
Human Superantibodies to 3CLpro Inhibit Replication of SARS-CoV-2 across Variants

 ,
,  ,
,  and
and
Abstract
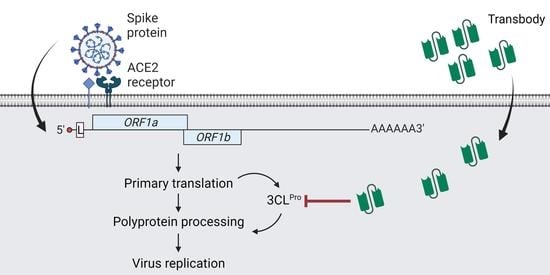
:
1. Introduction
2. Result
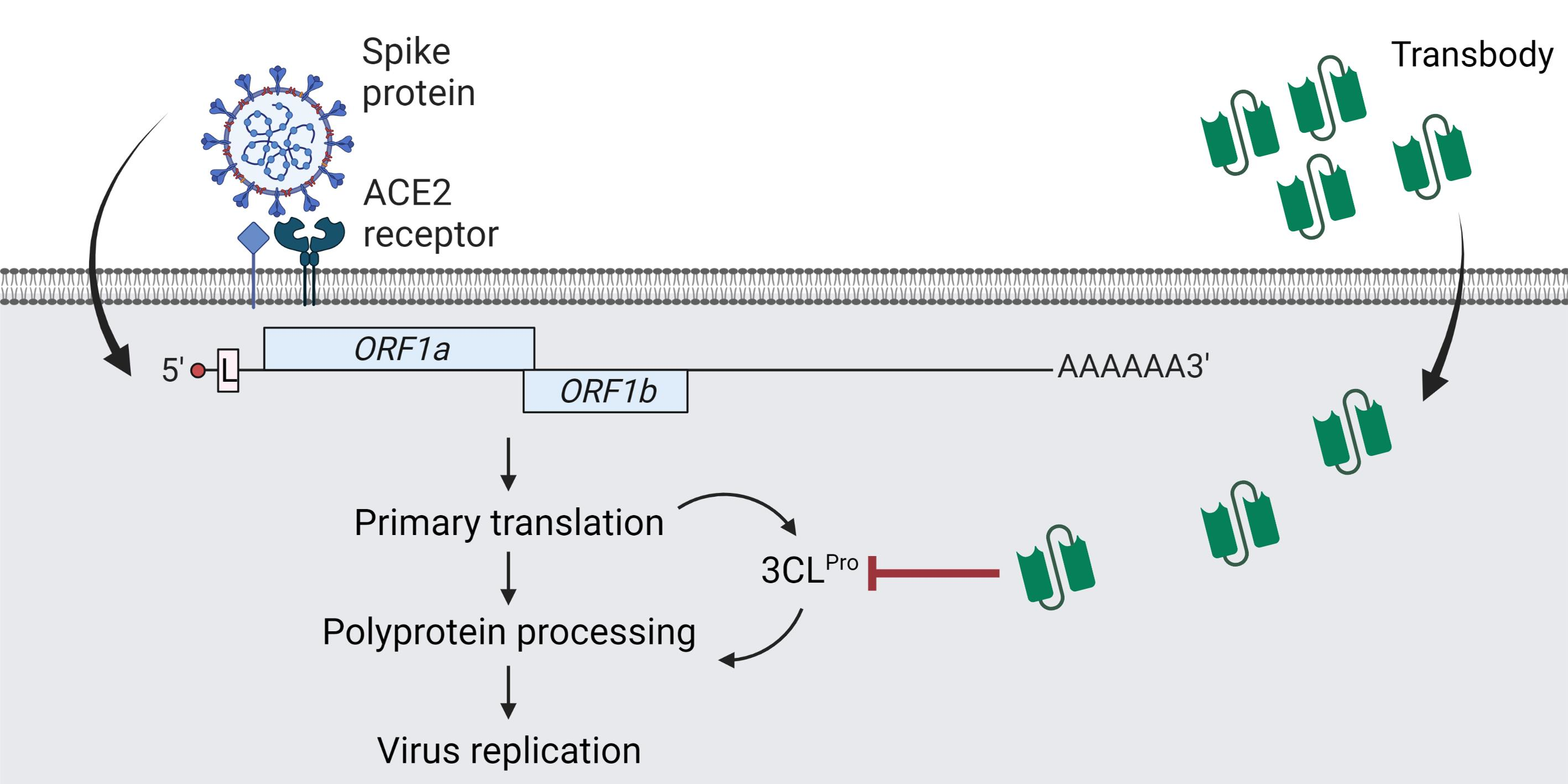
2.1. Production of Recombinant 3CLpro (r3CLpro) of SARS-CoV-2
2.2. Protease Activity of Recombinant 3CLpro Determined by Fluorescence Resonance Energy Transfer (FRET) Assay
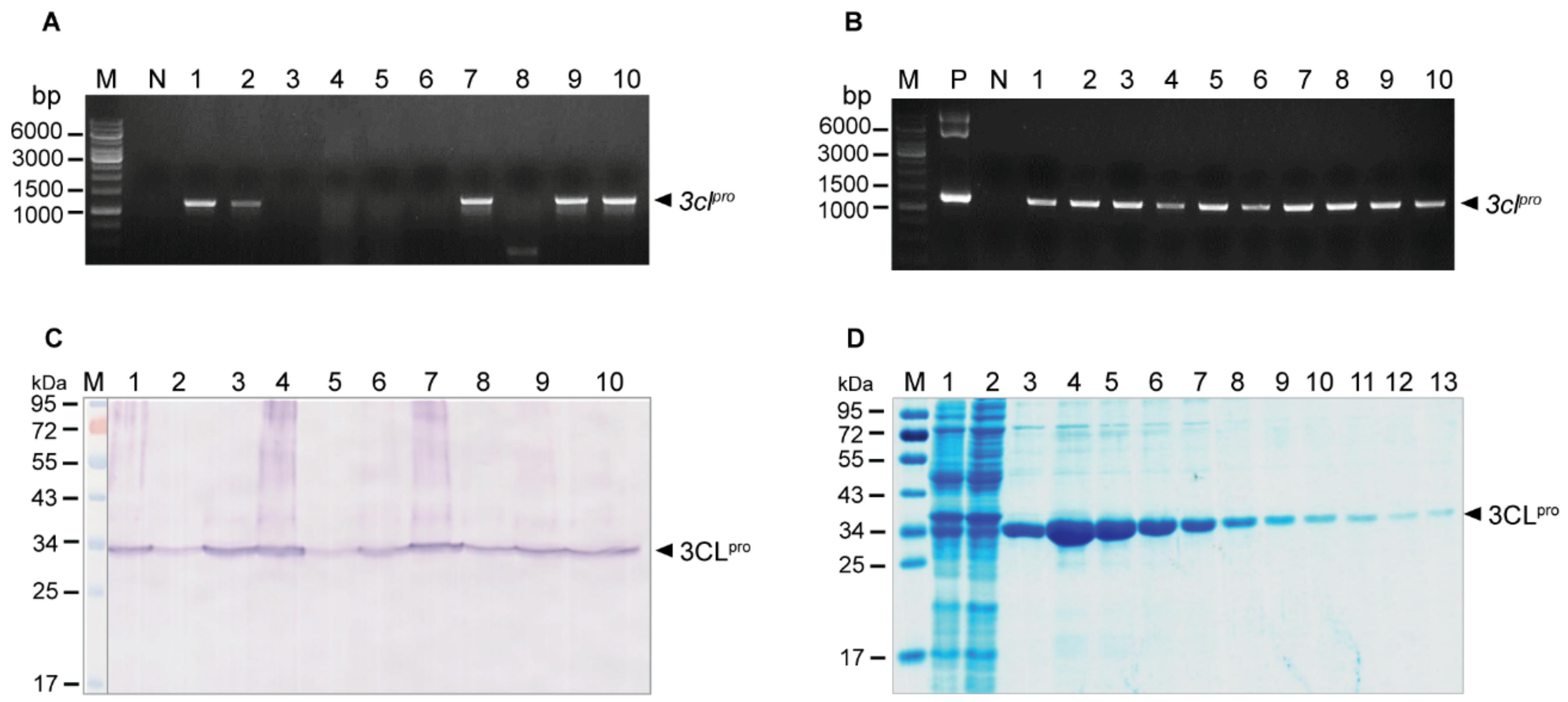
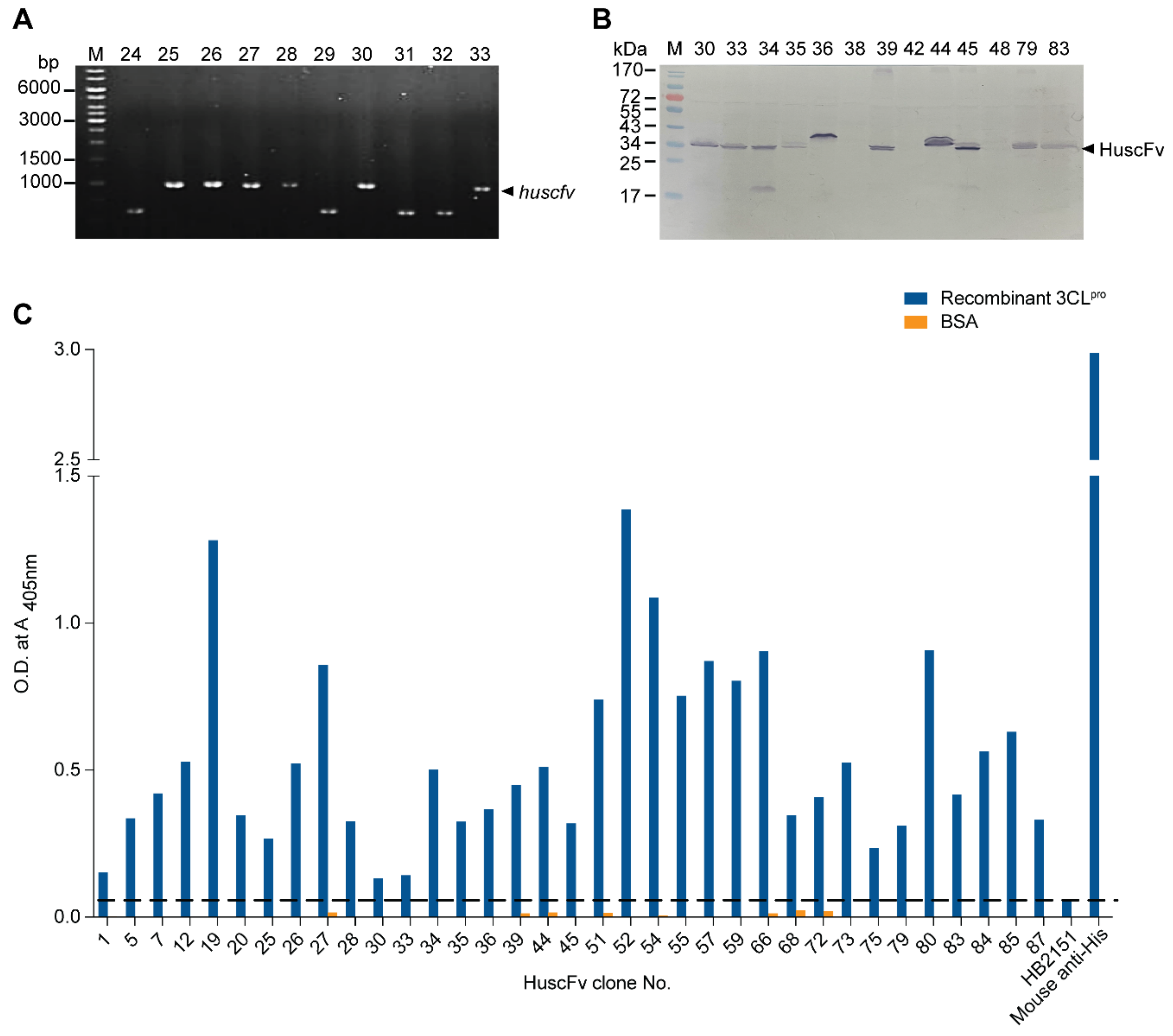
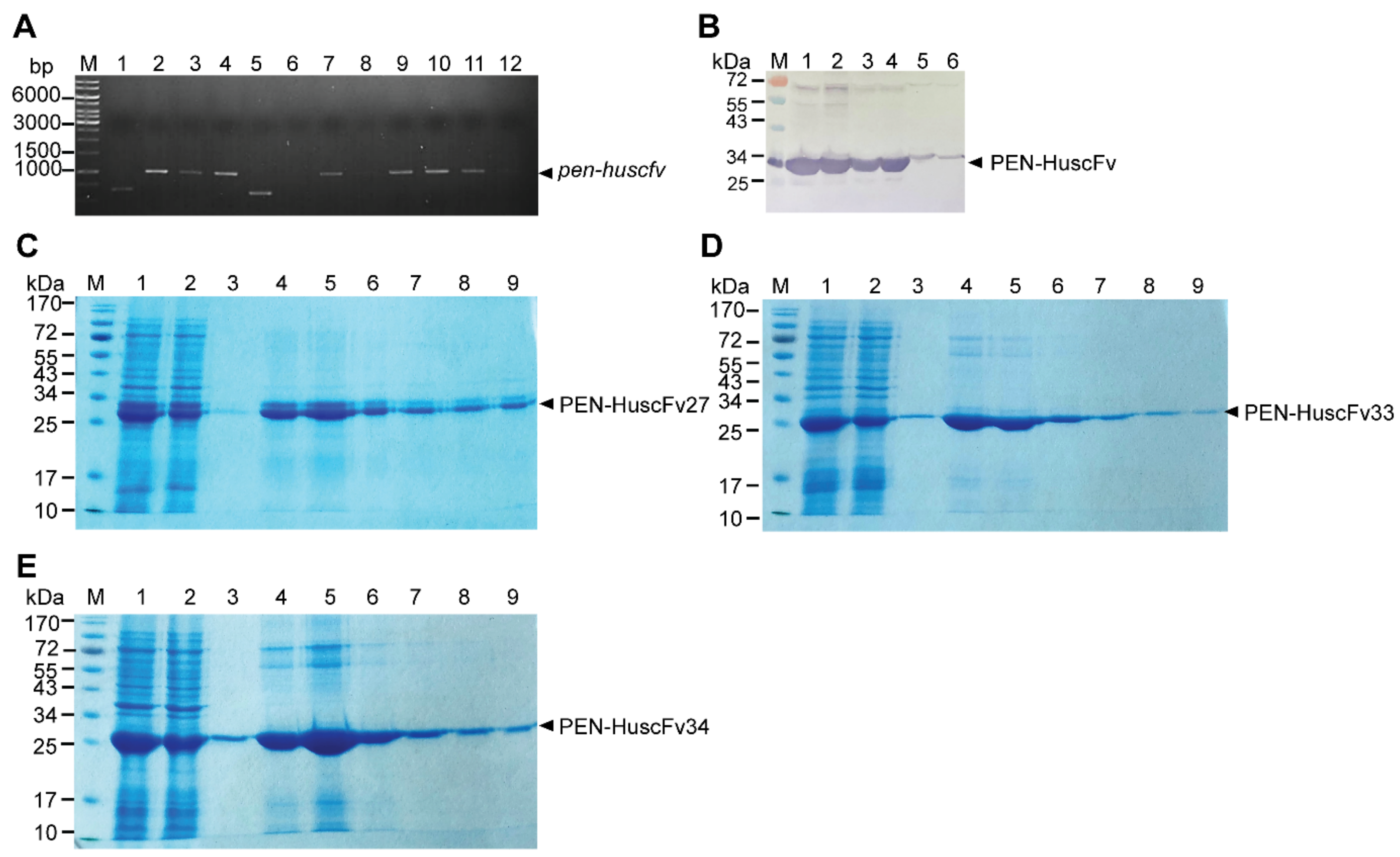
2.3. Human Single-Chain Antibodies (HuscFvs) to r3CLpro Prepared by Phage Display Technology
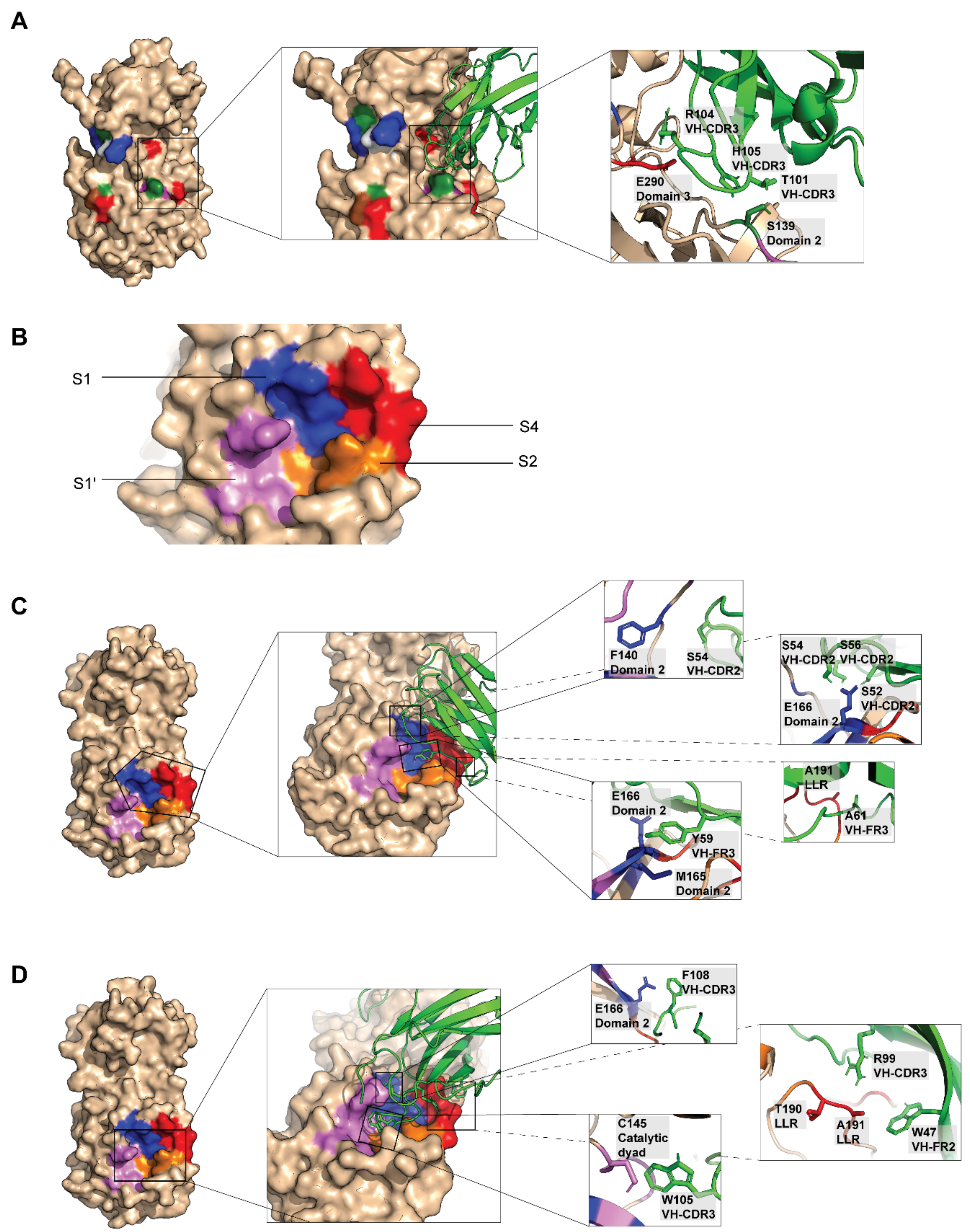
2.4. Computerized Simulation to Guide Selection of the HuscFv-Producing E. coli Clones
2.5. Cell-Penetrating Peptide (CPP)-Linked HuscFvs
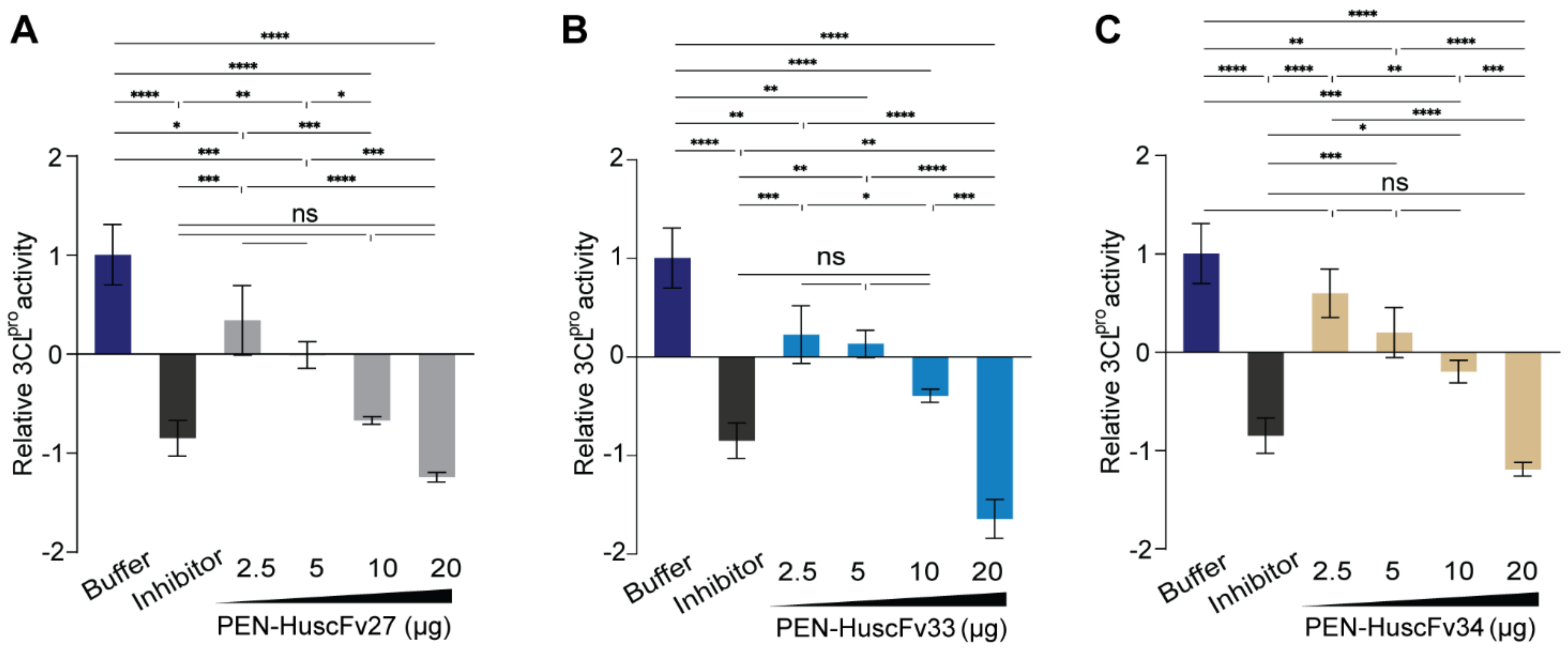
2.6. Inhibition of 3CLpro Enzymatic Activity by PEN-HuscFvs
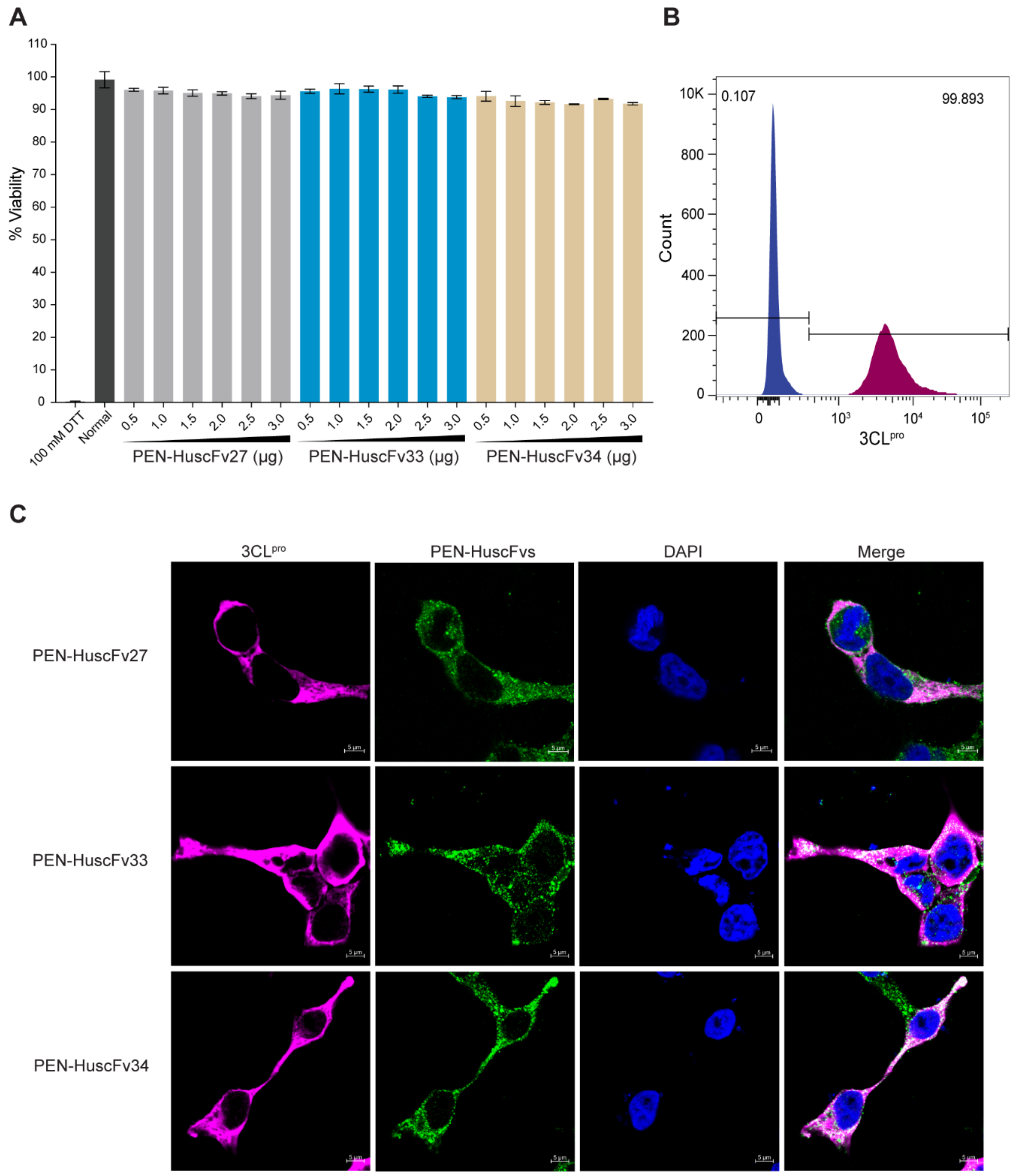
2.7. Biocompatibility of the PEN-HuscFvs to Human Cells
2.8. Mammalian Cells Expressing Intracellular 3CLpro
2.9. Cell-Penetrating Ability of the PEN-HuscFvs
2.10. Cell-Penetrable PEN-HuscFvs (Superantibodies) Inhibited Replication of SARS-CoV-2 Wild Type and Variants of Concern (VOC)
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cell Cultures and Virus Propagation
4.2. Production of Recombinant 3CLpro (r3CLpro)
4.3. Fluorescence Resonance Energy Transfer (FRET) Assay for Determining 3CLpro Enzymatic Activity and 3CLpro Inhibition Assay
4.4. HuscFv Phage Display Library
4.5. Production of HuscFvs to r3CLpro
