The Contribution of Endothelial Dysfunction in Systemic Injury Subsequent to SARS-Cov-2 Infection


 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
- (1)
- At the moment of platelet activation, arachidonic acid is converted to thromboxane A2, which is a potent pro-aggregatory and vasoconstrictive factor [9]. Then the platelets degranulate and finally undergo a conformational change assuming a starry shape, which is characteristic of their physical aggregation [10].
- (2)
- The activation of the coagulation cascade determines, through several pathways, the formation of thrombin that splits the fibrinogen into fibrin. This fibrillar protein polymerizes to form a “mesh” together with platelets at the wound site.
2. Major Molecular Targets of SARS-CoV-2
3. ACE2 Receptor Characterization
- Different catalytic activity: ACE is a dipeptidase that hydrolyses bound pairs of amino acids cleaving the C-terminal dipeptide from Ang I to form the octapeptide Ang II. ACE2 is a carboxypeptidase capable of breaking peptide bonds between amino acids at the level of terminal C residue and removing the residue from the decapeptide Ang I to form angiotensin-1–9.
- Several substrates and bond specificity: in particular, ACE binds and cleaves Ang I, Ang 1–9, and many bioactive peptides. In contrast, ACE2 cleaves Ang I, Ang II, apelin-13, and apelin-36 [45].
- A different expression in different tissues of the organism has been shown: ACE is more ubiquitous. In fact, this enzyme is expressed in the heart, lung, kidney, colon, small intestine, ovary, testis, prostate, liver, skeletal muscle, pancreas, and thyroid. In contrast, the expression of ACE2 is more specific and is limited in the heart, kidney, endothelial cells, and microvasculature [46,47].
