1. Introduction
1.1. SARS-CoV-2 Sequencing History in Lithuania
The first case of SARS-CoV-2 in Lithuania was detected in February 2020. At that time, the sequencing of SARS-CoV-2 in Lithuania had not yet been performed, therefore the first samples (one collected in February and two in March 2020) were sequenced at Charite Universitaetsmedizin, Berlin, Institute of Virology (Germany) and published on the world’s largest repository of SARS-CoV-2 genomes—GISAID (Global Initiative for Sharing All Influenza Data) (GISAID ID: EPI _ISL_416741; EPI _ISL_450496; EPI _ISL_450497). The first instance of SARS-CoV-2 sequencing in Lithuania was performed by researchers at the Lithuanian University of Health Sciences (GISAID ID: EPI _ISP_536398). During the following year, additional sequencing was performed by various initiatives of researchers, and in 2020, a total of 641 analyzed sequences were added to the GISAID database from various Lithuanian laboratories.
1.2. Sequencing as a National Surveillance Program
Sequencing as part of a national programme was implemented in February 2021 [
1]. As a result, more than 30,000 SARS-CoV-2 cases were analysed, and their gene sequences were uploaded to the GISAID database in 2021 [
2]. The main body of the national sequencing programme is the National Public Health Surveillance Laboratory (NPHSL), which is under the Ministry of Health (MOH) of Lithuania and was established to conduct laboratory testing of air, water, and food, and the quality other product, as well as providing the laboratory testing required for healthcare of the general public at a national level. In response to the COVID-19 pandemic, NPHSL was requested to organise and coordinate testing and detection for SARS-CoV-2 variants using next generation sequencing (NGS) technology. Currently, the NPHSL coordinates SARS-CoV-2 WGS testing for four Lithuanian institutions that are capable to conduct sequencing analysis: two third level hospitals, Vilnius University Hospital—Santaros klinikos, and the Hospital of Lithuanian University of Health Sciences—Kauno klinikos, and two universities, the Vilnius University and Lithuanian University of Health Sciences [
1].
The NPHSL is responsible for providing methodological guidelines for sample collection, preparation and testing, logistics, data analysis, aggregation, reporting, and publication. In collaboration with the Ministry of Health, sequencing data reports produced by the NPHSL enable rapid response to epidemiological COVID-19 changes in the country and provides important information on SARS-CoV-2 variant (VoC) occurrence and transmission routes [
1].
Currently, SARS-CoV-2 WGS is performed weekly, with most samples selected at random, paired with a small percentage of samples with targeted selection based on epidemiology, at the four forementioned institutions with NGS capabilities. Depending on the number of positive COVID-19 cases in different regions of Lithuania, prepared SARS-CoV-2 RNA-positive samples are collected from laboratories and distributed to sequencing centres by the NPHSL logistics team. SARS-CoV-2 WGS coordinated by NPHSL accountant for the SARS-CoV-2 sequencing of human samples for surveillance purpose. No other public or private sectors are included in this program.
At NPHSL, the collected sequencing results are combined with patient metadata and screened for “Variants of Concern,” “Variants of Interest,” and other variants that might contain concerning mutations. The data are shared with the National Public Health Centre, which provides contact tracing and control of emerging SARS-CoV-2 variants. NPHSL is responsible for reporting WGS data to the Tessy and GISAID databases and uploading the results to WHO/ECDC and other international institutions. Based on the sequencing results, NPHSL provides recommendations for testing and tracking strategies of potential carrier contacts to the Lithuanian Ministry of Health. As the total sequencing capacity in the country is insufficient for new variant detection before it reaches 2.5 percent outspread, in addition to WGS coordination, NPHSL biologists prepared approximately 300–600 SARS-CoV-2 positive samples per week to be sent-out to the ECDC reference laboratory for WGS testing.
Currently, all four Lithuanian sequencing centres responsible for SARS-CoV-2 NGS library preparation, quality control, sequencing, and genome assembly are using the available low-scale equipment. All centres have different WGS equipment, library preparation protocols, and analysis algorithms. The data obtained are entered into the National Data Management system Palantir [
3] and then uploaded to the GISAID database. Once the appropriate virus variants are identified, the data appear in the Palantir system, where regular updates and notifications are available to local epidemiologists. Currently, NPHSL is developing a new national WGS sequencing infrastructure for COVID-19 and the post-covid era that will allow it to meet the needs of the country.
1.3. Sample Selection Strategy in Lithuania
The primary objective of the national sequencing program is to representatively sequence SARS-CoV-2 cases for genomic surveillance in addition to routine surveillance. The sample list for the study is generated semi-automatically by retrieving information on positive samples from electronic health records. Efforts are being made to include samples from all regions of the country (10 counties). Outbreaks in certain regions result in more samples being added to the sequencing list. The downside of this strategy is that some samples fall within the sampling frame about a week before they are collected, which negatively impacts the timeliness of sequencing. The main goal of sequencing is to detect a new variant with a 95% confidence interval for the proportion of a given circulating variant when its relative proportion reaches 2.5%. In accordance with ECDC recommendations, the weakly sample size is estimated to be approximately 800 randomly selected samples at a relative 50% precision [
4].
In addition to the general sample list, certain samples are included as targeted sequencing. The goal is for these samples to comprise no more than 10% of all samples selected for sequencing while conserving sequencing resources. In the study group, a proportion of samples are collected non-randomly in the context of epidemiological investigations, suspected viruses with altered epidemiological characteristics, etc. Only some of the most informative samples from a single focus are included (not the entire family, group, etc.). For targeted sequencing, the following high priority criteria are used: individuals at higher zoonotic risk (mink farmers, etc.) and immunosuppressed individuals with disease onset for more than 2 months. The medium priority criteria are as follows: reinfections; travel-related cases, especially from regions with a high positive case count; outbreaks and other epidemiological investigation cases, etc.
After receiving the lists of selected samples, PCR laboratories select samples with PCR value below 30 to assure the quality of the analysis, from randomly selected samples and <32 of targeted sequencing. NPHSL couriers collect the RNA samples on dry ice and distribute them to sequencing laboratories. The average number of samples sequenced per week is about 800, depending on the number of positive cases. The average turnaround time is two weeks from positive PCR to sequencing results, variating between one to four weeks depending on the laboratory.
In order to understand why SARS-CoV-2 caused a global pandemic so fast, and to recognize how such viral advancements could be prevented in the future, here we discuss how different SARS-CoV-2 variants spread from the start of the pandemic to the start of February 2022 in Lithuania. Furthermore, we investigate what the main causes of the vast viral transmission were and how it was affected by various international movements.