4.6. Indirect Enzyme-Linked Immunosorbent Assay (Indirect ELISA)
4.7. Determination of Nucleotide and Amino Acid Sequences, Complementarity Determining Regions (CDRs) and Immunoglobulin Framework Regions (FRs) of the r3CLpro Bound HuscFvs
4.8. Computerized Homology Modeling and Intermolecular Docking for Determining Presumptive Residues and Domains of 3CLpro Bound by the HuscFvs
4.9. Production of Cell-Penetrable Antibodies (Penetratin Linked-HuscFvs, PEN-HuscFvs)
4.10. Preparation of Mammalian Cells That Expressed Intracellular r3CLpro
4.11. Biocompatibility of the PEN-HuscFvs to Human Cells
4.12. Determination of Cell-Penetrating Ability of the PEN-HuscFvs
4.13. HuscFv-Mediated Inhibition of the 3CLpro Catalytic Activity
4.14. HuscFv-Mediated Inhibition of the Authentic SARS-CoV-2 Replication
4.15. Quantitative RT-PCR
4.16. Plaque Forming Assay (PFA)
4.17. Focus-Forming Assay (FFA)
4.18. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, H.; Wei, L.; Niu, P. The novel coronavirus outbreak in Wuhan, China. Glob. Health Res. Policy 2020, 5, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): An update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Kyrou, I.; Randeva, H.S.; Spandidos, D.A.; Karteris, E. Not only ACE2-the quest for additional host cell mediators of SARS-CoV-2 infection: Neuropilin-1 (NRP1) as a novel SARS-CoV-2 host cell entry mediator implicated in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 21. [Google Scholar] [CrossRef]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J. Biol. Chem. 2021, 296, 100111. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, B.