- Inhibitor specificity: The ACE inhibitors act by blocking of the conversion of angiotensin I to angiotensin II and inhibiting the most important step in the RAS pathway. For this reason, they are widely used as a class of anti-hypertensive drugs. The ACE inhibitors have been linked to cases of hepatotoxicity. ACE2 cannot be inhibited to ACE inhibitors [48].
- The catalyzed peptides are preferably hydrolysed on the proline residues to the C-terminal group [50].
- Its enzymatic activity is regulated by chloride ions. The bond with the chloride induces conformational changes of the active site of the enzyme, which is responsible for modulation of the reactions [51].
Molecular Mechanisms of ACE-2-Covid 19 Interaction
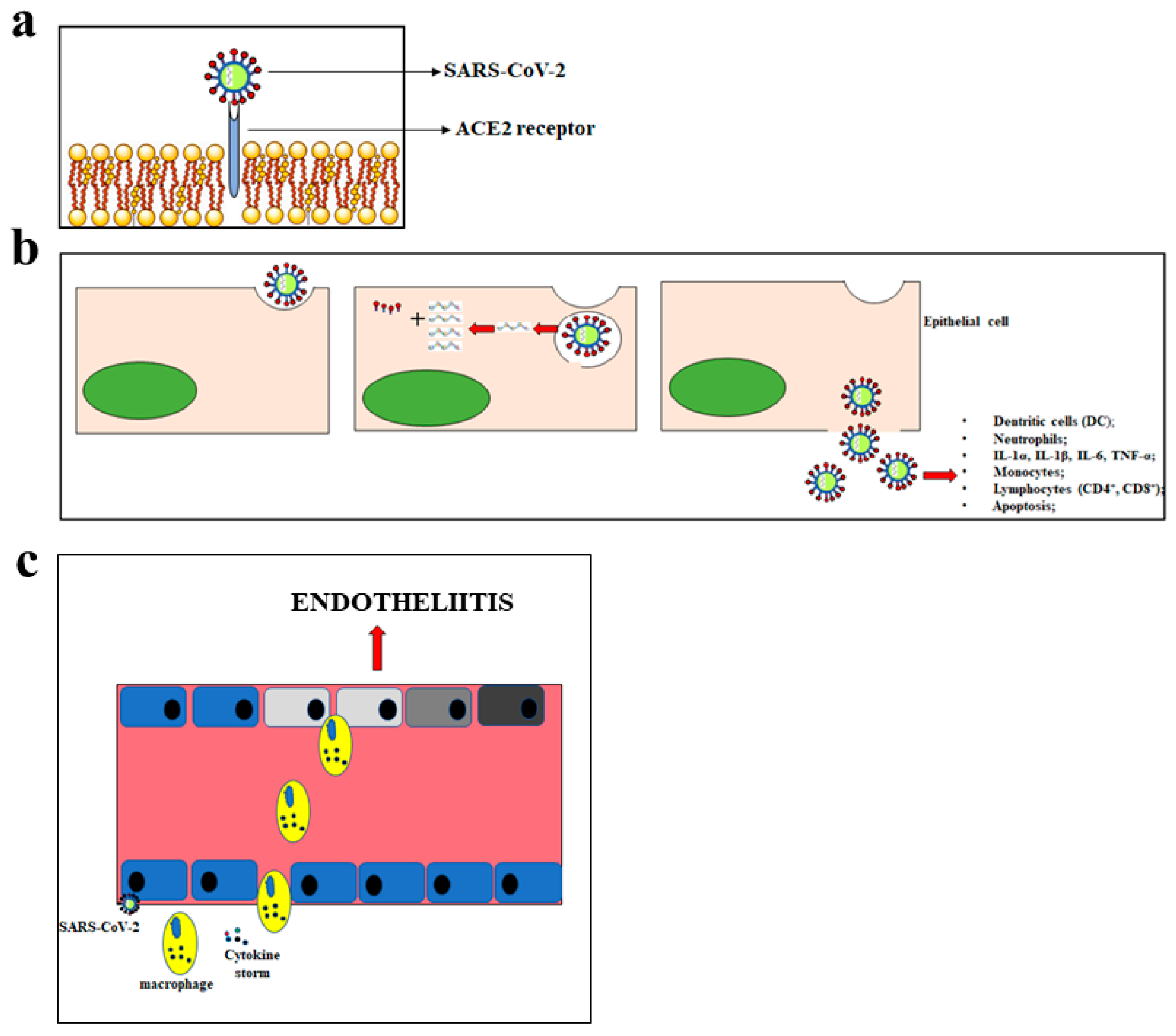
4. Endothelium Dysfunction and SARS-CoV-2
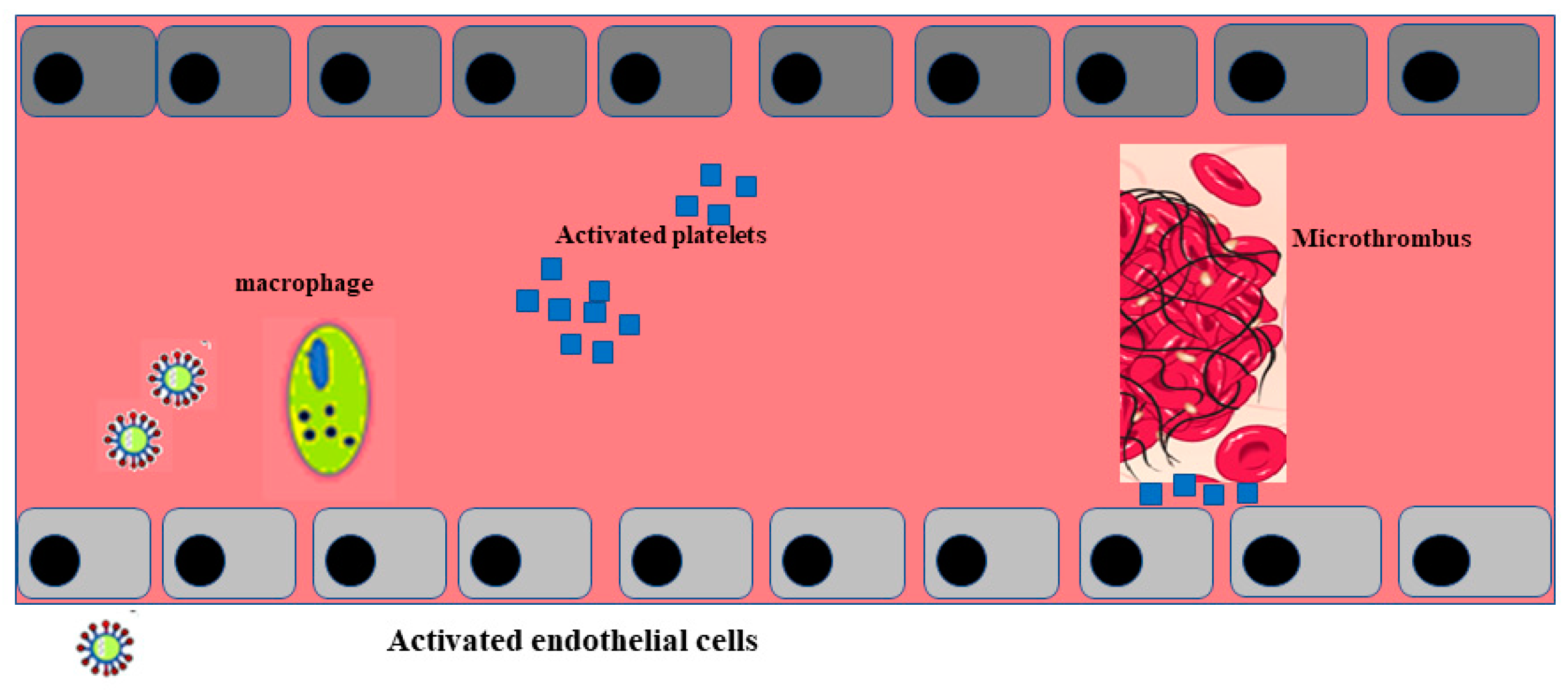
5. SARS-CoV-2 Related Vasculitis
6. Therapeutic Interventions in Endothelial Dysfunction and in Sars-Cov-2 Systemic Damage
- (1)
- Activation of GTPase enzymes triggering a self-repair process [101]. Following the dangerous stimuli, that alter the integrity of the endothelium, small enzymes with GTP-ases functions are activated, which, through their internal cross-talk, promote the hydrolysis of the GTP. These GTPases enzymes control the integrity of the endothelial barrier by stimulating the genesis of actin paracellular fibers and sealing some junctional gaps [101].