3. Results
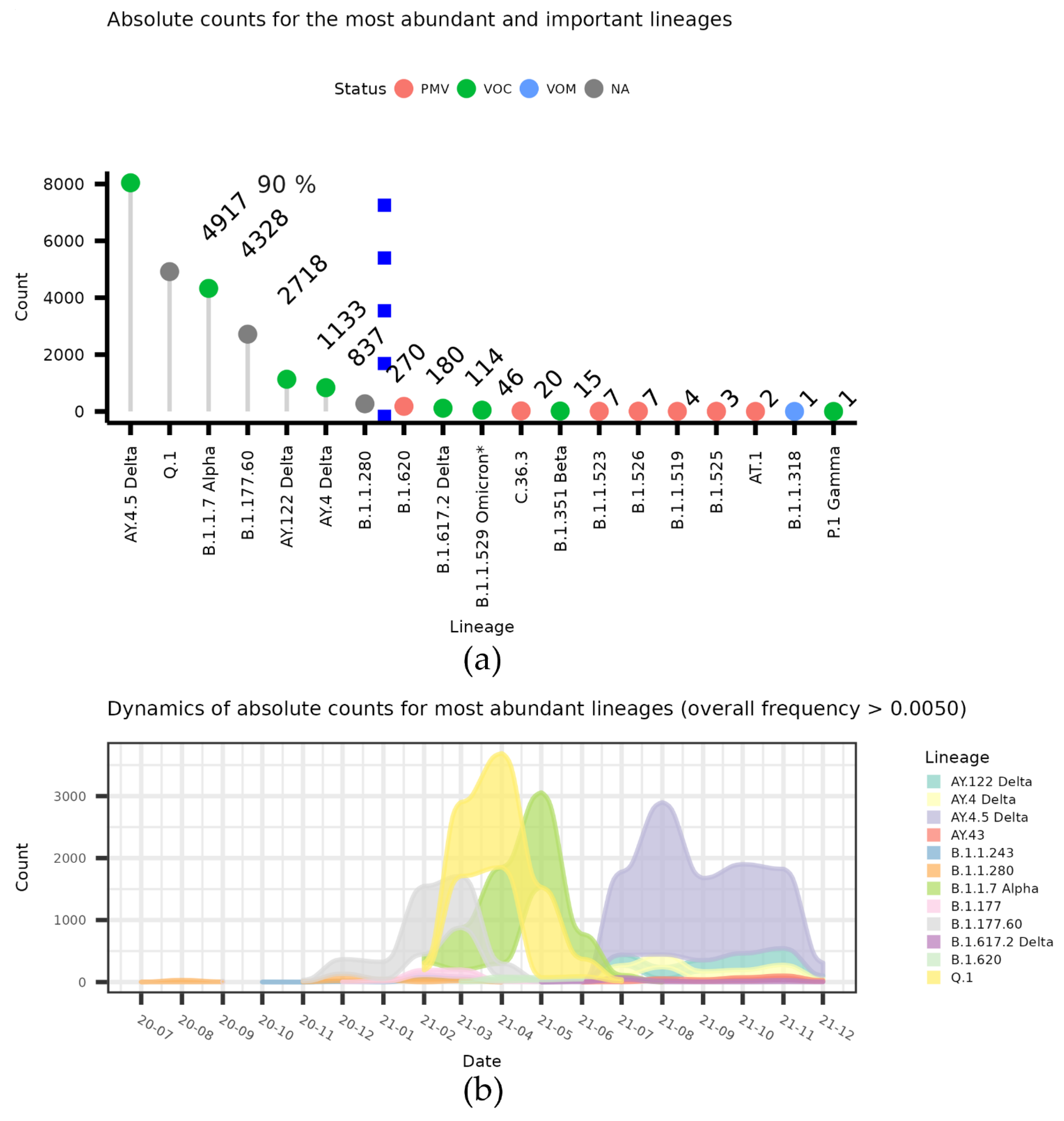
The dynamics of the lineages that make up at least 0.5 counts for the top strains that make up 90% of all Lithuanian sequences in GISAID data, together with other potentially important lineages, are indicated in
Figure 1a. The month-wise dynamics of the lineages that make up at least of 0.5% of all Lithuanian sequences in the GISAID data are depicted in the alluvium plots in
Figure 1b.
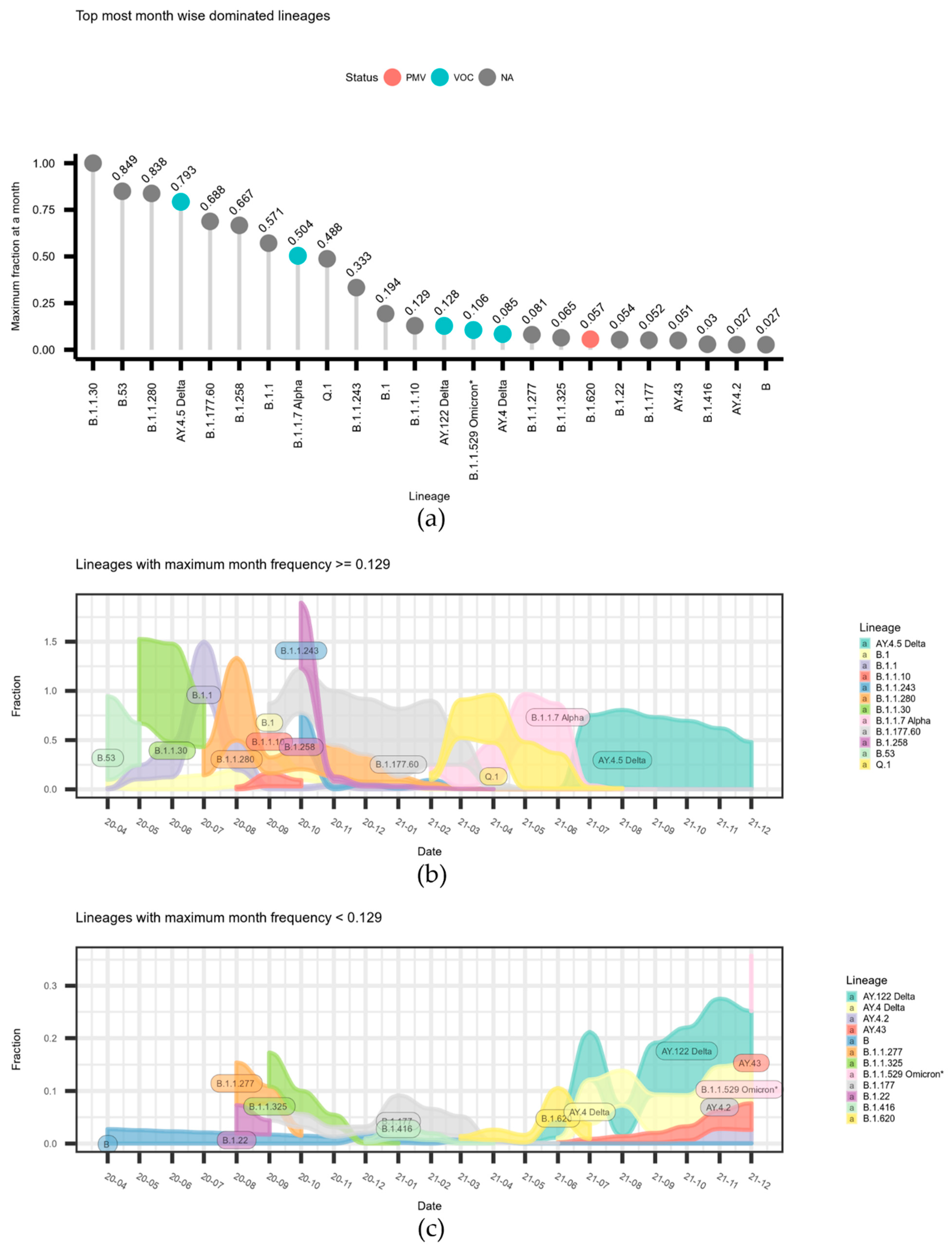
The data on relative month-wise frequency are given in
Figure 2. The data on the maximum monthly frequencies per lineages are given in
Figure 2a. The data on correspondingly month-wise dynamics are divided in two parts, for the most frequent and less frequent lineages, and are depicted in the
Figure 2a,b alluvium plots.
As we see in terms of the absolute numbers of COVID-19 spread (
Figure 1), Lithuania was dominated by several Lineages (most notably A.Y.4.5 and Q.1). However, until the autumn of 2020, there was no clear domination of one Lineage (
Figure 1a and
Figure 2a), and we were then faced with two clear waves: by the end of 2020, B.1.177.60 started a significant spread, and was then overtaken by B.1.1.7 and its localized version Q.1. Both slowly diminished, then a spike in numbers was caused by a Lithuanian Delta variant, A.Y.4.5. As we can see in
Figure 2a, the Delta variant A.Y.4.5 dominated over other lineages by the end of June 2021 and was further replaced by other Delta subtypes (
Figure 2c).