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004, 279, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Goyal, B.; Goyal, D. Targeting the dimerization of the main protease of coronaviruses: A potential broad-spectrum therapeutic strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef]
- Grottesi, A.; Besker, N.; Emerson, A.; Manelfi, C.; Beccari, A.R.; Frigerio, F.; Lindahl, E.; Cerchia, C.; Talarico, C. Computational studies of SARS-CoV-2 3CLpro: Insights from MD simulations. Int. J. Mol. Sci. 2020, 21, 5346. [Google Scholar] [CrossRef]
- Huang, C.; Wei, P.; Fan, K.; Liu, Y.; Lai, L. 3C-like proteinase from SARS coronavirus catalyzes substrate hydrolysis by a general base mechanism. Biochemistry 2004, 43, 4568–4574. [Google Scholar] [CrossRef]
- Shan, Y.F.; Li, S.F.; Xu, G.J. A novel auto-cleavage assay for studying mutational effects on the active site of severe acute respiratory syndrome coronavirus 3C-like protease. Biochem. Biophys. Res. Commun. 2004, 324, 579–583. [Google Scholar] [CrossRef]
- Hsu, M.F.; Kuo, C.J.; Chang, K.T.; Chang, H.C.; Chou, C.C.; Ko, T.P.; Shr, H.L.; Chang, G.G.; Wang, A.H.; Liang, P.H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005, 280, 31257–31266. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.; Schechter, I. Mapping the active site of papain with the aid of peptide substrates and inhibitors. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1970, 257, 249–264. [Google Scholar] [CrossRef]
