
Molecular Evolution of SARS-CoV-2 during the COVID-19 Pandemic


1
Biomedical Research Center (CINBIO), University of Vigo, 36310 Vigo, Spain
2
Department of Biochemistry, Genetics and Immunology, University of Vigo, 36310 Vigo, Spain
3
Galicia Sur Health Research Institute (IIS Galicia Sur), 36310 Vigo, Spain
*
Author to whom correspondence should be addressed.
Genes 2023, 14(2), 407; https://doi.org/10.3390/genes14020407
Submission received: 29 December 2022
/
Revised: 27 January 2023
/
Accepted: 2 February 2023
/
Published: 4 February 2023
(This article belongs to the Special Issue Feature Papers: Molecular Genetics and Genomics 2023)
Abstract
:The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) produced diverse molecular variants during its recent expansion in humans that caused different transmissibility and severity of the associated disease as well as resistance to monoclonal antibodies and polyclonal sera, among other treatments. In order to understand the causes and consequences of the observed SARS-CoV-2 molecular diversity, a variety of recent studies investigated the molecular evolution of this virus during its expansion in humans. In general, this virus evolves with a moderate rate of evolution, in the order of 10−3–10−4 substitutions per site and per year, which presents continuous fluctuations over time. Despite its origin being frequently associated with recombination events between related coronaviruses, little evidence of recombination was detected, and it was mostly located in the spike coding region. Molecular adaptation is heterogeneous among SARS-CoV-2 genes. Although most of the genes evolved under purifying selection, several genes showed genetic signatures of diversifying selection, including a number of positively selected sites that affect proteins relevant for the virus replication. Here, we review current knowledge about the molecular evolution of SARS-CoV-2 in humans, including the emergence and establishment of variants of concern. We also clarify relationships between the nomenclatures of SARS-CoV-2 lineages. We conclude that the molecular evolution of this virus should be monitored over time for predicting relevant phenotypic consequences and designing future efficient treatments.
1. Introduction
After the severe acute respiratory syndrome coronavirus (SARS-CoV) identified in China in 2002 [1] and the Middle East respiratory syndrome coronavirus (MERS-CoV) detected in 2012 in Saudi Arabia [2], the SARS-CoV-2 caused another severe respiratory disease in humans. The expansion of SARS-CoV-2 to humans started in late 2019 in Wuhan, China [3], and caused a worldwide pandemic with innumerable economic, social, and political consequences [4]. This virus was also transmitted from humans to other animals, such as mice, hamsters, cats, dogs, ferrets, and minks, among others [5,6]. Similarly to SARS-CoV, SARS-CoV-2 is a member of the Orthocoronavirinae subfamily, subgenus Sarbecovirus, and presents a positive sense single-stranded RNA genome with a length of approximately 29.9 kb [7,8]. During the pandemic, the genome of SARS-CoV-2 evolved, producing variants that displayed different infective and immunological properties [9,10], which caused a series of epidemiological waves around the world. Clearly, understanding the molecular evolution of SARS-CoV-2 is a key factor in predicting the future of the pandemic and designing more durable treatments, including vaccines and antiviral drugs [11,12,13]. Indeed, since SARS-CoV-2 is a newly emerging virus that infects humans with serious consequences, understanding its molecular adaptation to our species and treatments is required for public health [14].
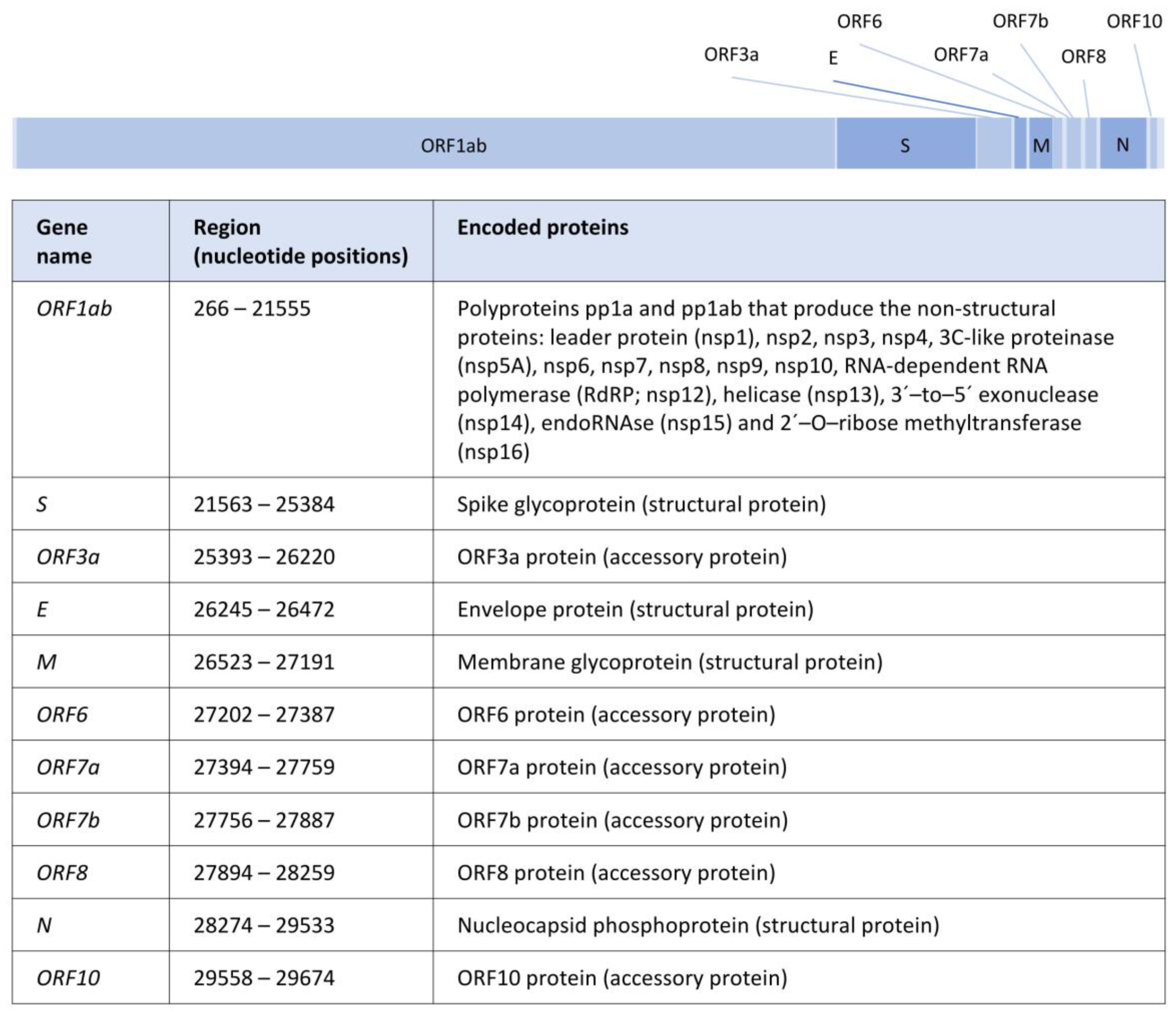
2. The SARS-CoV-2 Genome and Proteins
The almost 30 kb of the SARS-CoV-2 genome contains 11 genes that encode for 29 proteins, including nonstructural, structural, and accessory proteins [8,15] (Figure 1). These proteins are involved in the host cell recognition, entry and uncoating, replication and transcription, assembly, and release, among other functions (details below). In particular, the four genes coding for structural proteins, from 5′ to 3′, are the gene S (nucleotide positions 21563–25384, encoding the spike glycoprotein [16]), the gene E (nucleotide positions 26245–26472, which produces the viral envelope proteins [17]), the gene M (nucleotide positions 26523–27191, leading to the membrane M protein [17]), and the gene N (nucleotide positions 28274–29533, encoding nucleocapsid N proteins [18]).
Briefly (see also Figure 1), the spike glycoprotein binds the virus to the cell receptor; thus, its diversity should be considered in studies on the transmissibility of the virus and the design of certain therapies [19]. Within this protein, the S1 subunit includes a receptor-binding domain called RBD that contacts with the human angiotensin-converting enzyme 2 (ACE2) [16]. The viral envelope proteins encoded by the gene E are involved in the assembly and release of virions, as well as in ion transport and induction of host cell apoptosis [15,20], and also are often conserved in nearby coronaviruses [21]. The membrane M protein performs RNA packaging in the viral assembly by interacting with the N protein (see later) [22], and it is also conserved among related coronaviruses [21]. The nucleocapsid N proteins also participate in RNA packaging, providing stability to the viral assembly and transcription [20,23]. In addition, these proteins can antagonize antiviral interfering RNA and, by inhibition of cyclin-CDK, they can change the cell to the S phase where DNA duplication occurs [15]. The non-structural protein RNA-dependent RNA polymerase (RdRP) participates in the viral replication using a strand of RNA (template) to synthesize the new strand [24].
3. The Nomenclature and Evolutionary History of SARS-CoV-2 Lineages
A variety of SARS-CoV-2 lineages emerged during the expansion of this virus in humans, and their nomenclatures differ among authors or entities, producing some confusion. The most used nomenclature of SARS-CoV-2 lineages is the PANGO nomenclature [25]. It consists of two initial lineages (named with a letter, A and B) that produced sublineages represented by adding numerical sublevels (i.e., B.1 and B.1.6). Next, when the sublineages exceed three sublevels, a new letter is used instead of adding a fourth numerical sublevel (i.e., B.1.1.28.2 is named P.2 and B.1.1.7.7 is named Q.7) [26]. Another nomenclature was presented by Nextstrain [27], a project that phylogenetically classifies SARS-CoV-2 genomes available from the GISAID database [28]. The nomenclature of Nextstrain is based on the different clades that were detected over time. A third nomenclature was presented by GISAID. This nomenclature is based on letters assigned to molecular markers of clades of interest (i.e., the marker S-D614G was defined as clade G, and the subsequent marker S-A222V arising in clade G was defined as clade GV). These clades often display a direct association with PANGO lineages (i.e., GISAID GR corresponds to Nextstrain 20B and PANGO B.1.1.*). Next, WHO (World Health Organization) presented an additional classification of SARS-CoV-2 lineages and clades according to their relevance for the pandemic. In particular, WHO used the terms variant of concern (VOC), a variant of interest (VOI), and a variant under monitoring (VUM). This nomenclature of lineages and clades uses Greek letters (i.e., Alpha and Beta variants), and the number of considered variants increased with the real-time monitoring of the pandemic. A correspondence between PANGO, Nextstrain, GISAID, and WHO nomenclatures can be found in [26].