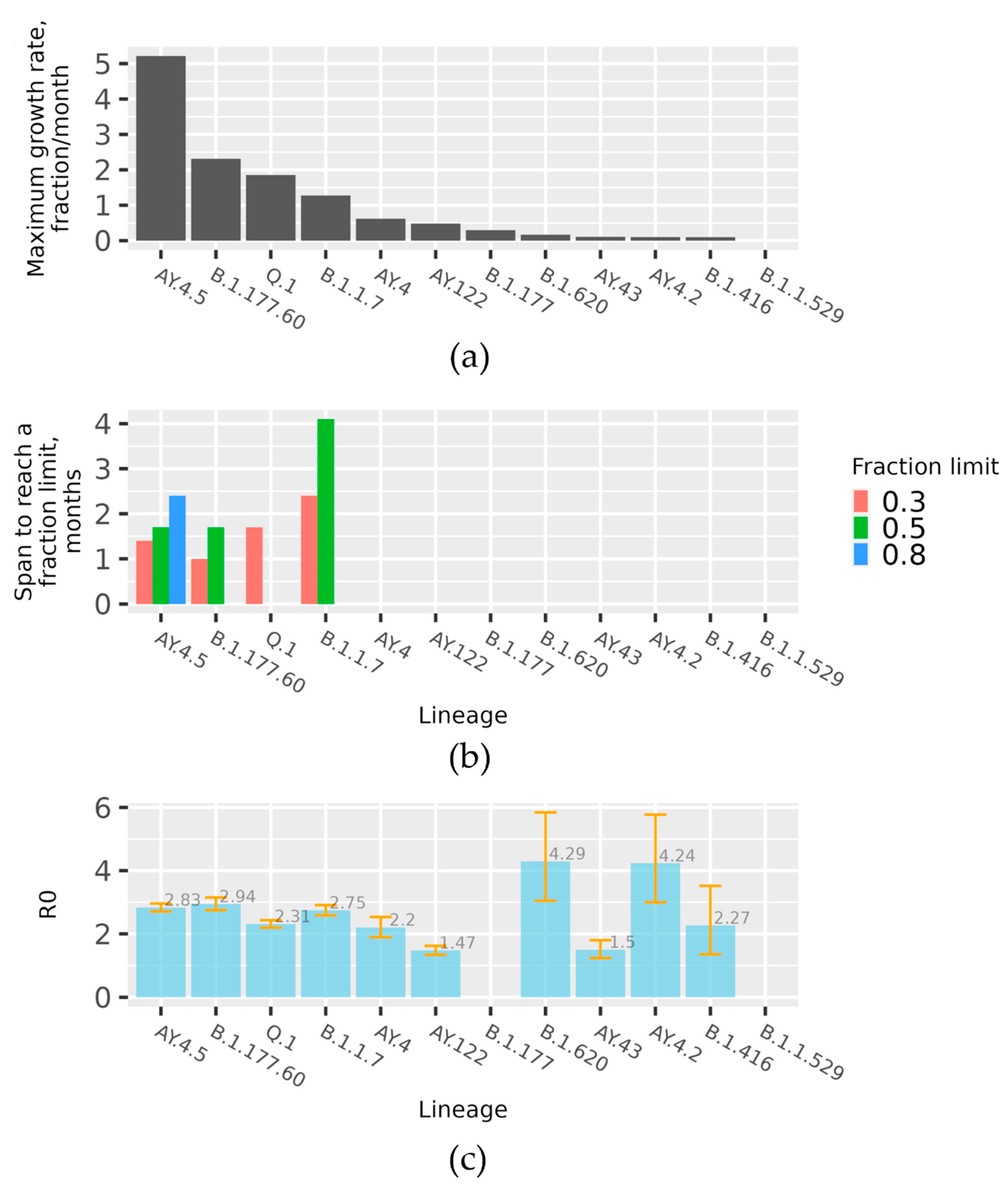
The data on the pace of spread for the lineages are represented in three aspects. The data on initial expansion rate of the month-wise fraction increase per lineages are given in
Figure 3a. The corresponding modelled R0 values based on absolute counts are given in
Figure 3c. Several lineages managed to reach a month-wise fraction of over 30%. The data on the time over which such lineages reached a certain month-wise fraction are given in
Figure 3b. As we see in
Figure 3a, the fastest observed spread was in the case of AY.4.5 and B.1.177.60 lineages, followed by Q.1. and B.1.17. Only one lineage, A.Y.4.5, managed to reach an 80% month-wise fraction (due to the limited study interval, data with Omicron Spread in early 2022 are not included), B.1.1.7 and B.1.177.60 reached 50% month-wise fraction, and Q.1 reached 30%.
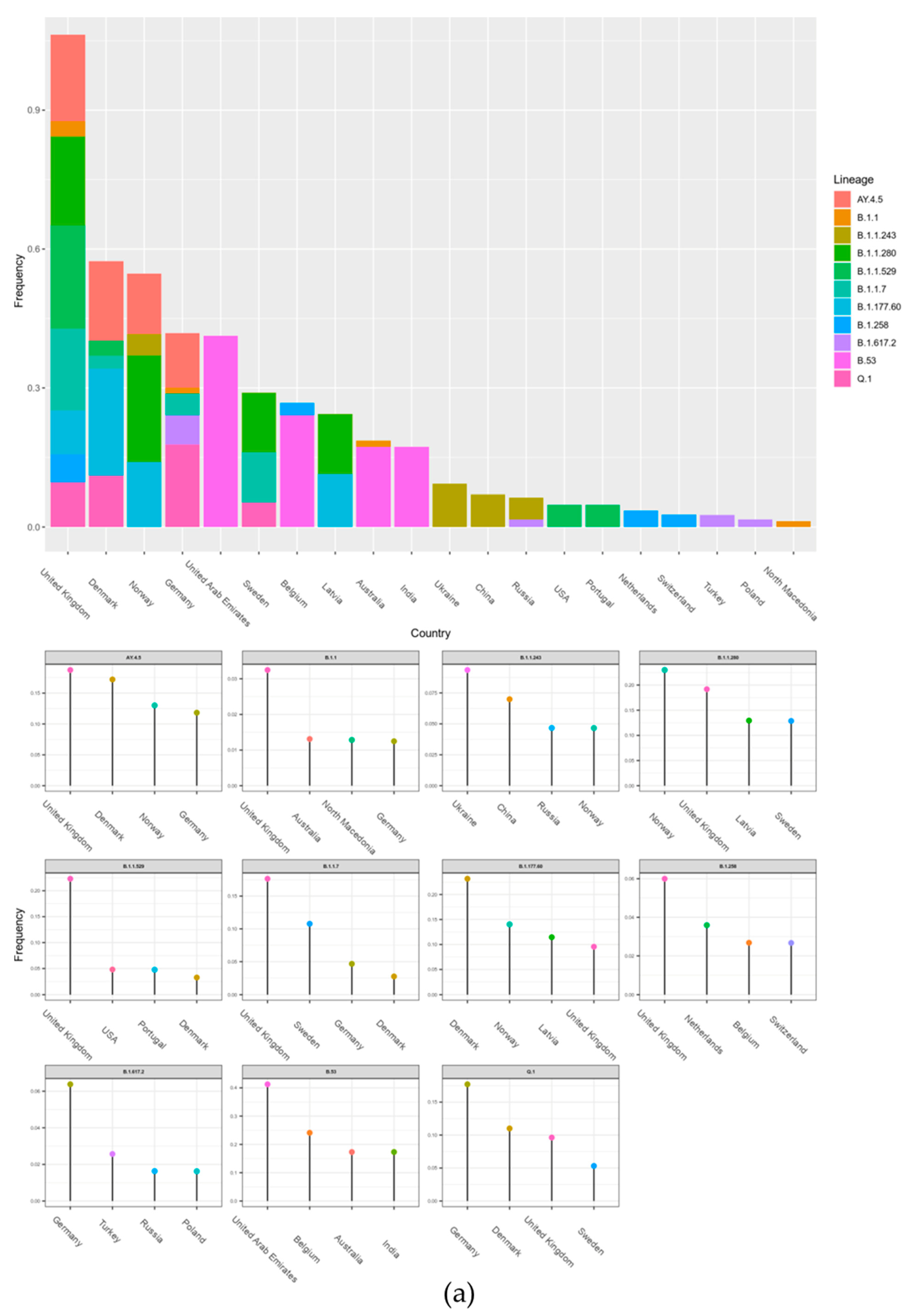
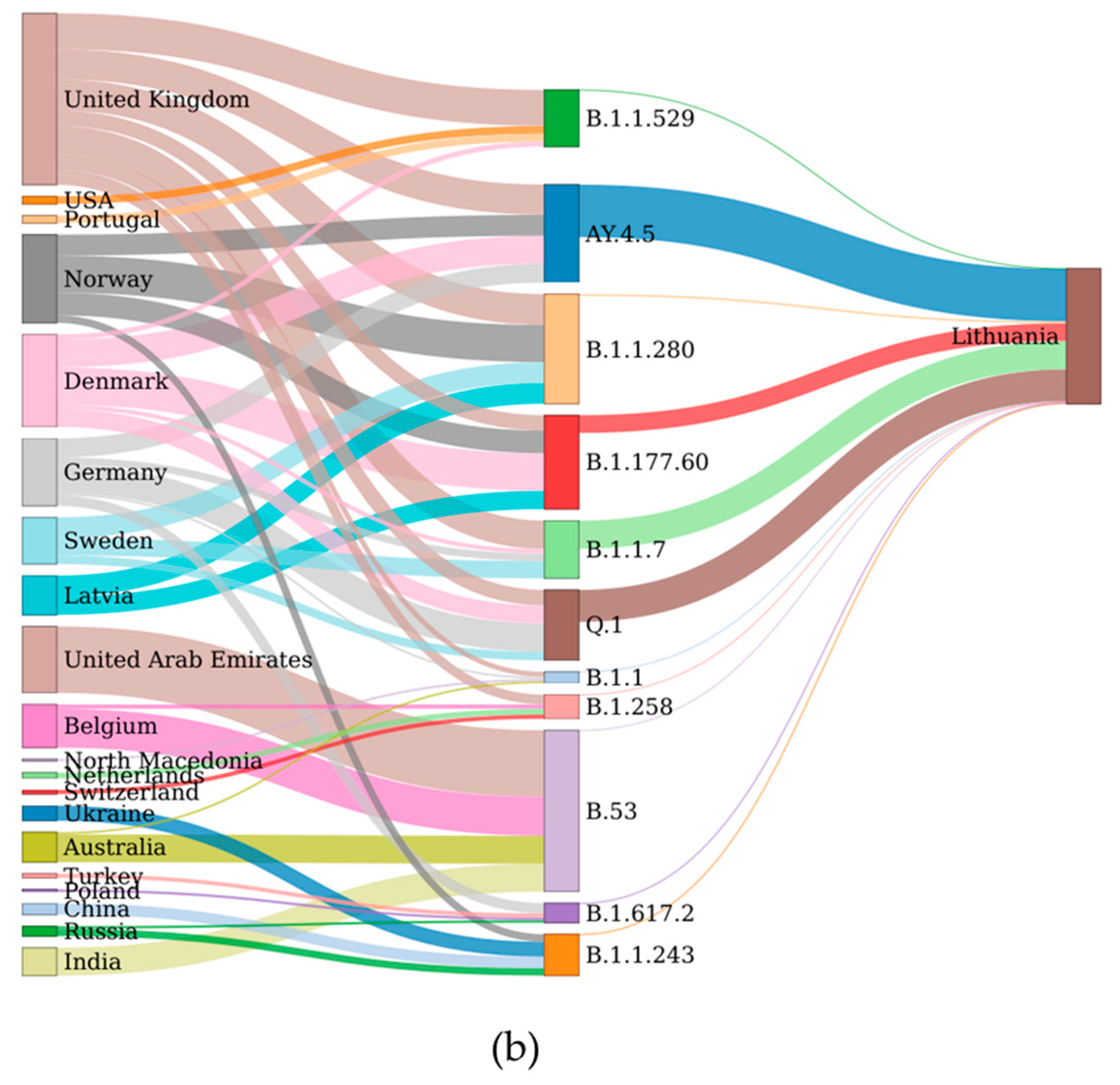
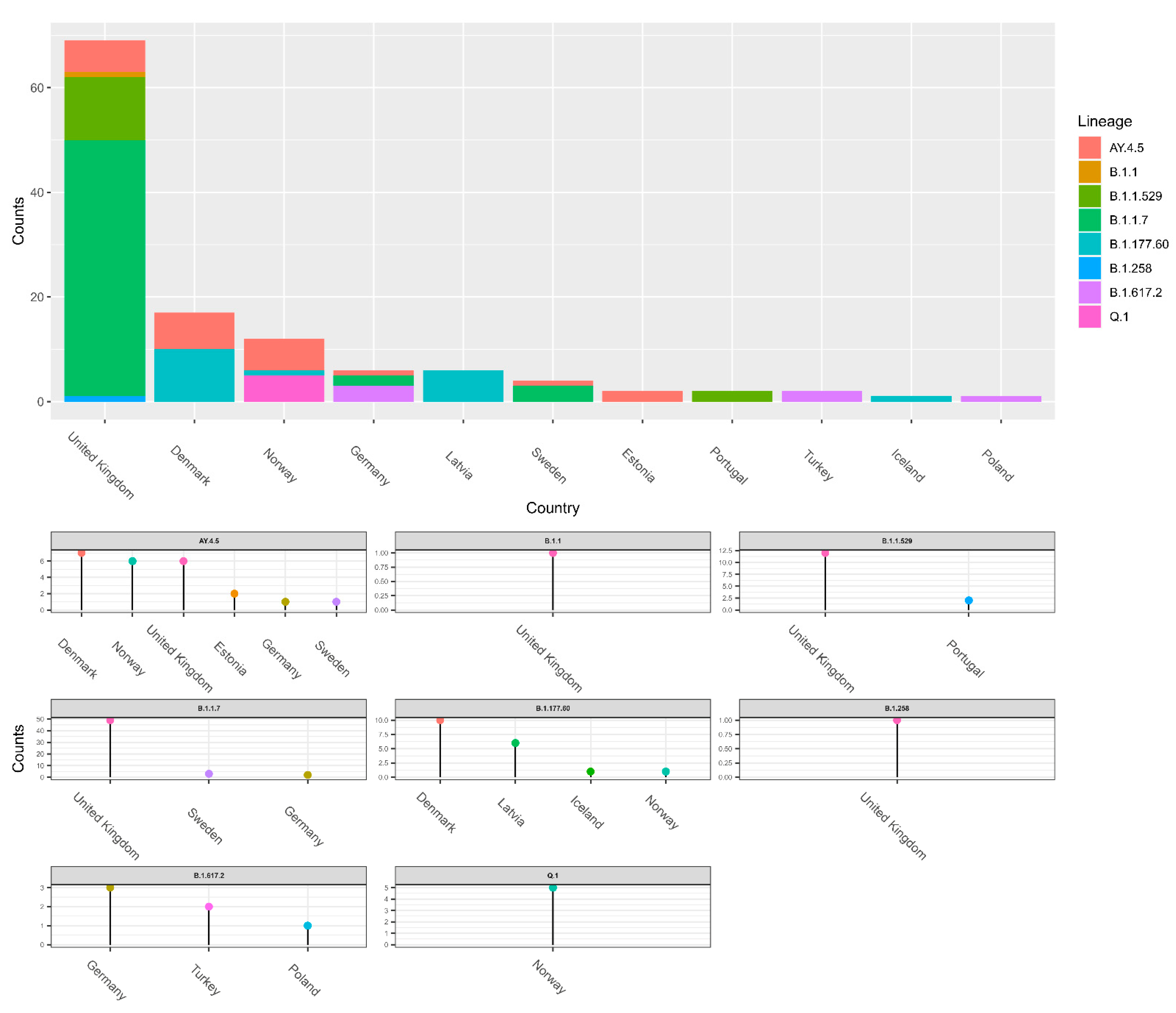
The data of potential transmission to Lithuania were evaluated in two ways. The results on the transmission cluster-based analysis are given in