- Kiemer, L.; Lund, O.; Brunak, S.; Blom, N. Coronavirus 3CLpro proteinase cleavage sites: Possible relevance to SARS virus pathology. BMC Bioinform. 2004, 5, 72. [Google Scholar] [CrossRef] [Green Version]
- Bacha, U.; Barrila, J.; Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry 2004, 43, 4906–4912. [Google Scholar] [CrossRef]
- Barrila, J.; Bacha, U.; Freire, E. Long-range cooperative interactions modulate dimerization in SARS 3CLpro. Biochemistry 2006, 45, 14908–14916. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, T.; Takemoto, C.; Kim, Y.T.; Wang, H.; Nishii, W.; Terada, T.; Shirouzu, M.; Yokoyama, S. SARS-CoV 3CL protease cleaves its C-terminal autoprocessing site by novel subsite cooperativity. Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [Google Scholar] [CrossRef] [Green Version]
- Novak, J.; Rimac, H.; Kandagalla, S.; Pathak, P.; Naumovich, V.; Grishina, M.; Potemkin, V. Proposition of a new allosteric binding site for potential SARS-CoV-2 3CL protease inhibitors by utilizing molecular dynamics simulations and ensemble docking. J. Biomol. Struct. Dyn. 2021, 39, 1–14. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Tan, K.P.; Wang, Y.M.; Lin, S.W.; Liang, P.H. Identification, synthesis and evaluation of SARS-CoV and MERS-CoV 3C-like protease inhibitors. Bioorg. Med. Chem. 2016, 24, 3035–3042. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, C.; Xin, L.; Ren, X.; Tian, L.; Ju, X.; Li, H.; Wang, Y.; Zhao, Q.; Liu, H.; et al. The development of coronavirus 3C-like protease (3CL(pro)) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711. [Google