- (2)
- Recovery of molecules that combat barrier breaking [102]. Some molecules can counteract any endothelial barrier failure by preventing its rupture. They include cAMP and cAMP derivatives that increase endothelial cell functions through the protein kinase A (PKA) pathway. In particular, caderin expression, which promotes the formation of paracellular adhering junctions in endothelial cells, is up-regulated [103]. Another important molecule involved in this process is phospholipid oxidized 1-palmitoyl-2-arachidonoyl-sn-glycer-3-phosphorylcholine (Oxpapc). Oxpapc limits the effects of proinflammatory agents, such as thrombin, participating in remodeling of endothelial cytoskeleton and favoring maintenance of the endothelial cytoskeleton by facilitating the assembly of tight and adherent junctions [104].
- (3)
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Akhmerov, A.; Marban, E. COVID-19 and the heart. Circ. Res. 2020, 126, 14431–14455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 5315–5338. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and important lessonsfrom the coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mohiddin, S.A.; Dimarco, A.; Patel, V.; Savvatis, K.; Marelli-Berg, F.M.; Madhur, M.S.; Tomaszewski, M.; Maffia, P.; D’Acquisto, F. COVID-19 and the cardiovascular system: Implications for risk assessment, diagnosis, and treatment options. Cardiovasc. Res. 2020, 116, 16661–16687. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline characteristics and outcomes of 1591 patients infected With SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunal, S.; Gupta, K.; Sharma, S.M.; Pathak, V.; Mittal, S.; Tarke, C. Cardiovascular system and COVID-19: Perspectives from a developing country. Monaldi Arch. Chest Dis. 2020, 90, 90. [Google Scholar] [CrossRef] [PubMed]
- Al-Ani, F.; Chehade, S.; Lazo-Langner, A. Thrombosis risk associated with COVID-19 infection. A scoping review. Thromb. Res. 2020, 192, 152–160. [Google Scholar] [CrossRef]
- Gąsecka, A.; Borovac, J.A.; Guerreiro, R.A.; Giustozzi, M.; Parker, W.; Caldeira, D.; Chiva-Blanch, G. Thrombotic Complications in Patients with COVID-19: Pathophysiological Mechanisms, Diagnosis, and Treatment. Cardiovasc. Drugs Ther. 2020, 1–15. [Google Scholar] [CrossRef]
- Parker, W.; Orme, R.C.; Hanson, J.; Stokes, H.M.; Bridge, C.M.; Shaw, P.A.; Sumaya, W.; Thorneycroft, K.; Petrucci, G.; Porro, B.; et al. Very-low-dose twice-daily aspirin maintains platelet inhibition and improves haemostasis during dual-antiplatelet therapy for acute coronary syndrome. Platelets 2019, 30, 148–157. [Google Scholar] [CrossRef]
- Paul, B.Z.; Jin, J.; Kunapuli, S.P. Molecular mechanism of thromboxane A(2)-induced platelet aggregation. Essential role for p2t(ac) and alpha(2a) receptors. J. Biol. Chem. 1999, 274, 291082–299114. [Google Scholar] [CrossRef] [Green Version]
- Briedé, J.J.; Tans, G.; Willems, G.M.; Hemker, H.C.; Lindhout, T. Regulation of Platelet Factor Va-dependent Thrombin Generation by Activated Protein C at the Surface of Collagen-adherent Platelets. J. Biol. Chem. 2001, 276, 7164–7168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liang, C. Innate recognition of microbial-derived signals in immunity and inflammation. Sci. China Life Sci. 2016, 59, 12101–12217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alijotas-Reig, J.; Esteve-Valverde, E.; Belizna, C.; Selva-O’Callaghan, A.; Pardos-Gea, J.; Quintana, A.; Mekinian, A.; Anunciacion-Llunell, A.; Miró-Mur, F. Immunomodulatory therapy for the management of severe COVID-19. Beyond the anti-viral therapy: A comprehensive review. Autoimmun. Rev. 2020, 19, 102569. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.P.; Viswanathan, S.; Wang, M.; Sun, L.Q.; Clark, G.C.; D’Elia, R. Current and future developments in the treatment of virus-induced hypercytokinemia. Future Med. Chem. 2017, 9, 169–178. [Google Scholar] [CrossRef]