Figure A1, and the results based on the TreeTime are given in
Figure 4. In both cases, the upper part of the figures represents aggregated data per country and the smaller graphs give details per different lineages. Additionally, a schematic view of the data shown in
Figure 4a is represented in
Figure 4b. As we see in
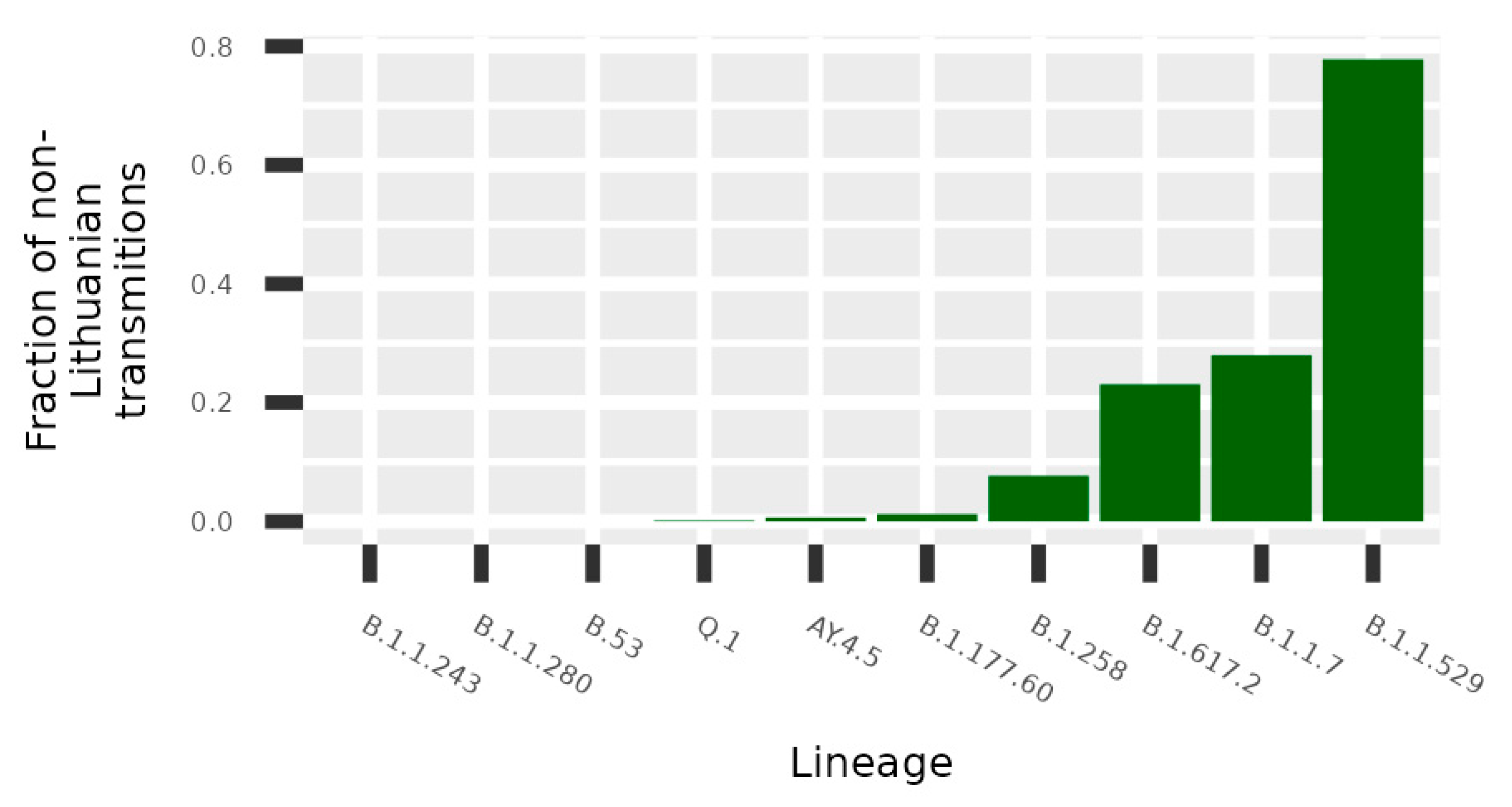
Figure 4, the United Kingdom, Denmark, and Norway are notable for the highest transmission intensities to Lithuania; however, the data indicate that some lineages originated from other countries: B.1.1.243 (Ukraine), B.53 (United Arab Emirates), and Q.1 and B.1.617.2 (Germany). Only the analysis based on transmission clusters enabled direct evaluation on rate of non-Lithuanian origin transmissions compared to local origin transmission (
Figure 5). The data indicate that the B.1.1.529, B.1.17, B.1.617.2, and B.1.258 lineages stand out as lineages with the highest fraction of potential foreign origin transmissions (listed in decreasing order).