Scholar] [CrossRef]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- Needle, D.; Lountos, G.T.; Waugh, D.S. Structures of the Middle East respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: Peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Resnick, S.J.; Iketani, S.; Hong, S.J.; Zask, A.; Liu, H.; Kim, S.; Melore, S.; Lin, F.Y.; Nair, M.S.; Huang, Y.; et al. Inhibitors of coronavirus 3CL proteases protect cells from protease-mediated cytotoxicity. J. Virol. 2021, 95, e02374-20. [Google Scholar] [CrossRef]
- Tahir Ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.L. Structural basis of SARS-CoV-2 3CL(pro) and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Chuck, C.P.; Chong, L.T.; Chen, C.; Chow, H.F.; Wan, D.C.; Wong, K.B. Profiling of substrate specificity of SARS-CoV 3CL. PLoS ONE 2010, 5, e13197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkeaw, K.; Sakolvaree, Y.; Srimanote, P.; Tongtawe, P.; Maneewatch, S.; Sookrung, N.; Tungtrongchitr, A.; Tapchaisri, P.; Kurazono, H.; Chaicumpa, W. Human monoclonal ScFv neutralize lethal Thai cobra, Naja kaouthia, neurotoxin. J. Proteom. 2009, 72, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, J.; Hu, T.; Chen, K.; Jiang, H.; Shen, X. Residues on the dimer interface of SARS coronavirus 3C-like protease: Dimer stability characterization and enzyme catalytic activity analysis. J. Biochem. 2008, 143, 525–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallin, R.F. A Practical Guide to ISO 10993-5: Cytotoxicity. Available online: https://www.mddionline.com/testing/practical-guide-iso-10993-5-cytotoxicity (accessed on 3 January 2022).