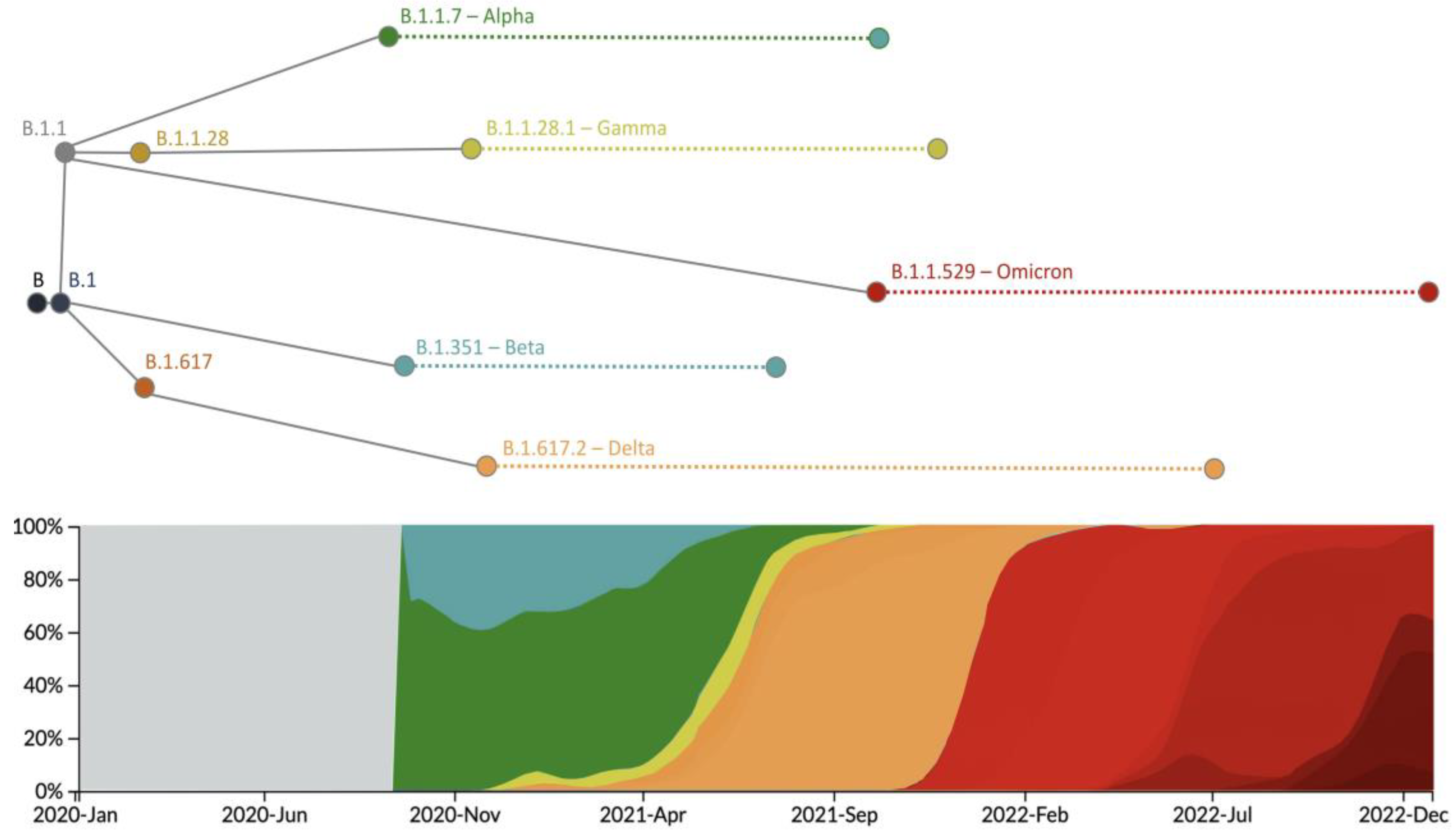
The SARS-CoV-2 genome was highly sequenced, providing a clear view of the evolutionary history of the main lineages of the virus, especially the VOC lineages (Alpha, Beta, Gamma, Delta, and Omicron). In Table 1 and Table 2, we show the mutations that define each VOC lineage, and in Figure 2, we illustrate their frequency over time and their main phylogenetic relationships. In 2020, the first VOC, the Alpha lineage (B.1.1.7), emerged after the accumulation of several genetic changes in the gene S [29,30] (Table 1), being responsible for an increase in transmissibility between 40% and 90%, respect to previous lineages [29,31]. Subsequently, the Beta lineage (B.1.351) appeared [32] with several mutations also in the gene S (Table 1), one of them (E484K) shared with the Alpha lineage. This genetic variation increased the affinity between the spike protein and the human ACE2 receptor with a subsequent increase in transmissibility up to 50% compared to previous lineages [31]. The lineage B.1.1.28 produced a third VOC, the Gamma variant (P.1) [31,33], which presented several genetic changes in the gene S (Table 1), some of them (N501Y, K417N, and E484K) shared with other previous VOCs, and that increased the affinity with ACE2 and, thus, the transmissibility [31]. In addition, Sabino et al. [34] indicated that this variant presents a higher rate of reinfection compared to previous variants. At the end of 2020, the Delta variant (B.1.617.2) emerged (Figure 2), displaying many additional mutations (Table 1) that produced an increase in transmissibility (higher binding stability with ACE2 [31]) and a rapid spread throughout the world, with greater severity and causing more ICU admissions and deaths [35]. In 2021 the Omicron variant (B.1.1.529 or BA) was detected [36,37]. It presented many mutations in the gene S (Table 2), especially in the coding region RBD, and some of them were shared with previous variants (K417N, T478K, E484K, N501Y, and D614G). A crucial aspect of this variant is its capacity to reduce immunity in vaccinated populations [32,38]. When Omicron becomes the dominant variant, its lineages, BA.1, BA.2, BA.3, BA.4, BA.5, and their descendants (i.e., BA.1.1, BA.2.12.1, BA.2.11, BA.2.75, and BA.4.6), emerged and increased in frequency. These lineages are derived from the accumulation of diverse mutations (Table 2) that increased infectivity and immune escape (i.e., BA.4 and BA.5 showed infectivity higher than BA.2, which, in turn, displayed higher infectivity than BA.1) [39]. Interestingly, multiple mutations were fixed in parallel in different VOCs through adaptation, considering their potential factoring in diverse major viral traits (Table 1 and Table 2, specific examples are illustrated in Table 3). For additional information about VOCs, the reader is referred to the reviews [31,36].
4. Evolutionary Mechanisms of SARS-CoV-2
4.1. The Mutation Process in SARS-CoV-2
A variety of viruses present high mutation rates, which, coupled with large population sizes, can result in a large genetic variability. Note that increasing genetic diversity is a key feature for the survival and pathogenesis of RNA viruses [57]. Thus, RNA viruses often present highly error-prone RNA polymerases that cause multiple mutations, but so far, SARS-CoV-2 has shown a moderate acquisition of mutations [58,59,60,61,62]. In particular, coronaviruses present a global mutation rate of around 10−6 per base and per infection cycle [61] (10−3 per site and year [62]), which varies among genes and where the highest rate in SARS-CoV-2 was detected in the S gene [63]. Mutations that occur in the viral surface of proteins are extremely important for generating antigenic variants that allow the virus to evade host immune surveillance, and they can also affect epidemiological and pathogenic characteristics, such as the basic reproductive number (R0), transmissibility, and mortality [64,65]. Indeed, certain mutations allow the virus to escape from the activity of antiviral treatments [66,67]. Interestingly, coevolving sites were detected in key SARS-CoV-2 proteins, such as the spike protein, probably as a consequence of protein stability, affinity, and interaction patterns under specific within-host pressures [43,68]. SARS-CoV-2 is a relatively recent virus infecting humans; thus, we believe that its genetic diversity will largely increase over time.
4.2. Recombination in the SARS-CoV-2
Recombination is a fundamental process in the evolution of multiple viruses and can produce new variants, better adapted to the immune system of the host and antiviral therapies [69]. Moreover, the identification of recombination is crucial to avoid biases in diverse phylogenetic analyses, such as phylogenetic tree reconstruction [70], ancestral sequence reconstruction [71], and detection of selection [72]. In RNA viruses, recombination can occur in the simultaneous infection (by two or more viruses) of the same host cell to produce a redistribution of mutations in the resulting recombinant genome [73]. Next, natural selection can operate upon these new genetic variants [74,75]. The recombination rate in the family Coronaviridae is relatively high [73]. However, this was not observed in the SARS-CoV-2, despite this coronavirus probably originating by recombination between other coronaviruses [76,77]. Multiple studies investigated the evidence of recombination along the genome of SARS-CoV-2, and most of them found that recombination events are scarce, perhaps because of a low recombination rate or because the overall low genetic diversity present in the virus made the detection of recombination difficult [78,79,80]. Thus, despite the fact that several studies found a lack of recombination [81,82], others could detect some evidence (details shown in Table 4). Concerning the latter, the detected recombination breakpoints mainly involved the genes ORF1ab and S [83,84]. Next, to our knowledge, in contrast with other RNA viruses [85,86,87], the population recombination rate in SARS-CoV-2 was not yet investigated. A key factor in obtaining clearer estimates of recombination is to overcome the huge computational burden required to analyze millions of currently available genome sequences [80]. In general, recombination is an evolutionary force that could change the course of the evolution of this virus by producing variants with improved transmissibility, infectivity, and resistance to therapies; thus, we believe that it should be seriously taken into account.
