CrossRef]
- Wiertsema, S.; Pvan Bergenhenegouwen, J.; Garssen, J.; Knippels, L.M.J. The Interplay between the Gut Microbiome and the Immune System in the Context of Infectious Diseases throughout Life and the Role of Nutrition in Optimizing Treatment Strategies. Nutrients 2021, 13, 886. [Google Scholar] [CrossRef]
- Sencio, V.; Machado, M.G.; Trottein, F. The lung–gut axis during viral respiratory infections: The impact of gut dysbiosis on secondary disease outcomes. Mucosal. Immunol. 2021, 14, 296–304. [Google Scholar] [CrossRef]
- Welp Allison, L.; Bomberger Jennifer, M. Bacterial Community Interactions During Chronic Respiratory Disease. Front. Cell. Infect. Microbiol. 2020, 10, 213. [Google Scholar] [CrossRef]
- Hanada, S.; Pirzadeh, M.; Carver, K.Y.; Deng, J.C. Respiratory Viral Infection-Induced Microbiome Alterations and Secondary Bacterial Pneumonia. Front. Immunol. 2018, 9, 2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Ma, W.T.; Pang, M.; Fan, Q.L.; Hua, J.L. The Commensal Microbiota and Viral Infection: A Comprehensive Review. Front. Immunol. 2019, 10, 1551. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Type | Sample Processed | Amplicons/Reads |
|---|---|---|
| NegA | 27 | 25 |
| PA | 30 | 26 |
| PSY | 27 | 13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolhe, R.; Sahajpal, N.S.; Vyavahare, S.; Dhanani, A.S.; Adusumilli, S.; Ananth, S.; Mondal, A.K.; Patterson, G.T.; Kumar, S.; Rojiani, A.M.; et al. Alteration in Nasopharyngeal Microbiota Profile in Aged Patients with COVID-19. Diagnostics 2021, 11, 1622. https://doi.org/10.3390/diagnostics11091622
Kolhe R, Sahajpal NS, Vyavahare S, Dhanani AS, Adusumilli S, Ananth S, Mondal AK, Patterson GT, Kumar S, Rojiani AM, et al. Alteration in Nasopharyngeal Microbiota Profile in Aged Patients with COVID-19. Diagnostics. 2021; 11(9):1622. https://doi.org/10.3390/diagnostics11091622
Chicago/Turabian StyleKolhe, Ravindra, Nikhil Shri Sahajpal, Sagar Vyavahare, Akhilesh S. Dhanani, Satish Adusumilli, Sudha Ananth, Ashis K. Mondal, G. Taylor Patterson, Sandeep Kumar, Amyn M. Rojiani, and et al. 2021. "Alteration in Nasopharyngeal Microbiota Profile in Aged Patients with COVID-19" Diagnostics 11, no. 9: 1622. https://doi.org/10.3390/diagnostics11091622