- Nunes Kochi, A.; Tagliari, A.P.; Forleo, G.B.; Fassini, G.M.; Tondo, C. Cardiac and arrhythmic complications in patients with COVID 19. J. Cardiovasc. Electrophysiol. 2020, 31, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: An updated analysis. Thromb. Res. 2020, 191, 148–150. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Warkentin, T.E.; Thachil, J.; van der Poll, T.; Levi, M. Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Thromb. Haemost. 2019, 17, 1989–1994. [Google Scholar] [CrossRef] [Green Version]
- Casini, A.; Fontana, P.; Glauser, F.; Robert-Ebadi, H.; Righini, M.; Blondon, M. Venous thrombotic risk related to SARS-CoV-2: Prevalence, recommendations and perspectives. Rev. Med. Suisse. 2020, 16, 9519–9554. [Google Scholar]
- Tang, N.; Bai, H.; Chen, X.; Gong, J.; Li, D.; Sun, Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J. Thromb. Haemost. 2020, 18, 1094–1099. [Google Scholar] [CrossRef]
- Hess, D.C.; Eldahshan, W.; Rutkowski, E. COVID-19-Related Stroke. Transl. Stroke Res. 2020, 11, 3223–3225. [Google Scholar] [CrossRef] [PubMed]
- Gavriilaki, E.; Brodsky, R.A. Severe COVID-19 infection and thrombotic microangiopathy: Success doesn’t come easily. Br. J. Haematol. 2020, 189, e227–e230. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.R.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Mani, J.S.; Johnson, J.B.; Steel, J.C.; Broszczak, D.A.; Neilsen, P.M.; Walsh, K.B.; Naiker, M. Natural product-derived phytochemicals as potential agents against coronaviruses: A review. Virus Res. 2020, 284, 197989. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belen-Apak, F.B.; Sarialioglu, F. The old but new: Can unfractioned heparin and low molecular weight heparins inhibit proteolytic activation and cellular internalization of SARS-CoV2 by inhibition of host cell proteases? Med. Hypotheses 2020, 142, 109743. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Ilmain, J.K.; Guo, X.; Bhabha, G.; Sanjana, N.E. The Spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, M. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 1–9. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef]
- Malik, Y.A. Properties of Coronavirus and SARS-CoV-2. Malays J. Pathol. 2020, 42, 3–11. [Google Scholar] [PubMed]
- Hulswit, R.J.; de Haan, C.A.; Bosch, B.J. Coronavirus Spike Protein and Tropism Changes. Adv. Virus Res. 2016, 96, 29–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R.A. Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Bhattacharya, M.; Lee, S.S. SARS-CoV-2 causing pneumonia-associated respiratory disorder (COVID-19): Diagnostic and proposed therapeutic options. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 4016–4026. [Google Scholar] [CrossRef]
- Stadnytskyi, V.; Bax, C.E.; Bax, A.; Anfinrud, P. The airborne lifetime of small speech droplets and their potential importance in SARS-CoV-2 transmission. Proc. Natl. Acad. Sci. USA 2020, 17, 11875–11877. [Google Scholar] [CrossRef]
- Lambert, D.W.; Clarke, N.E.; Turner, A.J. Not just angiotensinases: New roles for the angiotensin-converting enzymes. Cell Mol. Life Sci. 2010, 67, 89–98. [Google Scholar] [CrossRef]
- Perlot, T.; Penninger, J.M. ACE2-from the renin-angiotensin system to gut microbiota and malnutrition. Microbes Infect. 2013, 15, 8668–8673. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Roca-Ho, H.; Riera, M.; Palau, V.; Pascual, J.; Soler, M.J. Characterization of ACE and ACE2 Expression within Different Organs of the NOD Mouse. Int. J. Mol. Sci. 2017, 18, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Koibuchi, N.; Nishimatsu, H.; Higashikuni, Y.; Hirata, Y.; Kugiyama, K.; Nagai, R.; Sata, M. Candesartan ameliorates cardiac dysfunction observed in angiotensin-converting enzyme 2-deficient mice. Hypertens. Res. 2008, 31, 19531961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Sexton, D.J.; Skogerson, K.; Devlin, M.; Smith, R.; Sanyal, I.; Parry, T.; Kent, R.; Enright, J.; Wu, Q.-L.; et al. Novel peptide inhibitors of angiotensin-converting enzyme 2. J. Biol. Chem. 2003, 278, 155321–155540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvol, P.; Williams, T.A.; Soubrier, F. Peptidyl dipeptidase A: Angiotensin I-converting enzyme. Methods Enzymol. 1995, 248, 283–305. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Hikmet, F.; Méar, L.; Edvinsson, A.; Micke, P.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef]