4.3. The Rate of Molecular Evolution of the SARS-CoV-2
The overall rate of molecular evolution of SARS-CoV-2 ranges from 10−3 to 10−4 [81,110,111,112,113,114,115,116,117] (Table 5). However, different rates of evolution were identified among VOCs. In particular, the variant Alpha presented a rate of evolution of 8.47 × 10−3 (0.49–0.62 × 10−3), the variant Beta showed 1.71 × 10−3 (0.34–33.20 × 10−3), the variant Gamma showed 2.76 × 10−3 (1.21–13.23 × 10−3), and the variant Delta presented 1.54 × 10−3 (0.62–7.35 × 10−3) in substitutions per site and year (with 95% confidence interval) [118]. These rates were higher than those detected for non-VOC, which presented estimates of 0.53 × 10−3 (0.49–0.62 × 10−3) substitutions per site and year [118]. The rate of evolution in SARS-CoV-2 was also studied over time to evaluate the hypothesis of the molecular clock (constant rate of evolution over time [119]). Several authors [81,110,116] found that a relaxed molecular clock model fits better with the SARS-CoV-2 genome evolution when compared to a strict molecular clock model, suggesting that the rate of evolution changed over time. In this concern, Tay et al. [118] observed that the evolution of data collected at the beginning of the pandemic was better explained by a strict molecular clock model, while a relaxed molecular clock model could better fit with data collected more recently.
4.4. Molecular Adaptation in the SARS-CoV-2 Genome
The selection was widely investigated in SARS-CoV-2, especially using the traditional metric nonsynonymous/synonymous substitution rate ratio (dN/dS) [120]. At the global (whole genome) level, genetic signatures of negative (purifying) selection were observed, suggesting overall maintenance of the function of protein variants [62,121,122,123]. At the gene level, the negative selection was detected in most of the SARS-CoV-2 genes, including S [124,125,126,127,128], E [124,125,126,128], M [124,125,128], N [124,125,126,128], ORF6 [124,125,128], ORF7a [124,125], ORF7b [124], ORF8 [124,128], ORF10 [124], and also in regions, such as ORF1ab coding for nonstructural proteins [124,125,126,127,128]. By contrast, the positive (diversifying) selection was detected in several genes, including S [121,129], N [121,129], ORF3a [121,129], ORF8 [121,129], and in the coding regions of some non-structural proteins, such as nsp4 [121], nsp5A [128], nsp10 [128], and nsp13 [121]. Interestingly, some studies identified positive selection at the whole gene level in genes, for which other studies observed negative selection, for example, in ORF7a and ORF10 [128], suggesting that further analyses should be performed to clarify these findings. We highlight the positive selection detected in the gene S, especially in sites of the RBD, since those nonsynonymous changes can affect the binding of the spike protein with the ACE2 receptor (details below) [130]. Next, a number of studies detected positively selected sites (PSSs) in the genes S [20,122,123,131], N [20,122,123], ORF3a [123,126], ORF8 [123,126], and in the coding regions of the non-structural proteins nsp2 [129], nsp3 [129], nsp4 [129], nsp6 [123,129], nsp12 [129], and nsp13 [123,129]. Some of them were relevant, such as the PSS, associated with the D614G mutation in the gene S [108,122], which participates in the opening of the RBD [20] and produces an advantage for infectivity [48] and transmissibility [132]. In the same gene, a PSS associated with the L5F mutation facilitated the folding, assembly, and secretion of the virus [20], and PSSs associated with the mutations K417T, E484K, and L18F (also the N501Y mutation [133,134]) allowed escape from monoclonal antibodies [135,136,137,138]. Indeed, other detected PSSs were associated with the P681H mutation that increased the cleavage of spike protein with ACE2 [139] and the I82T mutation that increased the stability of the protein M [140]. In the gene ORF3a, a PSS associated with the Q57H mutation increased the affinity between the proteins ORF3a, spike, and M, favoring viral replication [141,142]. In the protein nsp6, a relevant PSS involved the L37F mutation that allowed the virus to reduce the cellular defense via autophagy regulation [42]. Concerning the region coding for nsp12 and nsp14 (RdRP), some PSSs were detected and related with the mutations P323 (nsp12), A394V, and I42V (nsp14) that were associated with promoting an increase in the mutation rate [121]. Of course, many negatively selected sites (NSSs) and coevolving sites were also identified along the genome and were associated with fundamental processes that should be maintained for viral replication [143].
Molecular Adaptation Induced by Therapies and Immune Systems
Genetic signatures of molecular adaptation in SARS-CoV-2 can also be observed as a consequence of the applied therapies and the immune systems. Concerning the latter, in the presence of the virus, the immune system operates through different mechanisms, such as CD8+ T and natural killer (NK) cells. Indeed, subepithelial dendritic cells and macrophages induce the differentiation of CD4+ T cells into memory T helper types Th1, Th17, and follicular T helper. The latter helps B cells to become plasma cells, promoting the production of IgM, IgA, and IgG antibodies [144].
Actually, the fixation of escape variants in virus populations was already observed [145,146], where multiple variants were able to evade therapeutic antibodies through escape mutations [145,146,147]. For example, the Beta and Gamma variants presented the mutation N439K that considerably increased the neutralizing activity of monoclonal antibodies and polyclonal serum [131,145,147]. Indeed, these variants often included the mutation K417N which also favors viral escape from diverse monoclonal antibodies, and the mutation N501Y (also observed in the Alpha and Omicron variants), which increases transmissibility through a higher affinity with ACE2 [147]. Therefore, therapeutic monoclonal antibodies, such as Casirivimab and Imdevimab, showed a smaller efficacy due to mutations, such as K417N and E484K (present in the Beta variant) for the former antibody, and mutations, such as L452R/Q (present in the Beta and Delta variants) for the latter [137,148]. Indeed, certain mutations observed in the Gamma and Omicron variants also reduced the efficacy of therapeutic monoclonal antibodies, such as Bamlanivimab (used to treat infection with the Gamma variant) [149] and Bamlanivimab, Etesevimab, Casirivimab, Imdevimab, and Regdanvimab (used against the Omicron variant) [67]. Moreover, the selection was not only detected to fix variants that escape from the recognition of monoclonal antibodies but also to fix the variants resistant to polyclonal serum and plasma. For example, Greaney et al. [147] identified mutations in the RBD region of the spike protein that reduced the efficacy of both types of antibodies.
Concerning vaccines, they produced strong selective pressures that caused the fixation of previously neutral or non-beneficial genetic changes. For example, some authors reported that the Novavax vaccine, which displayed an efficacy of 95.6% against the original SARS-CoV-2 variant, only presented an efficacy of 85.6% against the Alpha variant and 60% against the Beta variant [150]. Indeed, several studies showed that some mutations (i.e., E484K in the Beta variant and L452R in the Delta variant) caused resistance to vaccines based on mRNA (i.e., Pfizer and Moderna) and adenoviral vectors (i.e., Johnson & Johnson) [137,149,151]. In addition, the Gamma variant displayed resistance in Pfizer-vaccinated patients [149], and the Omicron variant showed escape in patients vaccinated with Pfizer, AstraZeneca, and Moderna [67,152]. Next, some studies detected mutations associated with resistance to Remdesivir, the most widespread antiviral treatment against SARS-CoV-2. These mutations in the gene S, which circulated at low frequency, were A97V [153], F480L/S/C [154], V557L [154], and E802D [155,156]. These findings suggest the need for continuous monitoring of SARS-CoV-2 molecular evolution for designing effective therapies against the variants circulating at every time. Indeed and despite the cited resistant mutants, vaccination largely reduces illness, hospitalization, and mortality [157,158]. In this concern, efforts should be made to provide access to vaccines in low-income countries, and there is a general need to improve the equitability of vaccination coverage worldwide [159].
5. Conclusions and Future Prospects
The SARS-CoV-2 pandemic promoted an impressive and convenient amount of works about this virus. Note that so far, more than 14 million SARS-CoV-2 genomes have been deposited in GISAID. This large amount of data allowed us to properly study the molecular evolution of this virus, identify the relationships between phenotypic (i.e., transmissibility and severity of the disease) and molecular (i.e., mutations) observations, and even predict which therapies are more appropriate at every time (for every variant). Samples of SARS-CoV-2 collected in humans overall presented genetic signatures of moderate mutation rate, little recombination, and diverse selective pressures (including positively selected sites, where some of them were promoted by treatments). Interestingly, several studies indicated that these evolutionary patterns could be changing over time, in particular toward increasing the rate of evolution (including a higher frequency of recombination events) and, consequently, the emergence and fixation of new variants. This trend could also be favored by the extremely large virus populations and the capacity of RNA viruses to adapt to new environments, such as those imposed by immune systems and therapies.
The pandemic of SARS-CoV-2 is far from over, and the virus continues circulating and evolving with sufficient capacity to produce variants presenting antiviral resistance. Therefore, monitoring SARS-CoV-2 evolution is extremely useful for designing treatments effective at each period of time. Moreover, we believe that efforts should be made to obtain a precise fitness landscape that includes the observed evolutionary trajectories of the virus, considering both mutation and recombination events, to better understand the relationships between those molecular changes and the observed phenotypic consequences. We also believe that in addition to the large amount of genomic data that is currently available for this virus, and that continues to increase, efforts should also be made to develop computational frameworks for the evolutionary analyses of such a large amount of data (see [160]). Future zoonoses events involving coronaviruses seem inevitable [11] and, in this concern, the knowledge that we can learn from the molecular evolution of SARS-CoV-2 could be useful to improve the prevention, anticipation, and management of future pandemics caused by similar viruses.
Author Contributions
Conceptualization, M.A. and L.D.G.-V.; Writing—Original Draft Preparation, M.A. and L.D.G.-V.; Writing—Review and editing, M.A. and L.D.G.-V.; Visualization, M.A. and L.D.G.-V.; Project Administration, M.A.; Funding Acquisition, M.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Spanish Ministry of Economy and Competitiveness and the Ministry of Science and Innovation through the Grant [PID2019-107931GA-I00/AEI/10.13039/501100011033].