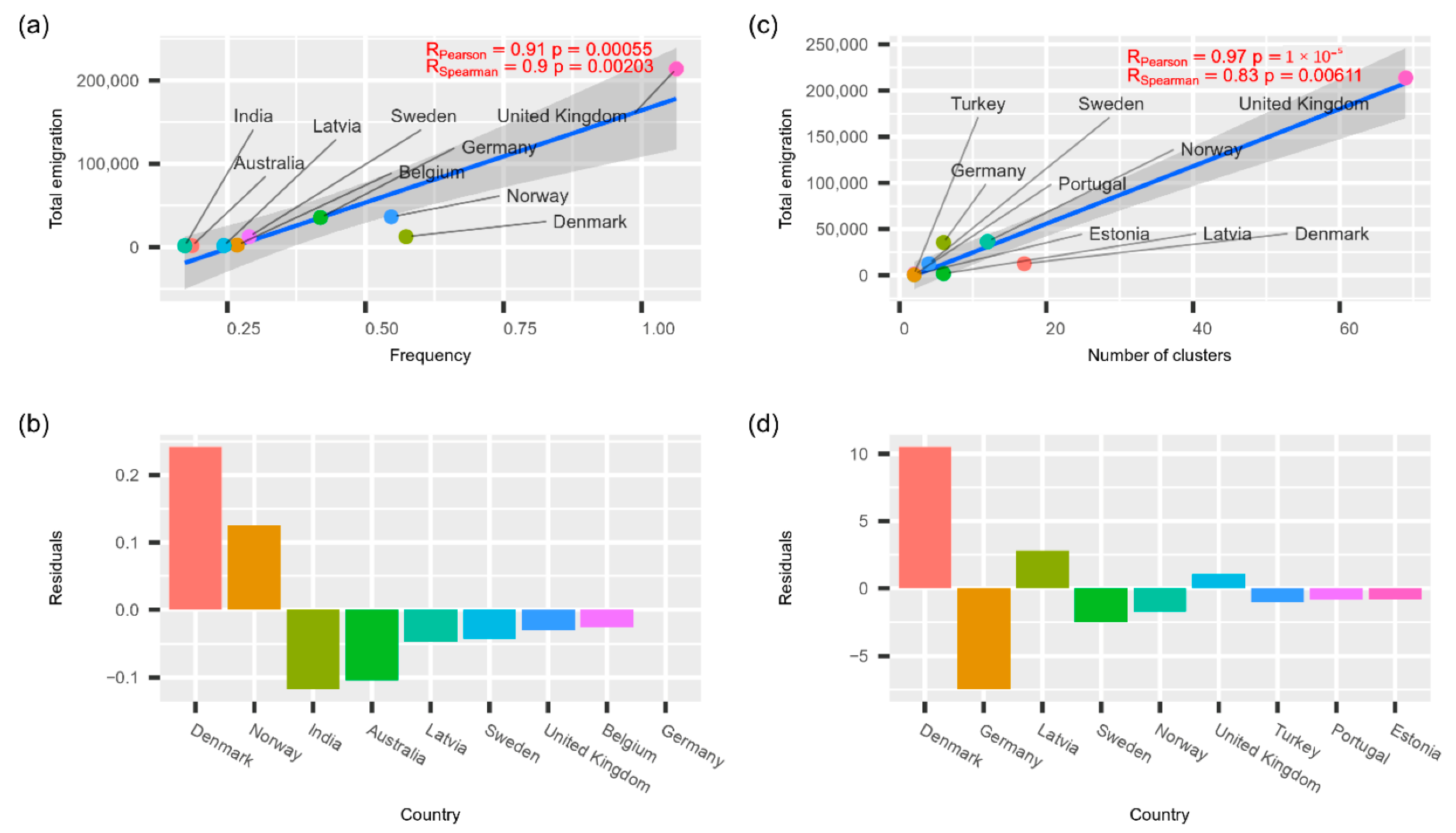
The results of correlational analysis between known emigration data and transmissions to Lithuania are given in
Figure 6. As we see in the figure, phylogeographical estimates of foreign transmission intensities are directly proportional to the total emigration intensity from Lithuania within the last ten years, and corresponding Pearson and Spearman correlation estimates are statistically significant. It is worth noticing that, based on the residuals of the linear models, Denmark and, to a lesser extent, Norway stand out as having higher transmission intensity to Lithuania than expected. The higher-than-expected transmission intensity from Germany is evident despite the way we analysed phylogeographic transmissions. However, higher-than-expected transmissions from Norway are only evident using transmission cluster-based estimates. Correspondingly, India and Australia are notable for lower-than-expected transmission intensities (based on the TreeTime GTR model), along with Germany (based on transmission cluster estimates).