- Labò, N.; Ohnuki, H.; Tosato, G. Vasculopathy and Coagulopathy Associated with SARS-CoV-2 Infection. Cells 2020, 9, 1583. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A peptidase in the renin–angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010, 128, 119–128. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of Biological Peptides by Human Angiotensin-converting Enzyme-related Carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [Green Version]
- Nadarajah, R.; Milagres, R.; Dilauro, M.; Gutsol, A.; Xiao, F.; Zimpelmann, J.; Kennedy, C.; Wysocki, J.; Batle, D.; Burns, K.D. Podocyte-specific overexpression of human angiotensin-converting enzyme 2 attenuates diabetic nephropathy in mice. Kidney Int. 2012, 82, 292–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuille-Dit-Bille, R.N.; Camargo, S.M.; Emmenegger, L.; Sasse, T.; Kummer, E.; Jando, J.; Hamie, Q.M.; Meier, C.F.; Hunziker, S.; Forras-Kaufmann, Z.; et al. Human intestine luminal ACE2 and amino acid transporter expression increased by ACE-inhibitors. Amino Acids 2015, 47, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, S.M.; Singer, D.; Makrides, V.; Huggel, K.; Pos, K.M.; Wagner, C.A.; Kuba, K.; Danilczyk, U.; Skovby, F.; Kleta, R.; et al. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology 2009, 136, 872–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Arthur, S.; Singh, S.; Sundaram, U. Cyclooxygenase pathway mediates the inhibition of Na-glutamine co-transporter B0AT1 in rabbit villus cells during chronic intestinal inflammation. PLoS ONE 2018, 13, e0203552. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-Dependent Entry of Severe Acute Respiratory Syndrome Coronavirus into Target Cells Expressing ACE2 with the Cytoplasmic Tail Deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef] [Green Version]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolaeindependent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Perrotta, F.; Matera, M.G.; Cazzola, M.; Bianco, A. Severe respiratory SARS-CoV2 infection: Does ACE2 receptor matter? Respir. Med. 2020, 168, 105996. [Google Scholar] [CrossRef] [PubMed]
- Qing, E.; Hantak, M.; Perlman, S.; Gallagher, T. Distinct Roles for Sialoside and Protein Receptors in Coronavirus Infection. mBio 2020, 11, e027641-9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.L.; Lan, P.D.; Thai, N.Q.; Nissley, D.A.; O’Brien, E.P.; Li, M.S. Does SARS-CoV-2 Bind to Human ACE2 More Strongly Than Does SARS-CoV? J. Phys. Chem. B 2020, 124, 7336–7347. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Ferreira, A.J.; Verano-Braga, T.; Bader, M. Angiotensin-converting enzyme2, angiotensin-(1–7) and Mas: New players of the renin-angiotensin system. J. Endocrinol. 2013, 216, R1–R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qaradakhi, T.; Gadanec, L.K.; McSweeney, K.R.; Tacey, A.; Apostolopoulos, V.; Levinger, I.; Rimarova, K.; Egom, E.E.; Rodrigo, L.; Kruzliak, P.; et al. The potential actions of angiotensin-converting enzyme II (ACE2) activator diminazene aceturate (DIZE) in various diseases. Clin. Exp. Pharmacol. Physiol. 2020, 47, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Narayan, R.K.; Kumari, C.; Faiq, M.; Kulandhasamy, M.; Kant, K.; Pareek, V. SARS-CoV-2 cell entry receptor ACE2 mediated endothelial dysfunction leads to vascular thrombosis in COVID-19 patients. Med Hypotheses 2020, 145, 110320. [Google Scholar] [CrossRef] [PubMed]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Teuwen, L.A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 21, 1–3. [Google Scholar] [CrossRef]
- Pagliaro, P.; Penna, C. ACE/ACE2 Ratio: A Key Also in 2019 Coronavirus Disease (Covid-19)? Front. Med. 2020, 7, 335. [Google Scholar] [CrossRef]