Acknowledgments
We thank the journal Genes for the invitation to contribute with this review article to the special issue “Feature Papers: Molecular Genetics and Genomics”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chan-Yeung, M.; Xu, R. SARS: Epidemiology. Respirology 2003, 8, S9–S14. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus–Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Bonotti, M.; Zech, S.T. The Human, Economic, Social, and Political Costs of COVID-19. In Recovering Civility during COVID-19; Springer: Berlin/Heidelberg, Germany, 2021; pp. 1–36. [Google Scholar] [CrossRef]
- Pan, T.; Chen, R.; He, X.; Yuan, Y.; Deng, X.; Li, R.; Yan, H.; Yan, S.; Liu, J.; Zhang, Y.; et al. Infection of wild-type mice by SARS-CoV-2 B. 1.351 variant indicates a possible novel cross-species transmission route. Signal Transduct. Target. Ther. 2021, 6, 420. [Google Scholar] [CrossRef] [PubMed]
- de Vries, R.D.; Rockx, B.; Haagmans, B.L.; Herfst, S.; Koopmans, M.P.; de Swart, R.L. Animal models of SARS-CoV-2 transmission. Curr. Opin. Virol. 2021, 50, 8–16. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The Species Severe Acute Respiratory Syndrome-Related Coronavirus: Classifying 2019-NCoV and Naming It SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Bai, C.; Zhong, Q.; Gao, G.F. Overview of SARS-CoV-2 Genome-Encoded Proteins. Sci. China Life Sci. 2022, 65, 280–294. [Google Scholar] [CrossRef]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The Emergence, Genomic Diversity and Global Spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Peacock, S.J.; et al. SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Singh, D.; Yi, S.V. On the Origin and Evolution of SARS-CoV-2. Exp. Mol. Med. 2021, 53, 537–547. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Bonilla, H.; Jagannathan, P.; Shafer, R.W. SARS-CoV-2 Antiviral Therapy. Clin. Microbiol. Rev. 2021, 34, e00109-21. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, L.; Poullikkas, T.; Eisenhower, S.; Monsanto, C.; Bakku, R.K.; Chen, M.-H.; Kalra, R.S. Viroinformatics-Based Analysis of SARS-CoV-2 Core Proteins for Potential Therapeutic Targets. Antibodies 2021, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Day, T.; Gandon, S.; Lion, S.; Otto, S.P. On the Evolutionary Epidemiology of SARS-CoV-2. Curr. Biol. 2020, 30, R849–R857. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and Functional Properties of SARS-CoV-2 Spike Protein: Potential Antivirus Drug Development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Abavisani, M.; Rahimian, K.; Mahdavi, B.; Tokhanbigli, S.; Mollapour Siasakht, M.; Farhadi, A.; Kodori, M.; Mahmanzar, M.; Meshkat, Z. Mutations in SARS-CoV-2 Structural Proteins: A Global Analysis. Virol. J. 2022, 19, 220. [Google Scholar] [CrossRef]
- Savastano, A.; Ibáñez de Opakua, A.; Rankovic, M.; Zweckstetter, M. Nucleocapsid Protein of SARS-CoV-2 Phase Separates into RNA-Rich Polymerase-Containing Condensates. Nat. Commun. 2020, 11, 6041. [Google Scholar] [CrossRef]
- Candido, K.L.; Eich, C.R.; de Fariña, L.O.; Kadowaki, M.K.; da Conceição Silva, J.L.; Maller, A.; Simão, R.C.G. Spike Protein of SARS-CoV-2 Variants: A Brief Review and Practical Implications. Braz. J. Microbiol. 2022, 53, 1133–1157. [Google Scholar] [CrossRef]
- Zhan, X.Y.; Zhang, Y.; Zhou, X.; Huang, K.; Qian, Y.; Leng, Y.; Yan, L.; Huang, B.; He, Y. Molecular Evolution of SARS-CoV-2 Structural Genes: Evidence of Positive Selection in the Spike Glycoprotein. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mohammad, T.; Choudhury, A.; Habib, I.; Asrani, P.; Mathur, Y.; Umair, M.; Anjum, F.; Shafie, A.; Yadav, D.K.; Hassan, M.I. Genomic Variations in the Structural Proteins of SARS-CoV-2 and Their Deleterious Impact on Pathogenesis: A Comparative Genomics Approach. Front. Cell. Infect. Microbiol. 2021, 11, 951. [Google Scholar] [CrossRef]
- Masters, P.S. Coronavirus Genomic RNA Packaging. Virology 2019, 537, 198–207. [Google Scholar] [CrossRef]
- Bai, Z.; Cao, Y.; Liu, W.; Li, J. The SARS-CoV-2 Nucleocapsid Protein and Its Role in Viral Structure, Biological Functions, and a Potential Target for Drug or Vaccine Mitigation. Viruses 2021, 13, 1115. [Google Scholar] [CrossRef] [PubMed]
- Hillen, H.S. Structure and Function of SARS-CoV-2 Polymerase. Curr. Opin. Virol. 2021, 48, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Alm, E.; Broberg, E.K.; Connor, T.; Hodcroft, E.B.; Komissarov, A.B.; Maurer-Stroh, S.; Melidou, A.; Neher, R.A.; O’Toole, Á.; Pereyaslov, D.; et al. Geographical and Temporal Distribution of SARS-CoV-2 Clades in the WHO European Region, January to June 2020. Eurosurveillance 2020, 25, 2001410. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Funk, T.; Pharris, A.; Spiteri, G.; Bundle, N.; Melidou, A.; Carr, M.; Gonzalez, G.; Garcia-Leon, A.; Crispie, F.; O’Connor, L.; et al. Characteristics of SARS-CoV-2 Variants of Concern B.1.1.7, B.1.351 or P.1: Data from Seven EU/EEA Countries, Weeks 38/2020 to 10/2021. Eurosurveillance 2021, 26, 2100348. [Google Scholar] [CrossRef]
- Choi, J.Y.; Smith, D.M. SARS-CoV-2 Variants of Concern. Yonsei Med. J. 2021, 62, 961–968. [Google Scholar] [CrossRef]
- Attwood, S.W.; Hill, S.C.; Aanensen, D.M.; Connor, T.R.; Pybus, O.G. Phylogenetic and Phylodynamic Approaches to Understanding and Combating the Early SARS-CoV-2 Pandemic. Nat. Rev. Genet. 2022, 23, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; de Lima Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef] [PubMed]
- Sabino, E.C.; Buss, L.F.; Carvalho, M.P.S.; Prete, C.A.; Crispim, M.A.E.; Fraiji, N.A.; Pereira, R.H.M.; Parag, K.V.; Peixoto, P.; Kraemer, M.U.G.; et al. Resurgence of COVID-19 in Manaus, Brazil, despite High Seroprevalence. Lancet 2021, 397, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.W.X.; Chiew, C.J.; Ang, L.W.; Mak, T.M.; Cui, L.; Toh, M.P.H.; Lim, Y.D.; Lee, P.H.; Lee, T.H.; Chia, P.Y.; et al. Clinical and Virological Features of SARS-CoV-2 Variants of Concern: A Retrospective Cohort Study Comparing B.1.1.7 (Alpha), B.1.315 (Beta), and B.1.617.2 (Delta). Clin. Infect. Dis. 2021, 75, ciab721. [Google Scholar] [CrossRef]
- Kannan, S.; Shaik Syed Ali, P.; Sheeza, A. Omicron (B.1.1.529)–Variant of Concern–Molecular Profile and Epidemiology: A Mini Review. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA. 1 to BA. 5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef]
- Lauring, A.S.; Tenforde, M.W.; Chappell, J.D.; Gaglani, M.; Ginde, A.A.; McNeal, T.; Ghamande, S.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; et al. Clinical severity of, and effectiveness of mRNA vaccines against, covid-19 from omicron, Delta, and Alpha SARS-CoV-2 variants in the United States: Prospective observational study. BMJ 2022, 376, e069761. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhi, H.; Teng, Y. The outbreak of SARS-CoV-2 Omicron lineages, immune escape, and vaccine effectivity. J. Med. Virol. 2023, 95, e28138. [Google Scholar] [CrossRef]
- Rajpal, V.R.; Sharma, S.; Kumar, A.; Chand, S.; Joshi, L.; Chandra, A.; Babbar, S.; Goel, S.; Raina, S.N.; Shiran, B. ‘Is omicron mild’? Testing this narrative with the mutational landscape of its three lineages and response to existing vaccines and therapeutic antibodies. J. Med. Virol. 2022, 94, 3521–3539. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G.; et al. Emergence of SARS-CoV-2 omicron lineages BA. 4 and BA. 5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary Analysis of SARS-CoV-2: How Mutation of Non-Structural Protein 6 (NSP6) Could Affect Viral Autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef] [PubMed]
- Kistler, K.E.; Huddleston, J.; Bedford, T. Rapid and parallel adaptive mutations in spike S1 drive clade success in SARS-CoV-2. Cell Host Microbe 2022, 30, 545–555. [Google Scholar] [CrossRef]
- McAuley, A.J.; Kuiper, M.J.; Durr, P.A.; Bruce, M.P.; Barr, J.; Todd, S.; Au, G.G.; Blasdell, K.; Tachedjian, M.; Lowther, S.; et al. Experimental and in silico evidence suggests vaccines are unlikely to be affected by D614G mutation in SARS-CoV-2 spike protein. npj Vaccines 2020, 5, 96. [Google Scholar] [CrossRef] [PubMed]
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Alshammary, H.; Amoako, A.A.; Awawda, M.H.; Beach, K.F.; Bermúdez-González, M.C.; Chernet, R.L.; et al. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. Lancet Microbe 2021, 2, e283–e284. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Patil, V.M.; Gupta, M.K. Mutation informatics: SARS-CoV-2 receptor-binding domain of the spike protein. Drug Discov. Today 2022, 27, 103312. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Kasry, A.; Amin, M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Discov. 2021, 10, 100086. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751. [Google Scholar] [CrossRef]
- Lista, M.J.; Winstone, H.; Wilson, H.D.; Dyer, A.; Pickering, S.; Galao, R.P.; De Lorenzo, G.; Cowton, V.M.; Furnon, W.; Suarez, N.; et al. The P681H Mutation in the Spike Glycoprotein of the Alpha Variant of SARS-CoV-2 Escapes IFITM Restriction and Is Necessary for Type I Interferon Resistance. J. Virol. 2022, 96, e01250-22. [Google Scholar] [CrossRef]
- Fratev, F. N501Y and K417N mutations in the spike protein of SARS-CoV-2 alter the interactions with Both hACE2 and human-derived antibody: A free energy of perturbation retrospective study. J. Chem. Inf. Model. 2021, 61, 6079–6084. [Google Scholar] [CrossRef]
- Yamamoto, M.; Tomita, K.; Hirayama, Y.; Inoue, J.I.; Kawaguchi, Y.; Gohda, J. SARS-CoV-2 Omicron spike H655Y mutation is responsible for enhancement of the endosomal entry pathway and reduction of cell surface entry pathway. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hu, B.; Chan, J.F.W.; Liu, H.; Liu, Y.; Chai, Y.; Shi, J.; Shuai, H.; Hou, Y.; Huang, X.; Yuen, T.T.-T.; et al. Spike mutations contributing to the altered entry preference of SARS-CoV-2 omicron BA.1 and BA.2. Emerg. Microbes Infect. 2022, 11, 2275–2287. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Triche, T.J.; Bard, J.D.; Biegel, J.A.; Judkins, A.R.; Gai, X. Spike Protein NTD mutation G142D in SARS-CoV-2 Delta VOC lineages is associated with frequent back mutations, increased viral loads, and immune evasion. medRxiv 2021. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J. Med. Virol. 2021, 93, 5638–5643. [Google Scholar] [CrossRef]