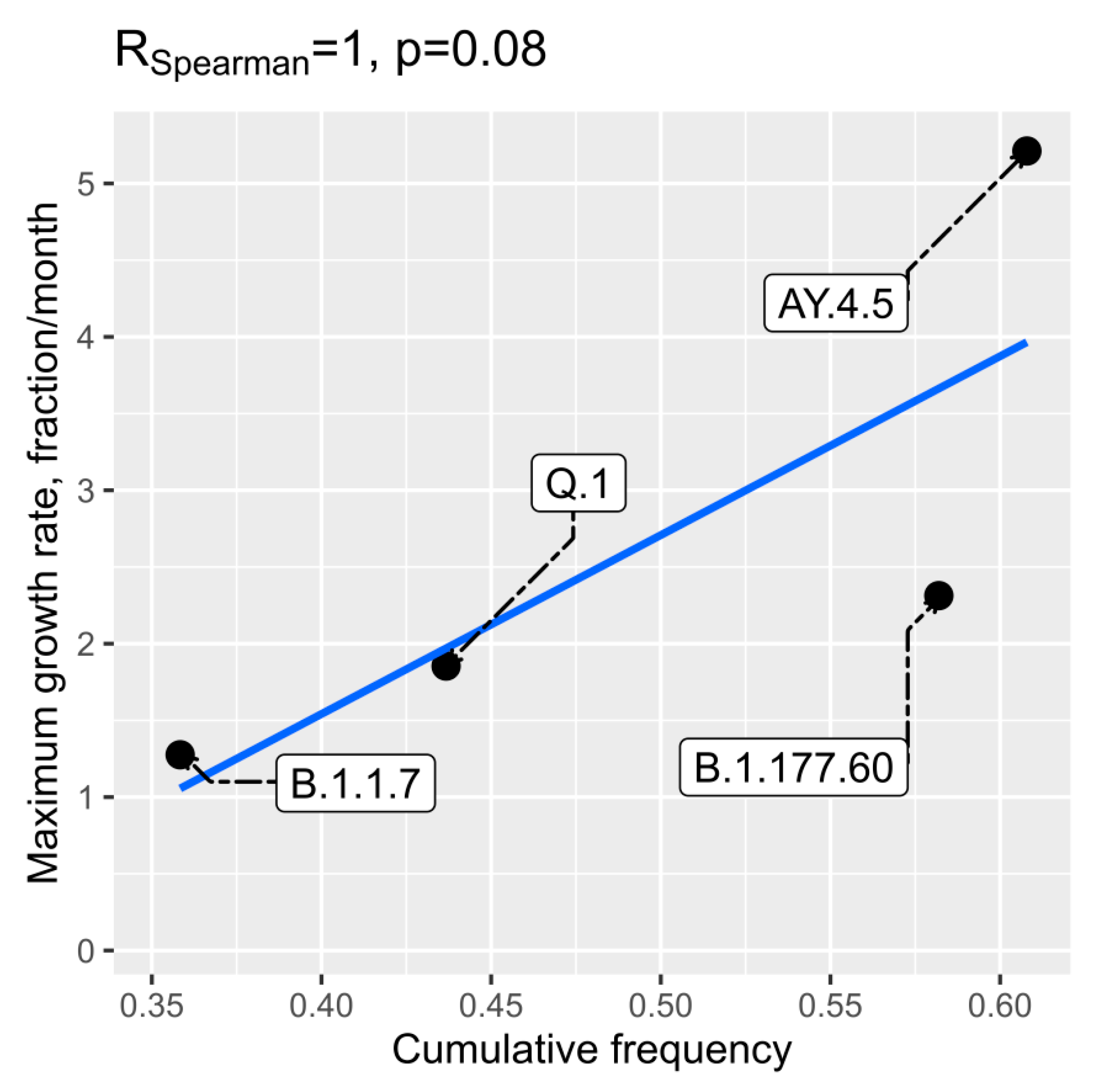
We also checked if higher evaluated R0 values or initial spread dynamics could be related to higher transmission intensities from abroad to Lithuania. Pearson and Spearman correlations between frequency of transmissions to Lithuania and R0 were low and statistically insignificant. However, we detected a weak correlation between initial maximum growth rate and frequency of transmissions to Lithuania as inferred from the time-scaled phylogenetic trees. The data on both of these two aspects were available only for four lineages and are given in
Figure 7. The P value for the Spearman correlation was 0.08 (R = 1).
4. Discussion
During the pandemic’s first year (2020), several viral lineages were distinguished and were all prevalent at the same time. By the end of the second year of the pandemic (2021), the viral advancement pattern had changes, showing one or more dominant lineages, instead of the high variety noted previously.
The first SARS-CoV-2 genomes that were sequenced in Germany (n = 3) had a very similar gene sequence to the Wuhan-hu-1 [
19] lineage, differing by an average of 4–5 nucleotides [
2,
20]. It is noteworthy that in one of the first cases in Lithuania that were sequenced, a strain with the S:D614 mutation was detected, which was later confirmed to have higher infectivity and promote a faster spread [
21,
22].
In April 2020, the first B.53 virus lineages were identified in several samples collected and analysed in Lithuania. This lineage was first detected in February 2020 in the United Arab Emirates and is characterized by S:Y28H, ORF:8S84L, and N:P344S mutations [
23]. According to GISAID data (February 2022), a total of 84 cases were registered worldwide, of which 73.0% were in Lithuania, 24.0% in the United Arab Emirates, 1.0% in India, 1.0% in Belgium, and 1.0% in Australia [
2,
23]. It is likely that this lineage was responsible for the first major outbreak that began to be monitored in the second half of March 2020. Due to the low amounts of sampling, it is difficult to evaluate the prevalence of this viral lineage; however, we have observed a wide spread of B.53 at the beginning of the pandemic. In the following spring and throughout the summer, two other Lithuanian SARS-CoV-2 lineages (B.1.1.30 and B.1.1) started to spread.
The second widely viral lineage was B.1.1.30, which spread during the period 18 May 2020–3 June 2020. All 13 cases of this lineage were registered in Lithuania, Vilnius district. With a gradual decrease in the number of cases with this lineage, Pango, classified as B.1.1, which is characterized by mutations in S:D614G, N:G204R, N:R203K, NSP2:T332I, and NSP12:P323L, spread at a similar time. It comes as no surprise that both the B.1.1.30 and B.1.1 lineages’ most intensive spread to Lithuania was from the United Kingdom (
Figure 4), which is the country that hosts most Lithuanian emigrants. The outbreak of this lineage was replaced by the vast spread of B.1.1.280. The viral lineage, which has been present in Lithuania for almost a year, was observed from 30 July 2020 to 12 July 2021, with the percentage of cases being 92.0% in Lithuania, 3.0% in Norway, and about 1.0% in Germany, the United Kingdom, and Latvia [
23]. These figures agree with the data in
Figure 4 indicating that the most intensive spread of this lineage to Lithuania was from Norway. This lineage was not related to the changes in the biological characteristics of the virus; therefore, it is likely that the epidemiological factors, such as long-term spread, were the main cause of the wide spread of this strain, rather than the altered viral characteristics.
The increase in variety of lineages could be related to summer holidays or similar travels. The first more-pronounced mutation of a single virus line was observed in late autumn, when the number of COVID-19 cases began to rise rapidly. The prevailing Lithuanian virus lineages resulted from the Spain-originated lineage B.1.177, which at that time was the dominating lineage in Europe. Currently, there is limited evidence of increased transmissibility or severity of the infections associated with the B.1.177 lineage [
24]. The fact that B.1.177 was defined by the S:A222V mutation in the Spike protein could also confer a significant advantage to the virus infectivity, favouring its rapid selection and dissemination. B.1.177.60 was the first lineage to over-dominate others, and in September of 2020 this lineage was observed in Lithuania for the first time. There is no evidence of any appreciable effect of A222V on the phenotype of the virus (i.e., infectivity and transmissibility), and thus it is generally assumed that its increase in abundance was random rather than a selection advantage [
25]. As we can see in
Figure 3, this lineage is notable for very fast initial spread rate—it is next to AY.4.5 (Delta sub-lineage), and within a little more than one month and a half this lineage reached 50% of all month-wise sequenced cases. Throughout the propagation period, eight sublines were identified, of which the most spread-out was the Lithuanian subline B.1.177.60, which had appeared quickly after the emergence of B.1.177, which had its last cases in Lithuania observed on 21st of November 2021. As we can see in
Figure 4, this lineage spread to Lithuania is linked with Nordic countries (Denmark, Norway). Since the lineage was discovered, n = 5162 sequences were produced from sampled cases worldwide. The mutations present in this sub-lineages spike protein were S:L18F and ORF3a:V77F, in addition to the conventional B.1.177 mutations ORF1b-P314L, S:A222V, S:D614G, ORF8:S84L, ORF10:A220V, and ORF10:V30L. Although B.1.177 has been introduced to Lithuania many times, from the profile of hereditary mutations we can say that the vast majority of cases caused by the currently circulating B.1.177 lineage originated from several larger outbreaks. It is the first virus lineage of Lithuanian origin that is widely distributed abroad (mainly in Northern Europe), with the total percentage of cases being 57.0% in Lithuania, 14.0% in Latvia, 12.0% in Denmark, 6.0% in Finland, and 3.0% in Estonia [
23]. Thus, the trend of local spread remained the same. In 2021, in February, the peak of this lineage was reached, and with the introduction of the first variant of concern (VOC) B.1.1.7, also known as Alpha, it began to advance this over-dominated lineage.