- Thomas, M.C.; Pickering, R.J.; Tsorotes, D.; Koitka, A.; Sheehy, K.; Bernardi, S.; Toffoli, B.; Nguyen-Huu, T.P.; Head, G.A.; Fu, Y.; et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE. Circ. Res. 2010, 107, 8888–8897. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.Y.; Chang, S.C.; Wu, H.Y.; Yu, T.C.; Wei, W.C.; Lin, S.; Chien, C.L.; Chang, M.F. Upregulation of the chemokine (CC motif) ligand 2 via a severe acute respiratory syndrome coronavirus spike-ACE2 signaling pathway. J. Virol. 2010, 84, 7703–7712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraga-Silva, R.A.; Sorg, B.S.; Wankhede, M.; Dedeugd, C.; Jun, J.Y.; Baker, M.B.; Li, Y.; Castellano, R.K.; Katovich, M.J.; Raizada, M.K.; et al. ACE2 activation promotes antithrombotic activity. Mol. Med. 2010, 16, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Hippisley-Cox, J.; Young, D.; Coupland, C.; Channon, K.M.; Tan, P.S.; Harrison, D.A.; Rowan, K.; Aveyard, P.; Pavord, I.D.; Watkinson, P.J. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: Cohort study including 8.3 million people. Heart 2020, 106, 15031–15511. [Google Scholar] [CrossRef] [PubMed]
- Kiseleva, R.Y.; Glassman, P.M.; Greineder, C.F.; Hood, E.D.; Shuvaev, V.V.; Muzykantov, V.R. Targeting therapeutics to endothelium: Are we there yet? Drug Deliv. Transl. Res. 2018, 8, 883–902. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Scicchiatano, M.; Carresi, C.; Scarano, F.; Bosco, F.; Nucera, S.; Ruga, S.; Zito, M.C.; et al. The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. Int. J. Mol. Sci. 2018, 19, 2693. [Google Scholar] [CrossRef] [Green Version]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Macrì, R.; Scicchitano, M.; Bosco, F.; Scarano, F.; Ruga, S.; et al. The Role of Endothelial Dysfunction in Peripheral Blood Nerve Barrier: Molecular Mechanisms and Pathophysiological Implications. Int. J. Mol. Sci. 2019, 20, 3022. [Google Scholar] [CrossRef] [Green Version]
- Keaney, J.; Campbell, M. The dynamic blood-brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef]
- Durak-Kozica, M.; Baster, Z.; Kubat, K.; Stępień, E. 3D visualization of extracellular vesicle uptake by endothelial cells. Cell Mol. Biol. Lett. 2018, 17, 23–57. [Google Scholar] [CrossRef] [Green Version]
- Radeva, M.Y.; Waschke, J. Mind the gap: Mechanisms regulating the endothelial barrier. Acta Physiol. 2018, 222. [Google Scholar] [CrossRef]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; Van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef]
- Short, K.R.; Kasper, J.; van der Aa, S.; Andeweg, A.C.; Zaaraoui-Boutahar, F.; Goeijenbier, M.; Richard, M.; Herold, S.; Becker, C.; Scott, D.; et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur. Respir. J. 2016, 47, 9549–9566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, S.M.; Darwish, I.; Lee, W.L. Endothelial activation and dysfunction in the pathogenesis of influenza A virus infection. Virulence 2013, 4, 537–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front. Immunol. 2020, 11, 2063. [Google Scholar] [CrossRef] [PubMed]
- Green, C.E.; Turner, A.M. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Green Turn. Respir. Res. 2017, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janga, H.; Cassidy, L.; Wang, F.; Spengler, D.; Oestern-Fitschen, S.; Krause, M.F.; Seekamp, A.; Tholey, A.; Fuchs, S. Site-specific and endothelial-mediated dysfunction of the alveolar-capillary barrier in response to lipopolysaccharides. J. Cell. Mol. Med. 2018, 22, 9829–9898. [Google Scholar] [CrossRef] [Green Version]
- Tuder, R.M.; Yoshida, T. Stress Responses Affecting Homeostasis of the Alveolar Capillary Unit. Proc. Am. Thorac. Soc. 2011, 8, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Kaur, U.; Acharya, K.; Mondal, R.; Singh, A.; Saso, L.; Chakrabarti, S.; Chakrabarti, S.S. Should ACE2 be given a chance in COVID-19 therapeutics: A semi-systematic review of strategies enhancing ACE2. Eur. J. Pharmacol. 2020, 887, 173545. [Google Scholar] [CrossRef]
- Varga, V.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schueqbach, R.A.; Ruschitzka, E.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 14171–14418. [Google Scholar] [CrossRef]