- Wu, H.; Xing, N.; Meng, K.; Fu, B.; Xue, W.; Dong, P.; Tang, W.; Xiao, Y.; Liu, G.; Luo, H.; et al. Nucleocapsid mutations R203K/G204R increase the infectivity, fitness, and virulence of SARS-CoV-2. Cell Host Microbe 2021, 29, 1788–1801. [Google Scholar] [CrossRef]
- Rahimi, A.; Mirzazadeh, A.; Tavakolpour, S. Genetics and Genomics of SARS-CoV-2: A Review of the Literature with the Special Focus on Genetic Diversity and SARS-CoV-2 Genome Detection. Genomics 2021, 113, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.W.; Capoferri, A.A.; Katusiime, M.G.; Patro, S.C.; Kearney, M.F. Low Genetic Diversity May Be an Achilles Heel of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 24614–24616. [Google Scholar] [CrossRef]
- Lokman, S.M.; Rasheduzzaman, M.; Salauddin, A.; Barua, R.; Tanzina, A.Y.; Rumi, M.H.; Hossain, M.I.; Siddiki, A.M.A.M.Z.; Mannan, A.; Hasan, M.M. Exploring the Genomic and Proteomic Variations of SARS-CoV-2 Spike Glycoprotein: A Computational Biology Approach. Infect. Genet. Evol. 2020, 84, 104389. [Google Scholar] [CrossRef]
- Tang, X.; Wu, C.; Li, X.; Song, Y.; Yao, X.; Wu, X.; Duan, Y.; Zhang, H.; Wang, Y.; Qian, Z.; et al. On the Origin and Continuing Evolution of SARS-CoV-2. Natl. Sci. Rev. 2020, 7, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Amicone, M.; Borges, V.; Alves, M.J.; Isidro, J.; Zé-Zé, L.; Duarte, S.; Vieira, L.; Guiomar, R.; Gomes, J.P.; Gordo, I. Mutation Rate of SARS-CoV-2 and Emergence of Mutators during Experimental Evolution. Evol. Med. Public Health 2022, 10, 142–155. [Google Scholar] [CrossRef]
- Chaw, S.M.; Tai, J.H.; Chen, S.L.; Hsieh, C.H.; Chang, S.Y.; Yeh, S.H.; Yang, W.S.; Chen, P.J.; Wang, H.Y. The Origin and Underlying Driving Forces of the SARS-CoV-2 Outbreak. J. Biomed. Sci. 2020, 27, 73. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, X.; Zhou, J.; Dong, Y.; Jiang, W.; Jiang, W. Rampant C-to-U Deamination Accounts for the Intrinsically High Mutation Rate in SARS-CoV-2 Spike Gene. RNA 2022, 28, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.A.; VanInsberghe, D.; Koelle, K. Insights from SARS-CoV-2 Sequences. Science 2021, 371, 466–467. [Google Scholar] [CrossRef]
- Ye, C.; Thornlow, B.; Kramer, A.; McBroome, J.; Hinrichs, A.; Corbett-Detig, R.; Turakhia, Y. Pandemic-Scale Phylogenetics. bioRxiv 2021. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced Sensitivity of SARS-CoV-2 Variant Delta to Antibody Neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.H.; Porrot, F.; Staropoli, I.; Lemoine, F.; et al. Considerable Escape of SARS-CoV-2 Omicron to Antibody Neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Priya, P.; Shanker, A. Coevolutionary forces shaping the fitness of SARS-CoV-2 spike glycoprotein against human receptor ACE2. Infect. Genet. Evol. 2021, 87, 104646. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in Viruses: Mechanisms, Methods of Study, and Evolutionary Consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef]
- Schierup, M.H.; Hein, J. Consequences of Recombination on Traditional Phylogenetic Analysis. Genetics 2000, 156, 879–891. [Google Scholar] [CrossRef]
- Arenas, M.; Posada, D. The Effect of Recombination on the Reconstruction of Ancestral Sequences. Genetics 2010, 184, 1133–1139. [Google Scholar] [CrossRef]
- Arenas, M.; Posada, D. The Influence of Recombination on the Estimation of Selection from Coding Sequence Alignments. In Natural Selection: Methods and Applications; Fares, M.A., Ed.; CRC Press: Boca Raton, FL, USA, 2014; Chapter 6; pp. 112–125. [Google Scholar]
- Simon-Loriere, E.; Holmes, E.C. Why Do RNA Viruses Recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- VanInsberghe, D.; Neish, A.; Lowen, A.C.; Koelle, K. Identification of SARS-CoV-2 Recombinant Genomes. biorXiv 2020. [Google Scholar] [CrossRef]
- Zhu, Z.; Meng, K.; Meng, G. Genomic recombination events may reveal the evolution of coronavirus and the origin of SARS-CoV-2. Sci. Rep. 2020, 10, 21617. [Google Scholar] [CrossRef]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.-P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through Recombination and Strong Purifying Selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. Evaluation of Methods for Detecting Recombination from DNA Sequences: Empirical Data. Mol. Biol. Evol. 2002, 19, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of Methods for Detecting Recombination from DNA Sequences: Computer Simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Arenas, M. Computational Analysis of Recombination in Viral Nucleotide Sequences. In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Oxford, UK, 2021; Volume 1, pp. 108–115. [Google Scholar]
- Nie, Q.; Li, X.; Chen, W.; Liu, D.; Chen, Y.; Li, H.; Li, D.; Tian, M.; Tan, W.; Zai, J. Phylogenetic and Phylodynamic Analyses of SARS-CoV-2. Virus Res. 2020, 287, 198098. [Google Scholar] [CrossRef]
- Richard, D.; Owen, C.J.; van Dorp, L.; Balloux, F. No Detectable Signal for Ongoing Genetic Recombination in SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jackson, B.; Rambaut, A.; Pybus, O.G.; Robertson, D.L.; Connor, T.; Loman, N.J. COG-UK Consortium (2021) Recombinant SARS-CoV-2 Genomes Involving Lineage B.1.1.7 in the UK. Virological. Available online: https://virological.org/t/recombinant-sars-cov-2-genomes-involving-lineage-b-1-1-7-in-the-uk/658 (accessed on 22 November 2022).
- Lacek, K.A.; Rambo-Martin, B.L.; Batra, D.; Zheng, X.; Sakaguchi, H.; Peacock, T.; Keller, M.; Wilson, M.M.; Sheth, M.; Davis, M.L.; et al. Identification of a Novel SARS-CoV-2 Delta-Omicron Recombinant Virus in the United States. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lopes, J.S.; Arenas, M.; Posada, D.; Beaumont, M.A. Coestimation of Recombination, Substitution and Molecular Adaptation Rates by Approximate Bayesian Computation. Heredity 2014, 112, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Arenas, M.; Lorenzo-Redondo, R.; Lopez-Galindez, C. Influence of Mutation and Recombination on HIV-1 in Vitro Fitness Recovery. Mol. Phylogenet. Evol. 2016, 94, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Castelhano, N.; Araujo, N.M.; Arenas, M. Heterogeneous Recombination among Hepatitis B Virus Genotypes. Infect. Genet. Evol. 2017, 54, 486–490. [Google Scholar] [CrossRef]
- Wertheim, J.O.; Wang, J.C.; Leelawong, M.; Martin, D.P.; Havens, J.L.; Chowdhury, M.A.; Pekar, J.E.; Amin, H.; Arroyo, A.; Awandare, G.A.; et al. Detection of SARS-CoV-2 Intra-Host Recombination during Superinfection with Alpha and Epsilon Variants in New York City. Nat. Commun. 2022, 13, 3645. [Google Scholar] [CrossRef] [PubMed]
- Combes, P.; Bisseux, M.; Bal, A.; Marin, P.; Latour, J.; Archimbaud, C.; Brebion, A.; Chabrolles, H.; Regagnon, C.; Lafolie, J.; et al. Evidence of Co-Infections during Delta and Omicron SARS-CoV-2 Variants Co-Circulation through Prospective Screening and Sequencing. Clin. Microbiol. Infect. 2022, 28, 1503. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Montagutelli, X.; Lemoine, F.; Donati, F.; Touret, F.; Bourret, J.; Prot, M.; Munier, S.; Attia, M.; Conquet, L.; et al. Rapid characterization of a Delta-Omicron SARS-CoV-2 recombinant detected in Europe. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Colson, P.; Fournier, P.E.; Delerce, J.; Million, M.; Bedotto, M.; Houhamdi, L.; Yahi, N.; Bayette, J.; Levasseur, A.; Fantini, J.; et al. Culture and Identification of a “Deltamicron” SARS-CoV-2 in a Three Cases Cluster in Southern France. J. Med. Virol. 2022, 94, 3739–3749. [Google Scholar] [CrossRef]
- Bolze, A.; Basler, T.; White, S.; Rossi, A.D.; Wyman, D.; Roychoudhury, P.; Greninger, A.L.; Hayashibara, K.; Beatty, M.; Shah, S.; et al. Evidence for SARS-CoV-2 Delta and Omicron Co-Infections and Recombination. Med 2022, 3, 848–859. [Google Scholar] [CrossRef]
- Karbalaei, M.; Keikha, M. Deltacron Is a Recombinant Variant of SARS-CoV-2 but Not a Laboratory Mistake. Ann. Med. Surg. 2022, 79, 104032. [Google Scholar] [CrossRef]
- Wang, L.; Gao, G.F. The “Wolf” Is Indeed Coming: Recombinant “Deltacron” SARS-CoV-2 Detected. China CDC Wkly. 2022, 4, 285–287. [Google Scholar] [CrossRef]
- Kreier, F. Deltacron: The Story of the Variant That Wasn’t. Nature 2022, 602, 19. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R.; Dhama, K. Recombinant SARS-CoV-2 Variants XD, XE, and XF: The Emergence of Recombinant Variants Requires an Urgent Call for Research–Correspondence. Int. J. Surg. 2022, 102, 106670. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, R.K.; Kandi, V.; Tuli, H.S.; Chakraborty, C.; Dhama, K. The Recombinant Variants of SARS-CoV-2: Concerns Continues amid COVID-19 Pandemic. J. Med. Virol. 2022, 94, 3506–3508. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Lan, W.; Wu, X.; Zhao, T.; Duan, B.; Yang, P.; Ren, Y.; Quan, L.; Zhao, W.; Seto, D.; et al. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct. Target. Ther. 2020, 7, 138. [Google Scholar] [CrossRef]
- Burel, E.; Colson, P.; Lagier, J.C.; Levasseur, A.; Bedotto, M.; Lavrard-Meyer, P.; Fournier, P.E.; La Scola, B.; Raoult, D. Sequential Appearance and Isolation of a SARS-CoV-2 Recombinant between Two Major SARS-CoV-2 Variants in a Chronically Infected Immunocompromised Patient. Viruses 2022, 14, 1266. [Google Scholar] [CrossRef] [PubMed]
- Escalera-Zamudio, M.; Castelán-Sánchez, H.G.; Delaye-Arredondo, L.J.; Gutiérrez, B. RE: Proposal to Redesignate B.1.631 as Recombinant Lineage XB. Virological. Available online: https://virological.org/t/re-proposal-to-redesignate-b-1-631-as-recombinant-lineage-xb/746 (accessed on 24 November 2022).