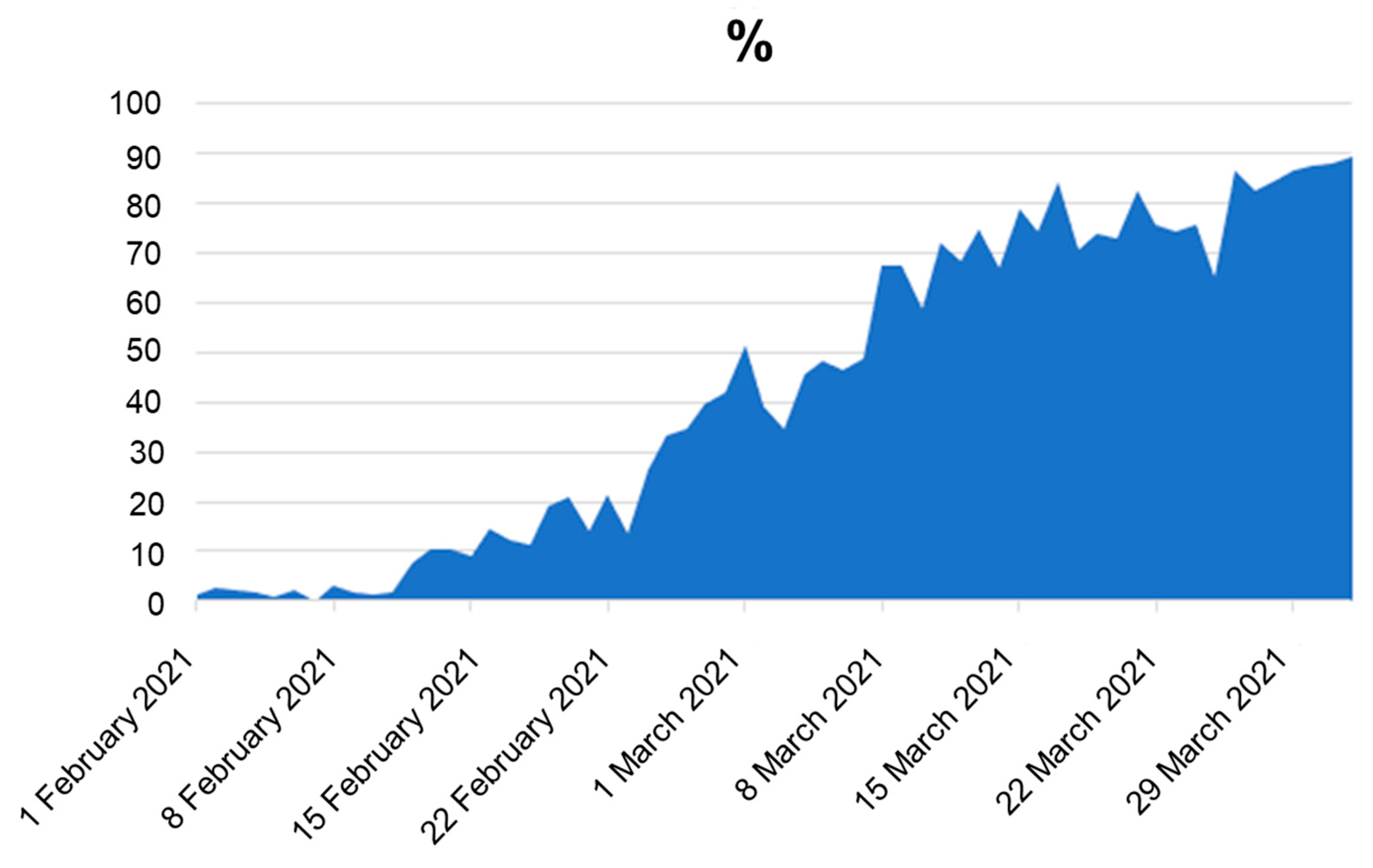
B.1.1.7 (Alpha variant) was first identified in the United Kingdom in late 2020 and was temporally associated with an increase in regional infections, and the spread to Lithuania is also linked with the United Kingdom. This variant contains more than a dozen mutations when compared with other circulating lineages, having several of them within the spike protein. Subsequently, it became the predominant variant in many countries, until the emergence of the Delta variant. In Lithuania, since the first case of B.1.1.7, a very rapid spread has been observed, with the number of sequenced samples of B.1.1.7 doubling in amount every two week in Lithuania, while the Alpha variant was distinguishable based on the failure to amplify the S gene fragment (S-dropout), caused by S:H69-70del [
26] (
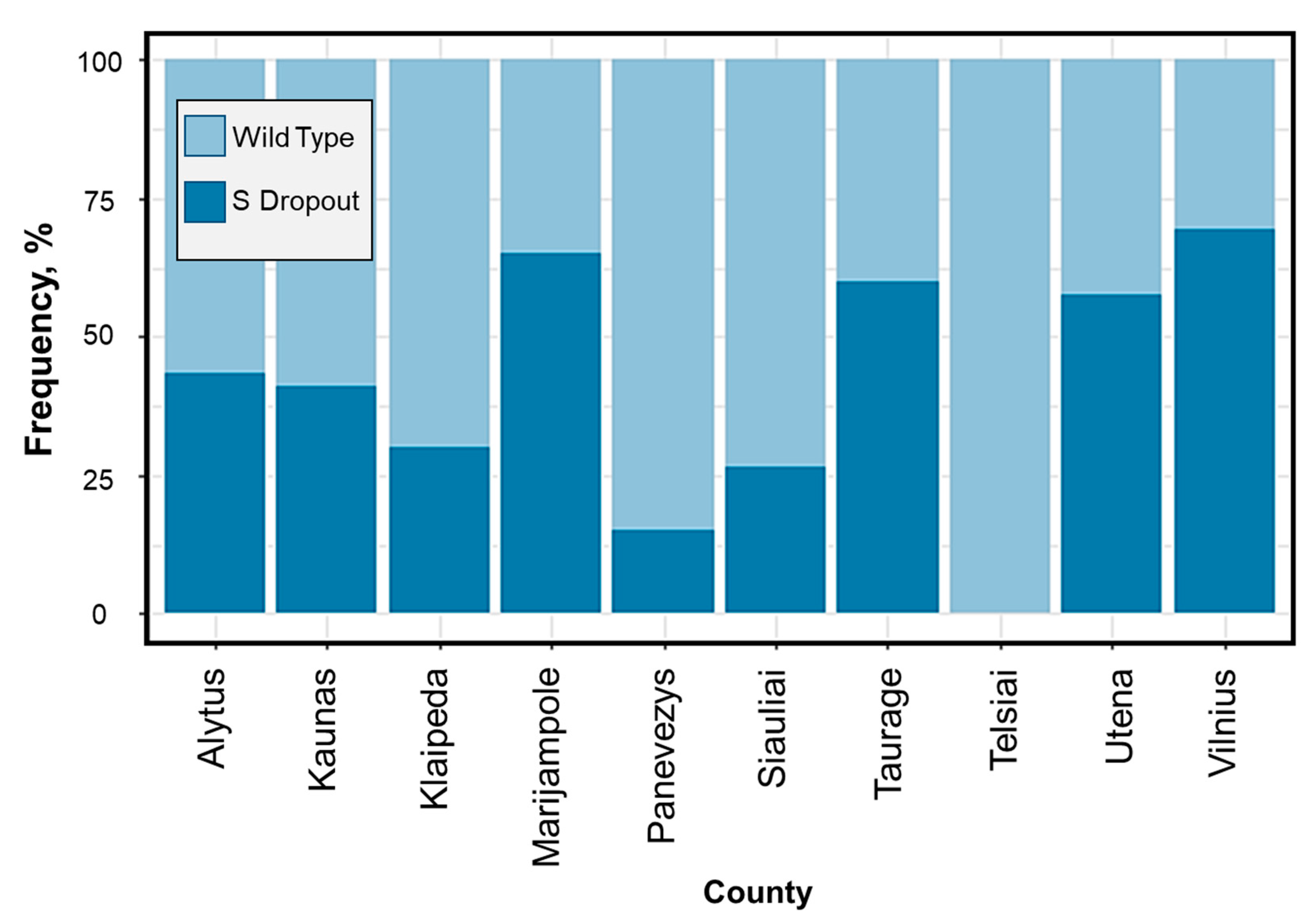
Figure A2). Due to the delay of sequencing results and being aware of the new VOC, the Lithuanian government decided to take a “snapshot” of detecting B.1.1.7 distribution across the country by collecting and analysing all samples at a high priority rate. According to the results of the research, it was observed that, in Vilnius, Utena, and Marijampole counties, this variant accounted for 70% or more of new cases at that time, and in the other part of Lithuania its prevalence was at least 25% (
Figure A3). According to the Lithuanian department of statistics, the regions of Lithuania where the B.1.1.7 virus lineage was predominating led in terms of both cases of illness and hospitalization in the country. At that time, infections with COVID-19 in Vilnius County accounted for 54% of all cases in Lithuania, and the number of cases diagnosed doubled since February. Thus, it is clear that this lineage has contributed significantly to the increase in the number of local infections. The situation was the opposite in western Lithuania, and it is likely that such regional differences were due to restrictions on movement between municipalities. Q.1 (Lithuanian sub-lineage) was pushed out of the country not long after its parent B.1.1.7, which spread rapidly due to frequent internal outbreaks. Q.1 is a local version of B.1.1.7 n. This lineage circulated and dominated before other lineages until the appearance of Delta in June 2021, which, one month later, accounted for 95% of all sequenced samples.