- Opal, S.M.; van der Poll, T. Endothelial barrier dysfunction in septic shock. J. Intern. Med. 2015, 277, 277–293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tecson, K.M.; McCullough, P.A. Endothelial dysfunction contributes to COVID-19-associated vascular inflammation and coagulopathy. Rev. Cardiovasc. Med. 2020, 21, 315–319. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Ladikou, E.E.; Sivaloganathan, A.H.; Kate, A.; Milne, M.; William, A.; Ramasamy, R.; Saad, R.; Stoneman, S.M.; Eziefula, A.C.; Cheevassut, T. Von Willebrand factor (vWF): Marker of endothelial damage and thrombotic risk in COVID-19? Clin. Med. 2020, 20, e178–e182. [Google Scholar] [CrossRef] [PubMed]
- Menter, T.; Haslbauer, J.D.; Nienhold, R.; Savic, S.; Hopfer, H.; Deigendesch, N.; Frank, S.; Turek, D.; Willi., N.; Pargger, H. Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology 2020, 77, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yang, M.; Wan, C.; Yi, L.-X.; Tang, F.; Zhu, H.-Y.; Yi, F.; Yang, H.-C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.A.; Cavaliere, L.F.; Grant-Kels, J.M. Cutaneous vasculitis: Diagnosis and management. Clin. Dermatol. 2006, 24, 414–429. [Google Scholar] [CrossRef]
- Freeman, E.E.; McMahon, D.E.; Fitzgerald, M.E.; Fox, L.P.; Rosenbach, M.; Takeshita, J.; French, L.E.; Thiers, B.H.; Hruza, G.J. The American Academy of Dermatology COVID-19 registry: Crowdsourcing dermatology in the age of COVID-19. J. Am. Acad. Dermatol. 2020, 83, 509–510. [Google Scholar] [CrossRef]
- Joob, B.; Wiwanitkit, V. COVID-19 can present with a rash and be mistaken for Dengue. J. Am. Acad. Dermatol. 2020, 82, e177. [Google Scholar] [CrossRef]
- Recalcati, S. Cutaneous manifestations in COVID-19: A first perspective. JEADV 2020, 34, e212–e213. [Google Scholar] [CrossRef]
- Mazzotta, F.; Troccoli, T.; Bonifazi, E. Acute acro-ischemia in the child at the time of covid-19. Eur. J. Pediatric Dermatol. 2020, 30, 71–74. [Google Scholar] [CrossRef]
- Criado, P.R.; Martinez Zugaib Abdalla, B.; Carvalho de Assis, I.; van Blarcum de Graaff Mello, C.; Cacciolari Caputo, G.; Campos Vieira, I. Are the cutaneous manifestations during or due to SARS-CoV-2 infection/COVID-19 frequent or not? Revision of possible pathophysiologic mechanisms. Inflamm. Res. 2020, 69, 745–756. [Google Scholar] [CrossRef]
- Castelnovo, L.; Capelli, F.; Tamburello, A.; Faggioli, P.M.; Mazzone, A. Symmetric cutaneous vasculitis in COVID-19 pneumonia. JEADV 2020, 34, e362–e363. [Google Scholar] [CrossRef] [PubMed]
- Suganya, N.; Bhakkiyalakshmi, E.; Sarada, D.V.L.; Ramkumar, K.M.R. Reversibility of endothelial dysfunction in diabetes: Role of polyphenols. Br. J. Nutr. 2016, 116, 223–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollace, V.; Tavernese, A.; Mollace, R. The essential role of a “healthy” relationship between caveolin-1 and endothelial nitric oxide synthase in counteracting vascular inflammation and atherothrombosis. Kardiol. Pol. 2020, 78, 96–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs-Kasa, A.; Kim, K.M.; Cherian-Shaw, M.; Black, S.M.; Fulton, D.J.; Verin, A.D. Extracellular adenosine-induced Rac1 activation in pulmonary endothelium: Molecular mechanisms and barrier-protective role. J. Cell Physiol. 2018, 233, 57365–57746. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, T.; Tian, Y.; Sarich, N.; Ke, Y.; Meliton, A.; Shah, A.S.; Andreasson, K.; Birukov, K.G.; Birukova, A.A. Regulation of lung endothelial permeability and inflammatory responses by prostaglandin A2: Role of EP4 receptor. Mol. Biol. Cell. 2017, 28, 16221–16635. [Google Scholar] [CrossRef]
- Zhang, J.; Lu, X.; Liu, M.; Fan, H.; Zheng, H.; Zhang, S.; Rahman, N.; Wołczyński, S.; Kretowski, A.; Li, X. Melatonin inhibits inflammasome-associated activation of endothelium and macrophages attenuating pulmonary arterial hypertension. Cardiovasc. Res. 2019, 116, 2156–2169. [Google Scholar] [CrossRef] [PubMed]
- Konya, V.; Üllen, A.; Kampitsch, N.; Theiler, A.; Philipose, S.; Parzmair, G.P.; Marsche, G.; Peskar, B.A.; Schuligoi, R.; Sattler, W.; et al. Endothelial E-type prostanoid 4 receptors promote barrier function and inhibit neutrophil trafficking. J. Allergy Clin. Immunol. 2013, 131, 532–540.e2. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Birukov, K.G. Oxidized Phospholipids in Healthy and Diseased Lung Endothelium. Cells 2020, 9, 981. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.; Shaaya, M.; Harijith, A.; Jacobson, J.R.; Karginov, A.; Natarajan, V. Sphingolipids Signaling in Lamellipodia Formation and Enhancement of Endothelial Barrier Function. Curr. Top. Membr. 2018, 82, 1–31. [Google Scholar]