- Lindh, E.; Smura, T.; Blomqvist, S.; Liitsola, K.; Vauhkonen, H.; Savolainen, L.; Ikonen, J.; Ronkainen, J.; Taskila, J.; Taskila, T.; et al. Genomic and Epidemiological Report of the Recombinant XJ Lineage SARS-CoV-2 Variant, Detected in Northern Finland, January 2022. Eurosurveillance 2022, 27, 2200257. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Ng, D.Y.M.; Liu, G.Y.Z.; Cheng, S.S.M.; Krishnan, P.; Chang, L.D.J.; Cheuk, S.S.Y.; Hui, M.M.Y.; Lam, T.T.Y.; Peiris, M.; et al. Recombinant BA.1/BA.2 SARS-CoV-2 Virus in Arriving Travelers, Hong Kong, February 2022. Emerg. Infect. Dis. 2022, 28, 1276–1278. [Google Scholar] [CrossRef]
- Liu, X.; Xiong, J.; Sun, Z.; Hu, J.; Thilakavathy, K.; Chen, M.; Zhao, Q.; Feng, Y.; Jiang, Q.; Xiong, C. Omicron: A chimera of two early SARS-CoV-2 lineages. Signal Transduct. Target. Ther. 2022, 7, 90. [Google Scholar] [CrossRef]
- Turkahia, Y.; Thornlow, B.; Hinrichs, A.; McBroome, J.; Ayala, N.; Ye, C.; Maio, N.D.; Haussler, D.; Lanfear, R.; Corbett-Detig, R. Pandemic-Scale Phylogenomics Reveals Elevated Recombination Rates in the SARS-CoV-2 Spike Region. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gutierrez, B.; Castelán Sánchez, H.G.; da Silva Candido, D.; Jackson, B.; Fleishon, S.; Houzet, R.; Ruis, C.; Delaye, L.; Faria, N.R.; Rambaut, A.; et al. Emergence and Widespread Circulation of a Recombinant SARS-CoV-2 Lineage in North America. Cell Host Microbe 2022, 30, 1112–1123. [Google Scholar] [CrossRef]
- Ignatieva, A.; Hein, J.; Jenkins, P.A. Ongoing Recombination in SARS-CoV-2 Revealed through Genealogical Reconstruction. Mol. Biol. Evol. 2022, 39, msac028. [Google Scholar] [CrossRef] [PubMed]
- Varabyou, A.; Pockrandt, C.; Salzberg, S.L.; Pertea, M. Rapid Detection of Inter-Clade Recombination in SARS-CoV-2 with Bolotie. Genetics 2021, 218, iyab074. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef] [PubMed]
- Taghizadeh, P.; Salehi, S.; Heshmati, A.; Houshmand, S.M.; InanlooRahatloo, K.; Mahjoubi, F.; Sanati, M.H.; Yari, H.; Alavi, A.; Jamehdar, S.A.; et al. Study on SARS-CoV-2 Strains in Iran Reveals Potential Contribution of Co-Infection with and Recombination between Different Strains to the Emergence of New Strains. Virology 2021, 562, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Bukin, Y.S.; Bondaryuk, A.N.; Kulakova, N.V.; Balakhonov, S.V.; Dzhioev, Y.P.; Zlobin, V.I. Phylogenetic Reconstruction of the Initial Stages of the Spread of the SARS-CoV-2 Virus in the Eurasian and American Continents by Analyzing Genomic Data. Virus Res. 2021, 305, 198551. [Google Scholar] [CrossRef]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- Engelbrecht, S.; Delaney, K.; Kleinhans, B.; Wilkinson, E.; Tegally, H.; Stander, T.; van Zyl, G.; Preiser, W.; de Oliveira, T. Multiple Early Introductions of SARS-CoV-2 to Cape Town, South Africa. Viruses 2021, 13, 526. [Google Scholar] [CrossRef]
- Ghafari, M.; du Plessis, L.; Raghwani, J.; Bhatt, S.; Xu, B.; Pybus, O.G.; Katzourakis, A. Purifying Selection Determines the Short-Term Time Dependency of Evolutionary Rates in SARS-CoV-2 and PH1N1 Influenza. Mol. Biol. Evol. 2022, 39, msac009. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Acciarri, C.; Galli, M.; Zehender, G. Early Phylogenetic Estimate of the Effective Reproduction Number of SARS-CoV-2. J. Med. Virol. 2020, 92, 675–679. [Google Scholar] [CrossRef] [Green Version]
- Pekar, J.; Worobey, M.; Moshiri, N.; Scheffler, K.; Wertheim, J.O. Timing the SARS-CoV-2 Index Case in Hubei Province. Science 2021, 372, 412–417. [Google Scholar] [CrossRef]
- Pipes, L.; Wang, H.; Huelsenbeck, J.P.; Nielsen, R. Assessing Uncertainty in the Rooting of the SARS-CoV-2 Phylogeny. Mol. Biol. Evol. 2021, 38, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Sallam, M.; Ababneh, N.A.; Dababseh, D.; Bakri, F.G.; Mahafzah, A. Temporal Increase in D614G Mutation of SARS-CoV-2 in the Middle East and North Africa. Heliyon 2021, 7, e06035. [Google Scholar] [CrossRef] [PubMed]
- Tay, J.H.; Porter, A.F.; Wirth, W.; Duchene, S. The Emergence of SARS-CoV-2 Variants of Concern Is Driven by Acceleration of the Substitution Rate. Mol. Biol. Evol. 2022, 39, msac013. [Google Scholar] [CrossRef] [PubMed]
- Bromham, L.; Penny, D. The Modern Molecular Clock. Nat. Rev. Genet. 2003, 4, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Del Amparo, R.; Branco, C.; Arenas, J.; Vicens, A.; Arenas, M. Analysis of Selection in Protein-Coding Sequences Accounting for Common Biases. Brief. Bioinform. 2021, 22, bbaa431. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhao, S.; Liu, Q.; Zhang, X.; Sha, T.; Su, Y.; Zhao, W.; Bao, Y.; Xue, Y.; Chen, H. Ongoing Positive Selection Drives the Evolution of SARS-CoV-2 Genomes. Genom. Proteom. Bioinform. 2022, in press. [CrossRef]
- Rochman, N.D.; Wolf, Y.I.; Faure, G.; Mutz, P.; Zhang, F.; Koonin, E.V. Ongoing Global and Regional Adaptive Evolution of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2104241118. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M.V. Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic. Front. Microbiol. 2020, 11, 550674. [Google Scholar] [CrossRef]
- Koçhan, N.; Eskier, D.; Suner, A.; Karakülah, G.; Oktay, Y. Different Selection Dynamics of S and RdRp between SARS-CoV-2 Genomes with and without the Dominant Mutations. Infect. Genet. Evol. 2021, 91, 104796. [Google Scholar] [CrossRef]
- Dearlove, B.; Lewitus, E.; Bai, H.; Li, Y.; Reeves, D.B.; Joyce, M.G.; Scott, P.T.; Amare, M.F.; Vasan, S.; Michael, N.L.; et al. A SARS-CoV-2 Vaccine Candidate Would Likely Match All Currently Circulating Variants. Proc. Natl. Acad. Sci. USA 2020, 117, 23652–23662. [Google Scholar] [CrossRef]
- Kumar, R.; Verma, H.; Singhvi, N.; Sood, U.; Gupta, V.; Singh, M.; Kumari, R.; Hira, P.; Nagar, S.; Talwar, C.; et al. Comparative Genomic Analysis of Rapidly Evolving SARS-CoV-2 Reveals Mosaic Pattern of Phylogeographical Distribution. mSystems 2020, 5, e00505-20. [Google Scholar] [CrossRef] [PubMed]
- Chiara, M.; Horner, D.S.; Gissi, C.; Pesole, G. Comparative Genomics Suggests Limited Variability and Similar Evolutionary Patterns between Major Clades of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; Cesare, M.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. Within-Host Genomics of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lo Presti, A.; Rezza, G.; Stefanelli, P. Selective Pressure on SARS-CoV-2 Protein Coding Genes and Glycosylation Site Prediction. Heliyon 2020, 6, e05001. [Google Scholar] [CrossRef] [PubMed]
- Berrio, A.; Gartner, V.; Wray, G.A. Positive Selection within the Genomes of SARS-CoV-2 and Other Coronaviruses Independent of Impact on Protein Function. PeerJ 2020, 8, e10234. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; de Oliveira, T.; Vermeulen, M.; van der Berg, K.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar] [CrossRef]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The Emergence and Ongoing Convergent Evolution of the SARS-CoV-2 N501Y Lineages. Cell 2021, 184, 5189–5200. [Google Scholar] [CrossRef]
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-terminal Domain Antigenic Mapping Reveals a Site of Vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347. [Google Scholar] [CrossRef]
- Upadhyay, V.; Patrick, C.; Lucas, A.; Mallela, K.M.G. Convergent Evolution of Multiple Mutations Improves the Viral Fitness of SARS-CoV-2 Variants by Balancing Positive and Negative Selection. Biochemistry 2022, 61, 963–980. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody Resistance of SARS-CoV-2 Variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Schmidt, F.; Weisblum, Y.; Muecksch, F.; Barnes, C.O.; Finkin, S.; Schaefer-Babajew, D.; Cipolla, M.; Gaebler, C.; Lieberman, J.A.; et al. MRNA Vaccine-Elicited Antibodies to SARS-CoV-2 and Circulating Variants. Nature 2021, 592, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Lubinski, B.; Fernandes, M.H.; Frazier, L.; Tang, T.; Daniel, S.; Diel, D.G.; Jaimes, J.A.; Whittaker, G.R. Functional evaluation of the P681H mutation on the proteolytic activation of the SARS-CoV-2 variant B.1.1.7 (Alpha) spike. Iscience 2022, 25, 103589. [Google Scholar] [CrossRef] [PubMed]