- Liu, H.; Ye, F.; Sun, Q.; Liang, H.; Li, C.; Li, S.; Lu, R.; Huang, B.; Tan, W.; Lai, L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzyme Inhib. Med. Chem. 2021, 36, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Franco-Paredes, C. Transmissibility of SARS-CoV-2 among fully vaccinated individuals. Lancet Infect. Dis. 2022, 22, 183–195. [Google Scholar] [CrossRef]
- Chaisri, U.; Chaicumpa, W. Evolution of therapeutic antibodies, influenza virus biology, influenza, and influenza immunotherapy. Biomed. Res. Int. 2018, 2018, 9747549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudinski, M.R.; Coates, E.E.; Novik, L.; Widge, A.; Houser, K.V.; Burch, E.; Holman, L.A.; Gordon, I.J.; Chen, G.L.; Carter, C.; et al. Safety, tolerability, pharmacokinetics, and immunogenicity of the therapeutic monoclonal antibody mAb114 targeting Ebola virus glycoprotein (VRC 608): An open-label phase 1 study. Lancet 2019, 393, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.M.; Hwang, Y.C.; Liu, I.J.; Lee, C.C.; Tsai, H.Z.; Li, H.J.; Wu, H.C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]
- Edara, V.V.; Hudson, W.H.; Xie, X.; Ahmed, R.; Suthar, M.S. Neutralizing antibodies against SARS-CoV-2 variants after infection and vaccination. JAMA 2021, 325, 1896–1898. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, X.; Yang, Y.; Hotez, P.J.; Du, L. Neutralizing antibodies for the treatment of COVID-19. Nat. Biomed. Eng. 2020, 4, 1134–1139. [Google Scholar] [CrossRef]
- Sadarangani, M.; Marchant, A.; Kollmann, T.R. Immunological mechanisms of vaccine-induced protection against COVID-19 in humans. Nat. Rev. Immunol. 2021, 21, 475–484. [Google Scholar] [CrossRef]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. N. Engl. J. Med. 2021, 384, 238–251. [Google Scholar] [CrossRef]
- Bux, J. Transfusion-related acute lung injury (TRALI): A serious adverse event of blood transfusion. Vox Sang. 2005, 89, 1–10. [Google Scholar] [CrossRef]
- Semple, J.W.; Rebetz, J.; Kapur, R. Transfusion-associated circulatory overload and transfusion-related acute lung injury. Blood 2019, 133, 1840–1853. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. WHO Recommends against the Use of Convalescent Plasma to Treat COVID-19. Available online: https://www.who.int/news/item/07-12-2021-who-recommends-against-the-use-of-convalescent-plasma-to-treat-covid-19 (accessed on 30 March 2022).
- Hwang, Y.C.; Lu, R.M.; Su, S.C.; Chiang, P.Y.; Ko, S.H.; Ke, F.Y.; Liang, K.H.; Hsieh, T.Y.; Wu, H.C. Monoclonal antibodies for COVID-19 therapy and SARS-CoV-2 detection. J. Biomed. Sci. 2022, 29, 1. [Google Scholar] [CrossRef]
- Spitalieri, P.; Centofanti, F.; Murdocca, M.; Scioli, M.G.; Latini, A.; Di Cesare, S.; Citro, G.; Rossi, A.; Orlandi, A.; Miersch, S.; et al. Two Different Therapeutic Approaches for SARS-CoV-2 in hiPSCs-Derived Lung Organoids. Cells 2022, 11, 1235. [Google Scholar] [CrossRef]
- Miersch, S.; Sharma, N.; Saberianfar, R.; Chen, C.; Caccuri, F.; Zani, A.; Caruso, A.; Case, J.B.; Diamond, M.S.; Amarasinghe, G.K.; et al. Ultrapotent and broad neutralization of SARS-CoV-2 variants by modular, tetravalent, bi-paratopic antibodies. Cell Rep. 2022, 39, 110905. [Google Scholar] [CrossRef]
- Annane, D.; Heming, N.; Grimaldi-Bensouda, L.; Frémeaux-Bacchi, V.; Vigan, M.; Roux, A.L.; Marchal, A.; Michelon, H.; Rottman, M.; Moine, P. Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: A proof-of-concept study. EClinicalMedicine 2020, 28, 100590. [Google Scholar] [CrossRef]
- Bottazzi, M.E.; Strych, U.; Hotez, P.J.; Corry, D.B. Coronavirus vaccine-associated lung immunopathology-what is the significance? Microbes Infect. 2020, 22, 403–404. [Google Scholar] [CrossRef]
- Sanchez-Zuno, G.A.; Matuz-Flores, M.G.; Gonzalez-Estevez, G.; Nicoletti, F.; Turrubiates-Hernandez, F.J.; Mangano, K.; Munoz-Valle, J.F. A review: Antibody-dependent enhancement in COVID-19: The not so friendly side of antibodies. Int. J. Immunopathol. Pharmacol. 2021, 35, 20587384211050199. [Google Scholar] [CrossRef]
- Narayan, R.; Tripathi, S. Intrinsic ADE: The dark side of antibody dependent enhancement during dengue infection. Front. Cell. Infect. Microbiol. 2020, 10, 580096. [Google Scholar] [CrossRef] [PubMed]
- Boumaza, A.; Gay, L.; Mezouar, S.; Bestion, E.; Diallo, A.B.; Michel, M.; Desnues, B.; Raoult, D.; La Scola, B.; Halfon, P.; et al. Monocytes and Macrophages, Targets of Severe Acute Respiratory Syndrome Coronavirus 2: The Clue for Coronavirus Disease 2019 Immunoparalysis. J. Infect. Dis. 2021, 224, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Wu, G. Potential 3-chymotrypsin-like cysteine protease cleavage sites in the coronavirus polyproteins pp1a and pp1ab and their possible relevance to COVID-19 vaccine and drug development. FASEB J. 2021, 35, e21573. [Google Scholar] [CrossRef]
- Glaab, E.; Manoharan, G.B.; Abankwa, D. Pharmacophore model for SARS-CoV-2 3CLpro small-molecule inhibitors and in vitro experimental validation of computationally screened inhibitors. J. Chem. Inf. Model. 2021, 61, 4082–4096. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. Methods Mol. Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chary, M.A.; Barbuto, A.F.; Izadmehr, S.; Hayes, B.D.; Burns, M.M. COVID-19: Therapeutics and Their Toxicities. J. Med. Toxicol. 2020, 16, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Bhattacharya, M.; Sharma, A.R. Emerging mutations in the SARS-CoV-2 variants and their role in antibody escape to small molecule-based therapeutic resistance. Curr. Opin. Pharmacol. 2022, 62, 64–73. [Google Scholar] [CrossRef]
- Mestrovic, T. First Detection of SARS-CoV-2 Remdesivir Resistance in an Immunocompromised Patient. Available online: https://www.news-medical.net/news/20211110/First-detection-of-SARS-CoV-2-remdesivir-resistance-in-an-immunocompromised-patient.aspx (accessed on 2 February 2022).