- Zheng, B.; Ye, L.; Zhouguang, W.; Zhu, S.; Wang, Q.; Shi, H.; Chen, D.; Wei, X.; Wang, Z.; Li, X.; et al. Epidermal growth factor attenuates blood-spinal cord barrier disruption via PI 3K/Akt/Rac1 pathway after acute spinal cord injury. J. Cell. Mol. Med. 2016, 20, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Oppedisano, F.; Maiuolo, J.; Gliozzi, M.; Musolino, V.; Carresi, C.; Nucera, S.; Scicchitano, M.; Scarano, F.; Bosco, F.; Macrì, R.; et al. The Potential for Natural Antioxidant Supplementation in the Early Stages of Neurodegenerative Disorders. Int. J. Mol. Sci. 2020, 21, 2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gliozzi, M.; Scicchitano, M.; Bosco, F.; Musolino, V.; Carresi, C.; Scarano, F.; Maiuolo, J.; Nucera, S.; Maretta, A.; Paone, A.; et al. Modulation of Nitric Oxide Synthases by Oxidized LDLs: Role in Vascular Inflammation and Atherosclerosis Development. Int. J. Mol. Sci. 2019, 20, 3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, Z.; Guan, X.; Zhi, B.Z. Resveratrol Attenuates Hypoxia-Reperfusion Injury Induced Rat Myocardium Microvascular Endothelial Cell Dysfunction Through Upregulating PI3K/Akt/SVV Pathways. Zhonghua Xin Xue Guan Bing Za Zhi 2014, 42, 670–674. [Google Scholar] [CrossRef]
- Mu, S.; Liu, Y.; Jiang, J.; Ding, R.; Li, X.; Li, X.; Ma, X. Unfractionated heparin ameliorates pulmonary microvascular endothelial barrier dysfunction via microtubule stabilization in acute lung injury. Respir. Res. 2018, 19, 220. [Google Scholar] [CrossRef] [PubMed]
- Slobodnick, A.; Shah, B.; Pillinger, M.H.; Krasnokutsky, S. Colchicine: Old and new. Am. J. Med. 2015, 128, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cure, C.; Kucuk, A.; Cure, E. Colchicine may not be effective in COVID-19 infection; it may even be harmful? Clin. Rheumatol. 2020. (In press) [CrossRef]
- Kollias, A.; Kyriakoulis, K.G.; Dimakakos, E.; Poulakou, G.; Stergiou, G.S.; Syrigos, K. Thromboembolic risk and anticoagulant therapy in COVID-19 patients: Emerging evidence and call for action. Br. J. Haematol. 2020, 189, 846–847. [Google Scholar] [CrossRef]
- Poterucha, T.J.; Libby, P.; Goldhaber, S.Z. More than an anticoagulant: Do heparins have direct anti-inflammatory effects? Thromb. Haemost. 2017, 117, 437–444. [Google Scholar] [CrossRef]
- Thachil, J. The versatile heparin in COVID-19. Int. Soc. Thromb. Haemost. 2020. (In Press) [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Delabranche, X.; Helms, J.; Meziani, F. Immunohaemostasis: A new view on haemostasis during sepsis. Ann. Intensive Care 2017, 7, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.P.; Darbousset, R.; Schoenwaelder, S.M. Thromboinflammation: Challenges of therapeutically targeting coagulation and other host defense mechanisms. Blood 2019, 133, 906–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 2001634. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiuolo, J.; Mollace, R.; Gliozzi, M.; Musolino, V.; Carresi, C.; Paone, S.; Scicchitano, M.; Macrì, R.; Nucera, S.; Bosco, F.; et al. The Contribution of Endothelial Dysfunction in Systemic Injury Subsequent to SARS-Cov-2 Infection. Int. J. Mol. Sci. 2020, 21, 9309. https://doi.org/10.3390/ijms21239309
Maiuolo J, Mollace R, Gliozzi M, Musolino V, Carresi C, Paone S, Scicchitano M, Macrì R, Nucera S, Bosco F, et al. The Contribution of Endothelial Dysfunction in Systemic Injury Subsequent to SARS-Cov-2 Infection. International Journal of Molecular Sciences. 2020; 21(23):9309. https://doi.org/10.3390/ijms21239309
Chicago/Turabian StyleMaiuolo, Jessica, Rocco Mollace, Micaela Gliozzi, Vincenzo Musolino, Cristina Carresi, Sara Paone, Miriam Scicchitano, Roberta Macrì, Saverio Nucera, Francesca Bosco, and et al. 2020. "The Contribution of Endothelial Dysfunction in Systemic Injury Subsequent to SARS-Cov-2 Infection" International Journal of Molecular Sciences 21, no. 23: 9309. https://doi.org/10.3390/ijms21239309