- Marques-Pereira, C.; Pires, M.N.; Gouveia, R.P.; Pereira, N.N.; Caniceiro, A.B.; Rosário-Ferreira, N.; Moreira, I.S. SARS-CoV-2 Membrane Protein: From Genomic Data to Structural New Insights. Int. J. Mol. Sci. 2022, 23, 2986. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tian, C.; Liu, P.; Guo, D.; Zheng, W.; Huang, X.; Zhang, Y.; Liu, L. Effects of SARS-CoV-2 Mutations on Protein Structures and Intraviral Protein-Protein Interactions. J. Med. Virol. 2021, 93, 2132–2140. [Google Scholar] [CrossRef] [PubMed]
- Omotoso, O.E. Contributory Role of SARS-CoV-2 Genomic Variations and Life Expectancy in COVID-19 Transmission and Low Fatality Rate in Africa. Egypt. J. Med. Hum. Genet. 2020, 21, 72. [Google Scholar] [CrossRef]
- Morales, A.C.; Rice, A.M.; Ho, A.T.; Mordstein, C.; Mühlhausen, S.; Watson, S.; Cano, L.; Young, B.; Kudla, G.; Hurst, L.D. Causes and Consequences of Purifying Selection on SARS-CoV-2. Genome Biol. Evol. 2021, 13, evab196. [Google Scholar] [CrossRef]
- Azkur, A.K.; Akdis, M.; Azkur, D.; Sokolowska, M.; van de Veen, W.; Brüggen, M.C.; O’Mahoni, L.; Gao, Y.; Nadeau, K.; Akdis, C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy 2020, 75, 1564–1581. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; da Silva Filipe, A.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete mapping of mutations to the SARS-CoV-2 spike receptor-binding domain that escape antibody recognition. Cell Host Microbe 2021, 29, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive Mapping of Mutations in the SARS-CoV-2 Receptor-Binding Domain That Affect Recognition by Polyclonal Human Plasma Antibodies. Cell Host Microbe 2021, 29, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective Mapping of Viral Mutations That Escape Antibodies Used to Treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 Variants B.1.351 and P.1 Escape from Neutralizing Antibodies. Cell 2021, 184, 2384–2393. [Google Scholar] [CrossRef]
- Mahase, E. Covid-19: Novavax Vaccine Efficacy Is 86% against UK Variant and 60% against South African Variant. BMJ 2021, 372, n296. [Google Scholar] [CrossRef]
- Tada, T.; Zhou, H.; Samanovic, M.I.; Dcosta, B.M.; Cornelius, A.; Herati, R.S.; Mulligan, M.J.; Landau, N.R. Neutralization of SARS-CoV-2 Variants by mRNA and Adenoviral Vector Vaccine-Elicited Antibodies. Front. Immunol. 2022, 13, 797589. [Google Scholar] [CrossRef]
- Tada, T.; Zhou, H.; Dcosta, B.M.; Samanovic, M.I.; Chivukula, V.; Herati, R.S.; Hubbard, S.R.; Mulligan, M.J.; Landau, N.R. Increased resistance of SARS-CoV-2 Omicron variant to neutralization by vaccine-elicited and therapeutic antibodies. EBioMedicine 2022, 78, 103944. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F.; McConnell, S.; Casadevall, A. Very Low Levels of Remdesivir Resistance in SARS-COV-2 Genomes after 18 Months of Massive Usage during the COVID19 Pandemic: A GISAID Exploratory Analysis. Antivir. Res. 2022, 198, 105247. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus Susceptibility to the Antiviral Remdesivir (GS-5734) Is Mediated by the Viral Polymerase and the Proofreading Exoribonuclease. mBio 2018, 9, e00221-18. [Google Scholar] [CrossRef] [Green Version]
- Szemiel, A.M.; Merits, A.; Orton, R.J.; MacLean, O.A.; Pinto, R.M.; Wickenhagen, A.; Lieber, G.; Turnbull, M.L.; Wang, S.; Furnon, W.; et al. In Vitro Selection of Remdesivir Resistance Suggests Evolutionary Predictability of SARS-CoV-2. PLoS Pathog. 2021, 17, e1009929. [Google Scholar] [CrossRef]
- Gandhi, S.; Klein, J.; Robertson, A.J.; Peña-Hernández, M.A.; Lin, M.J.; Roychoudhury, P.; Lu, P.; Fournier, J.; Ferguson, D.; Mohamed Bakhash, S.A.K.; et al. De Novo Emergence of a Remdesivir Resistance Mutation during Treatment of Persistent SARS-CoV-2 Infection in an Immunocompromised Patient: A Case Report. Nat. Commun. 2022, 13, 1547. [Google Scholar] [CrossRef]
- Huang, Y.Z.; Kuan, C.C. Vaccination to reduce severe COVID-19 and mortality in COVID-19 patients: A systematic review and meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2022, 26, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Watson, O.J.; Barnsley, G.; Toor, J.; Hogan, A.B.; Winskill, P.; Ghani, A.C. Global impact of the first year of COVID-19 vaccination: A mathematical modelling study. Lancet Infect. Dis. 2022, 22, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.R.; Galvani, A.P. The global impact of disproportionate vaccination coverage on COVID-19 mortality. Lancet Infect. Dis. 2022, 22, 1254–1255. [Google Scholar] [CrossRef] [PubMed]
- Morel, B.; Barbera, P.; Czech, L.; Bettisworth, B.; Hübner, L.; Lutteropp, S.; Serdari, D.; Kostaki, E.G.; Mamais, I.; Kozlov, A.M.; et al. Phylogenetic Analysis of SARS-CoV-2 Data Is Difficult. Mol. Biol. Evol. 2021, 38, 1777–1791. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Illustration of the SARS-CoV-2 genome structure. Scaled representation of genes along the SARS-CoV-2 genome, including a table with the nucleotide positions between each ORF (Open Reading Frame) and the corresponding encoded proteins according to https://www.ncbi.nlm.nih.gov/nuccore/1798174254 (accessed on 20 November 2022). Structural protein-coding genes are shown in dark blue.
Figure 1.
Illustration of the SARS-CoV-2 genome structure. Scaled representation of genes along the SARS-CoV-2 genome, including a table with the nucleotide positions between each ORF (Open Reading Frame) and the corresponding encoded proteins according to https://www.ncbi.nlm.nih.gov/nuccore/1798174254 (accessed on 20 November 2022). Structural protein-coding genes are shown in dark blue.

Figure 2.
Evolutionary history of main SARS-CoV-2 VOCs. This illustration shows the emergence, establishment, and genetic relationships of the main VOCs according to current knowledge. It also includes a graph describing the predominance of every VOC over time, which we obtained with Nextstrain based on data from GISAID [27,28].
Figure 2.
Evolutionary history of main SARS-CoV-2 VOCs. This illustration shows the emergence, establishment, and genetic relationships of the main VOCs according to current knowledge. It also includes a graph describing the predominance of every VOC over time, which we obtained with Nextstrain based on data from GISAID [27,28].


{kind=link}
{kind=link}
Table 1.
Mutations for each variant of concern presented by WHO to date (excluding Omicron, shown in Table 2). The presented main amino acid (Aa) and deletion (Del) mutations include their position in the corresponding protein or the entire genome, respectively [40]. Mutations present in different VOCs (including Omicron, Table 2), which constitute parallel mutations, are shown in italics.
Table 1.
Mutations for each variant of concern presented by WHO to date (excluding Omicron, shown in Table 2). The presented main amino acid (Aa) and deletion (Del) mutations include their position in the corresponding protein or the entire genome, respectively [40]. Mutations present in different VOCs (including Omicron, Table 2), which constitute parallel mutations, are shown in italics.
| VOC (PANGO Lineage) | Detected Mutations that Define VOCs (Except Omicron, Table 2) at Each Gene | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ORF1a | ORF1b | S | ORF3a | E | M | ORF6 | ORF7a | ORF7b | ORF8 | N | |
| Alpha (B.1.1.7) | Aa: T1001I A1708D I2230T Del: 3675–3677 | Aa: P314L | Aa: P9L V483F E484K F486I Q498R N501Y A570D D614G P681H T716I S982A D1118H Del: H69 V70 | Aa: Q27* R52I Y73C S84L | Aa: D3L R203K G204R S235F | ||||||
| Beta (B.1.351) | Aa: T265I K1655N K3353R Del: 3675–3677 | Aa: P314L | Aa: S13I D80A D215G, K417N E484K N501Y D614G A701V A879S Del: L242 A243 L244 | Aa: Q57H S171L | Aa: P71L | Aa: S84L | Aa: T205I | ||||
| Gamma (P.1) | Aa: S1188L K1795Q Del: 3675–3677 | Aa: P314L E1264D | Aa: L18F T20N P26S R78M D138Y R190S K417T E484K N501Y D614G H655Y T1027I V1176F | Aa: S253P | Aa: S84L E92K | Aa: P80R R203K G204R | |||||
| Delta (B.617.2) | Aa: P314L G662S P1000L | Aa: T19R T95I G142D E156G L452R T478K R567I D614G H655Y P681R D950N T1117I Del: F157 R158 | Aa: S26L | Aa: I82T | Aa: V82A, T120I | Aa: S84L Del: 119–120 | Aa: D63G R203M D377Y | ||||
Table 2.
Mutations for each lineage of the Omicron variant of concern. The presented main amino acid (Aa), nucleotide (Nt), insertion (Ins), and deletion (Del) mutations include their position in the corresponding protein or the entire genome, respectively [39,40,41]. Mutations present in different VOCs (including those shown in Table 1), which constitute parallel mutations, are shown in italics.
Table 2.