- Coghlan, A. Super-Antibodies Break the Cell Barrier; NewScientist: London, UK, 2004. [Google Scholar]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Kaewchim, K.; Glab-Ampai, K.; Mahasongkram, K.; Chulanetra, M.; Seesuay, W.; Chaicumpa, W.; Sookrung, N. Engineered fully human single-chain monoclonal antibodies to PIM2 kinase. Molecules 2021, 26, 6436. [Google Scholar] [CrossRef]
- Poungpair, O.; Pootong, A.; Maneewatch, S.; Srimanote, P.; Tongtawe, P.; Songserm, T.; Tapchaisri, P.; Chaicumpa, W. A human single chain transbody specific to matrix protein (M1) interferes with the replication of influenza A virus. Bioconjug. Chem. 2010, 21, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Dong-din-on, F.; Songserm, T.; Pissawong, T.; Srimanote, P.; Thanongsaksrikul, J.; Thueng-in, K.; Moonjit, P.; Lertwatcharasarakul, P.; Seesuay, W.; Chaicumpa, W. Cell penetrable human scFv specific to middle domain of matrix protein-1 protects mice from lethal influenza. Viruses 2015, 7, 154–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thueng-in, K.; Thanongsaksrikul, J.; Srimanote, P.; Bangphoomi, K.; Poungpair, O.; Maneewatch, S.; Choowongkomon, K.; Chaicumpa, W. Cell penetrable humanized-VH/V(H)H that inhibit RNA dependent RNA polymerase (NS5B) of HCV. PLoS ONE 2012, 7, e49254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thueng-in, K.; Thanongsaksrikul, J.; Jittavisutthikul, S.; Seesuay, W.; Chulanetra, M.; Sakolvaree, Y.; Srimanote, P.; Chaicumpa, W. Interference of HCV replication by cell penetrable human monoclonal scFv specific to NS5B polymerase. MAbs 2014, 6, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Jittavisutthikul, S.; Thanongsaksrikul, J.; Thueng-In, K.; Chulanetra, M.; Srimanote, P.; Seesuay, W.; Malik, A.A.; Chaicumpa, W. Humanized-VHH transbodies that inhibit HCV protease and replication. Viruses 2015, 7, 2030–2056. [Google Scholar] [CrossRef] [Green Version]
- Jittavisutthikul, S.; Seesuay, W.; Thanongsaksrikul, J.; Thueng-In, K.; Srimanote, P.; Werner, R.G.; Chaicumpa, W. Human transbodies to HCV NS3/4A protease inhibit viral replication and restore host innate immunity. Front. Immunol. 2016, 7, 318. [Google Scholar] [CrossRef] [Green Version]
- Glab-Ampai, K.; Malik, A.A.; Chulanetra, M.; Thanongsaksrikul, J.; Thueng-In, K.; Srimanote, P.; Tongtawe, P.; Chaicumpa, W. Inhibition of HCV replication by humanized-single domain transbodies to NS4B. Biochem. Biophys. Res. Commun. 2016, 476, 654–664. [Google Scholar] [CrossRef]
- Glab-Ampai, K.; Chulanetra, M.; Malik, A.A.; Juntadech, T.; Thanongsaksrikul, J.; Srimanote, P.; Thueng-In, K.; Sookrung, N.; Tongtawe, P.; Chaicumpa, W. Human single chain-transbodies that bound to domain-I of non-structural protein 5A (NS5A) of hepatitis C virus. Sci. Rep. 2017, 7, 15042. [Google Scholar] [CrossRef]
- Teimoori, S.; Seesuay, W.; Jittavisutthikul, S.; Chaisri, U.; Sookrung, N.; Densumite, J.; Saelim, N.; Chulanetra, M.; Maneewatch, S.; Chaicumpa, W. Human transbodies to VP40 inhibit cellular egress of Ebola virus-like particles. Biochem. Biophys. Res. Commun. 2016, 479, 245–252. [Google Scholar] [CrossRef]
- Seesuay, W.; Phanthong, S.; Densumite, J.; Mahasongkram, K.; Sookrung, N.; Chaicumpa, W. Human Transbodies to Reverse Transcriptase Connection Subdomain of HIV-1 Gag-Pol Polyprotein Reduce Infectiousness of the Virus Progeny. Vaccines 2021, 9, 893. [Google Scholar] [CrossRef]
- Seesuay, W.; Jittavisutthikul, S.; Sae-Lim, N.; Sookrung, N.; Sakolvaree, Y.; Chaicumpa, W. Human transbodies that interfere with the functions of Ebola virus VP35 protein in genome replication and transcription and innate immune antagonism. Emerg. Microbes Infect. 2018, 7, 1–15. [Google Scholar] [CrossRef]
- Brooks, N.; Esparon, S.; Pouniotis, D.; Pietersz, G.A. Comparative immunogenicity of a cytotoxic T cell epitope delivered by penetratin and TAT cell penetrating peptides. Molecules 2015, 20, 14033–14050. [Google Scholar] [CrossRef]
- Brooks, N.A.; Pouniotis, D.S.; Tang, C.K.; Apostolopoulos, V.; Pietersz, G.A. Cell-penetrating peptides: Application in vaccine delivery. Biochim. Biophys. Acta 2010, 1805, 25–34. [Google Scholar] [CrossRef]
- Sinha, S.; Tam, B.; Wang, S.M. RBD Double Mutations of SARS-CoV-2 Strains Increase Transmissibility through Enhanced Interaction between RBD and ACE2 Receptor. Viruses 2021, 14, 1. [Google Scholar] [CrossRef]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3CLpro | HuscFv27 | Interactive Bond | ||||
|---|---|---|---|---|---|---|
| Residue | Domain | Function of the Residue | Residue | Domain | ||
| Glu288 | III | Arg104 | VH-CDR3 | Hydrogen and Electrostatic | ||
| Asp289 | III | Arg104 | VH-CDR3 | Hydrogen and Electrostatic | ||
| Glu 290 | III | Homodimerization to form active enzyme | Arg104 | VH-CDR3 | Hydrogen and Electrostatic | |
| Lys5 | N-finger | Interact with Glu166 of another monomer to keep the S1 pocket in correct orientation | Ser103 | VH-CDR3 | Hydrogen | |
| Lys236 | III | Gly42 | VH-FR2 | Hydrogen | ||
| Ala285 | III | Asp108 | VH-CDR3 | Hydrogen | ||
| Asn274 | III | Gln39 | VH-FR2 | Hydrogen | ||
| Gln273 | III | Gly42 | VH-FR2 | Hydrogen | ||
| Met276 | III | Tyr95 | VH-FR3 | Hydrogen | ||
| Ser139 | II | Conserved residue in proximity of the protease active site | Thr101 | VH-CDR3 | Hydrogen | |
| Asp289 | III | Arg104 | VH-CDR3 | Hydrogen | ||
| Glu290 | III | Homodimerization to form active enzyme | Arg104 | VH-CDR3 | Hydrogen | |
| Leu287 | III | Arg104 | VH-CDR3 | Hydrogen | ||
| Glu290 | III | Homodimerization to form active enzyme | His105 | VH-CDR3 | Hydrogen | |
| Ser284 | III | Asp108 | VH-CDR3 | Hydrogen | ||
| Gly278 | III | Gln235 | VL-FR4 | Hydrogen | ||
| Gly138 | II | Gly102 | VH-CDR3 | Hydrogen | ||
| Glu288 | III | Phe107 | VH-CDR3 | Hydrogen | ||
| Tyr126 | II | Tyr100 | VH-CDR3 | Hydrophobic | ||
| Tyr126 | II | Gly102 | VH-CDR3 | Hydrophobic | ||
| Tyr126 | II | Ser103 | VH-CDR3 | Hydrophobic | ||
| Leu286 | III | Ala97 | VH-CDR3 | Hydrophobic | ||
| Ala285 | III | Tyr95 | VH-FR3 | Hydrophobic | ||
| Lys137 | II | His105 | VH-CDR3 | Hydrophobic | ||
| 3CLpro | HuscFv33 | Interactive bond | ||||
| Residue | Domain | Function of the Residue | Residue | Domain | ||
| Asn51 | I | Asp62 | VH-FR3 | Hydrogen | ||
| Lys137 | II | Gly102 | VH-CDR3 | Hydrogen | ||
| Glu166 | II | Homodimerization to form active enzyme | Tyr59 | VH-FR3 | Hydrogen | |
| Asp197 | II | Ser108 | VH-CDR3 | Hydrogen | ||
| Pro168 | II | Thr35 | VH-FR2 | Hydrogen | ||
| Glu166 | II | Homodimerization to form active enzyme | Ser52 | VH-CDR2 | Hydrogen | |
| Gly170 | II | Ser52 | VH-CDR2 | Hydrogen | ||
| Glu166 | II | Homodimerization to form active enzyme | Ser54 | VH-CDR2 | Hydrogen | |
| Glu166 | II | Homodimerization to form active enzyme | Ser56 | VH-CDR2 | Hydrogen | |
| Gln189 | LLR | Lys65 | VH-FR3 | Hydrogen | ||
| Leu50 | I | Lys65 | VH-FR3 | Hydrogen and Hydrophobic | ||
| Asn238 | III | Arg107 | VH-CDR3 | Hydrogen | ||
| Asp197 | LLR | Ser108 | VH-CDR3 | Hydrogen | ||
| Phe140 | II | Homodimerization to form active enzyme | Ser54 | VH-CDR2 | Hydrogen | |
| Asp197 | LLR | Gly102 | VH-CDR3 | Hydrogen | ||
| Thr199 | LLR | Ser104 | VH-CDR3 | Hydrogen | ||
| Ala194 | LLR | Trp47 | VH-FR2 | Hydrogen and Hydrophobic | ||
| Ala193 | LLR | Trp47 | VH-FR2 | Hydrophobic | ||
| Ala191 | LLR | Substrate binding | Ala61 | VH-FR3 | Hydrophobic | |
| Leu286 | III | Pro105 | VH-CDR3 | Hydrophobic | ||
| Met165 | II | Substrate binding | Tyr59 | VH-FR3 | Hydrophobic | |
| 3CLpro | HuscFv34 | Interactive bond | ||||
| Residue | Domain | Function of the Residue | Residue | Domain | ||
| Glu288 | III | Lys177 | VL-FR2 | Electrostatic | ||
| Ser46 | I | Gly101 | VH-CDR3 | Hydrogen | ||
| Ser46 | I | Asp102 | VH-CDR3 | Hydrogen | ||
| Gly170 | II | Asp109 | VH-CDR3 | Hydrogen | ||
| Pro168 | II | Trp47 | VH-FR2 | Hydrogen and Hydrophobic | ||
| Asn142 | II | Lys98 | VH-CDR3 | Hydrogen | ||
| Thr190 | LLR | Substrate binding | Arg99 | VH-CDR3 | Hydrogen | |
| Ser46 | I | Gly104 | VH-CDR3 | Hydrogen | ||
| Glu166 | II | Homodimerization to form active enzyme | Phe108 | VH-CDR3 | Electrostatic | |
| Ala285 | III | Ala178 | VL-FR2 | Hydrophobic | ||
| Ala285 | III | Pro179 | VH-FR2 | Hydrophobic | ||
| His172 | II | Val110 | VH-CDR3 | Hydrophobic | ||
| Ala191 | LLR | Substrate binding | Trp47 | VH-FR2 | Hydrophobic | |
| Ala193 | LLR | Trp47 | VH-FR2 | Hydrophobic | ||
| Cys145 | II | Catalytic dyad | Trp105 | VH-CDR3 | Hydrophobic | |
| Clone No. | EC50 (nM) | ||||
|---|---|---|---|---|---|
| Wuhan | α (B.1.1.7) | β (B.1.351) | δ (B.1.617.2) | Omicron (B.1.1.529) | |
| PEN-HuscFv27 | 692.5 | 778.1 | 1058 | 582.8 | 2694 |
| PEN-HuscFv33 | 491.4 | 696.7 | 297 | 478.1 | 1048 |
| PEN-HuscFv34 | 539.5 | 735.9 | 878.3 | 158.1 | 800.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glab-ampai, K.; Kaewchim, K.; Saenlom, T.; Thepsawat, W.; Mahasongkram, K.; Sookrung, N.; Chaicumpa, W.; Chulanetra, M. Human Superantibodies to 3CLpro Inhibit Replication of SARS-CoV-2 across Variants. Int. J. Mol. Sci. 2022, 23, 6587. https://doi.org/10.3390/ijms23126587
Glab-ampai K, Kaewchim K, Saenlom T, Thepsawat W, Mahasongkram K, Sookrung N, Chaicumpa W, Chulanetra M. Human Superantibodies to 3CLpro Inhibit Replication of SARS-CoV-2 across Variants. International Journal of Molecular Sciences. 2022; 23(12):6587. https://doi.org/10.3390/ijms23126587
Chicago/Turabian StyleGlab-ampai, Kittirat, Kanasap Kaewchim, Thanatsaran Saenlom, Watayagorn Thepsawat, Kodchakorn Mahasongkram, Nitat Sookrung, Wanpen Chaicumpa, and Monrat Chulanetra. 2022. "Human Superantibodies to 3CLpro Inhibit Replication of SARS-CoV-2 across Variants" International Journal of Molecular Sciences 23, no. 12: 6587. https://doi.org/10.3390/ijms23126587