Mutations for each lineage of the Omicron variant of concern. The presented main amino acid (Aa), nucleotide (Nt), insertion (Ins), and deletion (Del) mutations include their position in the corresponding protein or the entire genome, respectively [39,40,41]. Mutations present in different VOCs (including those shown in Table 1), which constitute parallel mutations, are shown in italics.
| Omicron (PANGO Lineage) | Detected Mutations that Define Omicron Lineages at Each Gene | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ORF1a | ORF1b | S | ORF3a | E | M | ORF6 | ORF7b | ORF8 | N | |
| BA.1 | Aa: K856R L2084I A2710T T3255I P3395H I3758V Del: S2083 3674-3676 | Aa: P314L I1566V | Aa: A67V T95I G142D L212I G339D S371L S373P S375F K417N N440K G446S S477N T478K E484A Q493R G496S Q498R N501Y Y505H T547K D614G H655Y N679K P681H N764K D796Y N856K Q954H N969K L981F Del: 69–70 143–145 211 Ins: 214EPE | Aa: T9I | Aa: D3G Q19E A63T | Aa: S84L | Aa: P13L R203K G204R Del: 31–33 | |||
| BA.2 | Aa: S135R T842I G1307S L3027F T3090I L3201F T3255I P3395H Del: 3675–3677 | Aa: P314L R1315C I1566V T2163I | Aa: T19I L24S G142D V213G G339D S371F S373P S375F T376A D405N R408S K417N N440K S477N T478K E484A Q493R Q498R N501Y Y505H D614G H655Y N679K P681H N764K D796Y Q954H N969K Del: 25–27 | Aa: T223I | Aa: T9I | Aa: Q19E A63T | Aa: D61L | Aa: S84L | Aa: P13L R203K G204R S413R Del: 31–33 | |
| BA.3 | Aa: S153R G1307S T3090I T3255I P3395H A3657V Del: 3675–3677 | Aa: P314L I1566V | Aa: A67V T95I G142D L212I G339D S371F S373P S375F D405N K417N N440K G446S S477N T478K E484A Q493R Q498R N501Y Y505H D614G H655Y N679K P681H N764K D796Y Q954H N969K Del: 69–70 143–145 211 | Aa: T223I | Aa: T9I | Aa: Q19E A63T | Aa: S84L | Aa: P13L R203K G204R S413R Del: 31–33 | ||
| BA.4 | Aa: S135R T842I G1307S L3027F T3090I T3255I P3395H Del: 141–143 3675–3677 Nt: G12160A | Aa: P314L R1315C I1566V T2163I | Aa: T19I L24S G142D V213G G339D S371F S373P S375F T376A D405N R408S K417N N440K G446S L452R S477N T478K E484A F486V Q498R N501Y Y505H T547K D614G H655Y N679K P681H N764K D796Y Q954H N969K Del: 25–27 69–70 | Aa: T223I | Aa: T9I | Aa: Q19E A63T | Aa: D61L | Aa: L11F | Aa: P13L P151S R203K G204R S413R Del: 31–33 | |
| BA.5 | Aa: S135R T842I G1307S L1507F L3027F T3090I T3255I P3395H Del: 3675–3677 Nt: G12160A | Aa: P314L R1315C I1566V T2163I | Same mutations as BA.4 | Aa: T223I | Aa: T9I | Aa: D3N Q19E A63T | Aa: P13L R203K G204R S413R Del: 31–33 | |||
Table 3.
Parallel mutations observed among variants of concern. The table indicates the mutations that are observed in different variants of concern, with their main corresponding consequences when documented (references).
Table 3.
Parallel mutations observed among variants of concern. The table indicates the mutations that are observed in different variants of concern, with their main corresponding consequences when documented (references).
| Gene | Mutation (s) | Consequences | References |
|---|---|---|---|
| ORF1a | Del: 3675–3677 | Deletion located in the protein nsp6, which is important for the synthesis of RNA. This deletion removes amino acids from a transmembrane loop A very similar deletion was observed in the VOC Omicron BA.1 (deletion 3674–3676), which was associated with a favoring effect about increasing mutability | [42,43] |
| ORF1b | P314L | This mutation is in linkage disequilibrium with the D614G mutation of the S gene | [44] |
| S | E484K | This mutation induces escape to monoclonal antibodies and reduces the neutralizing capacity of convalescent and post-vaccination polyclonal sera | [45] |
| Q498R | This mutation increases the affinity between SARS-CoV-2 RBD and human ACE2 | [46] | |
| N501Y | This mutation increases the affinity between SARS-CoV-2 RBD and human ACE2 | [47] | |
| D614G | This mutation increases infectivity by changing the formation of the spike protein to a competent state for binding with ACE2 | [48] | |
| P681H | This mutation confers resistance to type I interferons and reduces dependence on endosomal cathepsins favoring cell entry | [49] | |
| K417N | This mutation reduces the activity of human and commercial antibodies but also reduces the affinity of the spike protein with human ACE2 | [50] | |
| H655Y | This mutation increases the fusogenicity with human cell membrane by using cathepsin-mediated entry and reduces the entry using the serine transmembrane protease 2 | [51,52] | |
| T95I | - | - | |
| G142D | Mutation associated with immune evasion, back mutations, and increased viral load | [53] | |
| L452R | This mutation increases the infectivity and fusogenicity and promotes viral replication | [54] | |
| T478K | This mutation increases the electrostatic potential of the spike protein at human ACE2 binding and can also play a role in immune escape | [55] | |
| ORF8 | S84L | - | - |
| N | R203K, G204R | These mutations increase infectivity, fitness and virulence | [56] |
Table 4.
Main detected recombination events in SARS-CoV-2 as a function of the variants involved. The table shows the main published recombination events (each line in the second column) involving VOCs and non-VOCs (first column). Note that recombination between some variants was not detected so far (i.e., between Alpha and Delta). The breakpoint interval is shown in nucleotides.
Table 4.
Main detected recombination events in SARS-CoV-2 as a function of the variants involved. The table shows the main published recombination events (each line in the second column) involving VOCs and non-VOCs (first column). Note that recombination between some variants was not detected so far (i.e., between Alpha and Delta). The breakpoint interval is shown in nucleotides.
| Variants Involved | Breakpoint Interval/Gene or Genes Involved | Additional Information | References |
|---|---|---|---|
| Alpha (VOC) and Epsilon (VOI) | Inside the genes S, N, and ORF8 [88] | - | [88] |
| Delta (VOC) and Omicron (VOC) | 19220–21618/ORF1ab and S [89] 22034–22194/S [89,90,91] 22035–22577/S [84] 22204–22578/S [92] 22218–22586/S [89] 25469–25584/ORF3a [90,91] | Recombinant named “Deltacron” or “Deltamicron”. The concern about this chimera lies in the combination of the high transmissibility of Omicron and the virulence and severe disease caused by Delta [90,93] | [84,89,90,91,92,93,94,95,96,97,98] |
| Alpha (VOC) and B.1.160 | 17109–18877/ORF1ab [90,91] 25710–27972/ORF3a, E, M, ORF6, ORF7a, ORF7b [90,91] | Recombination occurred among parents of these variants | [90,91,99] |
| Various lineages and Alpha (VOC) | 21255–21764/ORF1ab, S [83] 6528–6954/ORF1ab [83] 24914–28651/S, ORF3a, E, M, ORF6, ORF7a, ORF7b, ORF8, N [83] 21575–23063/S [83] 11396–21991/ORF1ab, S [83] Window nt 3267–5388 gene ORF1ab [83] 12534–21765/ORF1ab, S [83] 26801–27972/M, ORF6, ORF7a, ORF7b, ORF8 [83] 6954–10870/ORF1ab [83] 22775–22778/S [100] | - | [83,100] |
| Various lineages and Omicron (VOC) | 13296–15240/ORF1ab [101] 20055–21618/ORF1ab, S [102] | - | [101,102] |
| Omicron parents | 21593–23118/S [103] | - | [98,103] |
| Not involving VOCs | Inside the gene S [104] 22775–22778/S [105] 19408–19411/ORF1ab [105] | - | [76,104,105,106,107,108,109] |
Table 5.
The rate of molecular evolution of SARS-CoV-2 identified in diverse studies. The rates of evolution are shown in substitutions per site and per year and include the statistical confidence in terms of confidence interval (CI), highest posterior density interval (HDPI), or Bayesian confidence interval (BCI) at 95% significance of the estimation.
Table 5.
The rate of molecular evolution of SARS-CoV-2 identified in diverse studies. The rates of evolution are shown in substitutions per site and per year and include the statistical confidence in terms of confidence interval (CI), highest posterior density interval (HDPI), or Bayesian confidence interval (BCI) at 95% significance of the estimation.
| Rate of Evolution (Substitutions/Site/Year) | Statistical Confidence (95%) | References |
|---|---|---|
| 7.31 × 10−4 | 5.95 × 10−4–8.68 × 10−4 (CI) | [11,110] |
| 1.1 × 10−3 | 7.03 × 10−4–1.5 × 10−3 (CI) | [111] |
| 8 × 10−4 | – | [112] |
| 1 × 10−4–1.4 × 10−3 | – | [113] |
| 7.8 × 10−4 | 1.1 × 10−4–1.5 × 10−3 (HPDI) | [114] |
| 9.9 × 10−4 | 6.29 × 10−4–1.35 × 10−3 (BCI) | [81] |
| 7.9 × 10−4 | 6.64 × 10−4–9.27 × 10−4 (HPDI) | [115] |
| 3.547 × 10−4 | 1.112 × 10−4–5.969 × 10−4 (CI) | [116] |
| 6.5 × 10−3 | 4.9 × 10−3–8.0 × 10−3 (HPDI) | [117] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
González-Vázquez, L.D.; Arenas, M. Molecular Evolution of SARS-CoV-2 during the COVID-19 Pandemic. Genes 2023, 14, 407. https://doi.org/10.3390/genes14020407
AMA Style
González-Vázquez LD, Arenas M. Molecular Evolution of SARS-CoV-2 during the COVID-19 Pandemic. Genes. 2023; 14(2):407. https://doi.org/10.3390/genes14020407
Chicago/Turabian StyleGonzález-Vázquez, Luis Daniel, and Miguel Arenas. 2023. "Molecular Evolution of SARS-CoV-2 during the COVID-19 Pandemic" Genes 14, no. 2: 407. https://doi.org/10.3390/genes14020407
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.