The Delta lineage accounted for more than 90% of all virus lineage detections for 6 months. During this period, AY.4, AY.122, AY.111, and AY.36—different delta sub-lineages—were the most observed, of which variants of sub-lineage AY.4 accounted for the largest percentage during the whole period. The Delta AY.4.5 local lineage out-dominated other Delta sub-lineages. In contrast to the case with Q.1 and its parental lineages, AY.4.5 differs from its predecessor lineages by a mutation in the S protein, S:D1259Y. Even though this mutation has not been fully characterized, there are possibilities that it provided infectivity advantages for the virus [
27]. This could be supported by a slightly higher R0 value compared to A.Y.4. However, after its peak, the fraction of AY.4.5 cases started slowly decreasing, while other Delta sub-lineages started to increase. Hypothetically, if the Omicron variant would have never arisen, we would have different Delta sub-lineages coexisting at a similar measure.
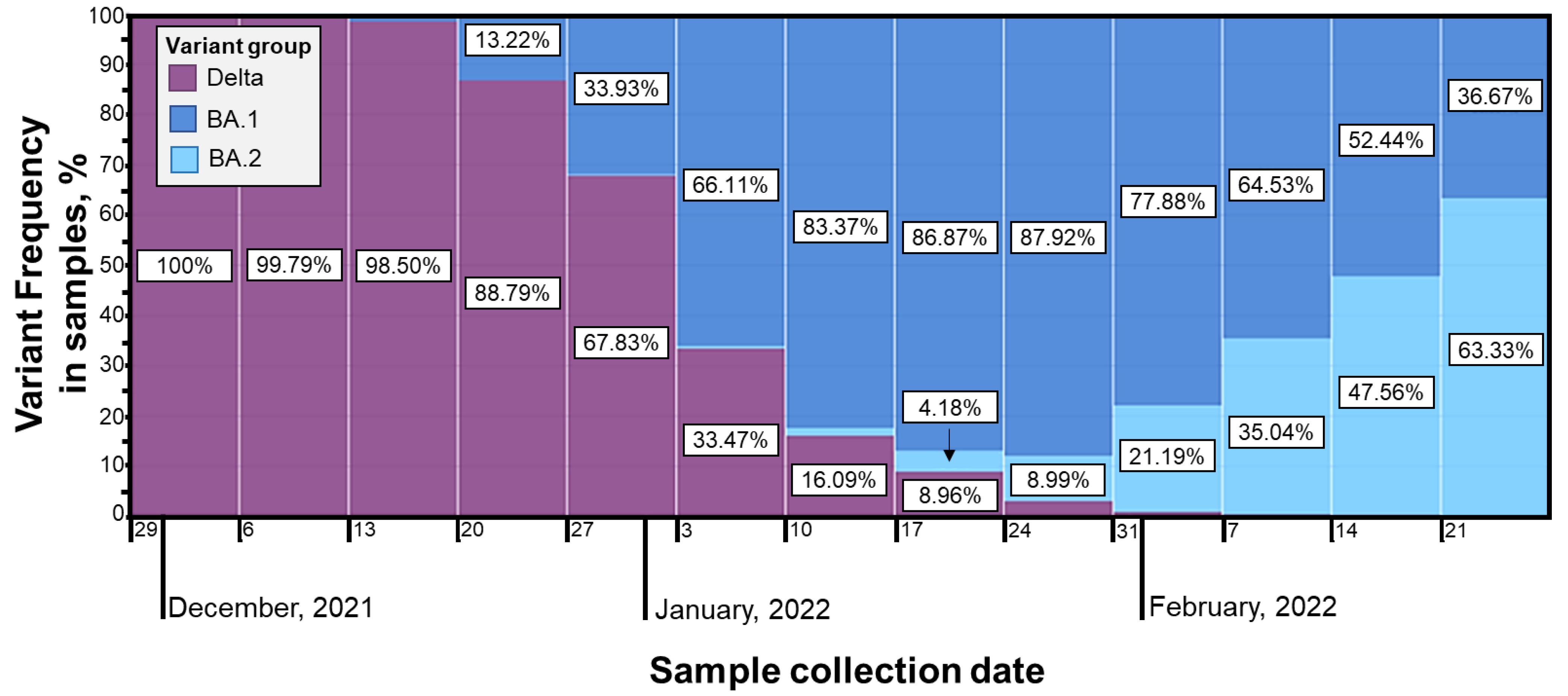
Omicron was first identified in December 2021 and quickly became the dominant strain. The BA.1 Omicron subtype was the fastest growing in Lithuania, following with a vast increase in BA.2, observed at the end of January and increasing by about 10% each week, leading to rapid over-domination [
28] (
Figure A4).
Many lineages of SARS-CoV-2 imported to Lithuania from abroad did not spread throughout the country, as shown in
Figure 2 and
Figure 5; Q.1 and its paternal lineage B.1.1.7 being the exceptions. At the beginning of its arrival/detection, Q.1 rapidly started to spread, and the number of cases grew even faster than the parental Alpha variant (
Figure 3). However, it was noted that B.1.1.7 became more prevalent and replaced the previously dominating Q.1 sub-lineage. It is highly likely that the lifting of certain pandemic and travel restrictions in the country is the reason behind the spread. Therefore, we can conclude that travel restrictions have a considerable influence on the spread of various SARS-CoV-2 sub-variants. However, it is worth mentioning that it has a minimal effect on the viral sub-lineages that were imported the most, such as B.1.1.529, B.1.617.2, and B.1.258, due to the fact that they did not contain distinguished traits that could increase their ability to spread. At the time the Delta variant became present in Lithuania, the AY.4.5 sub-lineage started to spread rapidly, influenced by large local outbreaks; however, since July the number of cases has declined compared to other sub-lineages. AY.4.5 is unlikely to have a significant biological advantage, with additional mutations in S:E156G and S:D1259Y in the SARS-CoV-2 genome. Large initial growth at the beginning of the lineages presence was determined as a result of the founder effect and other Delta mutations.
While analysing the variety of lineages, we noticed that increased traveling during the summer holidays and the easing of quarantine restrictions affected the diversity of the different lineages. We decided to verify this hypothesis by comparing the number of emigrants in the country with countries from which different virus lineages were usually imported by relying on the assumption that the countries that most Lithuanian people have emigrated to would import the highest number of different lineages. This notion was confirmed, and we discovered that the amount of emigration significantly correlated with the amount of different lineages present. However, Denmark was an exception, as emigration to this country from Lithuania is rather small, but the number of observed transmission clusters was rather high. In the present study, we did not investigate this further; however, this instance could be related to the pandemic restrictions and their early easing in Denmark [
29].
Lithuania and other countries that recently joined the EU have high emigration rates. We could expect that countries with the most abundant Lithuanian diaspora could be the most prominent sources of COVID-19 transmissions to Lithuania. The data in
Figure 4 suggest that this presumption is correct. Both methods used to detect international spread showed that the intensity of spread to Lithuania is directly proportional to the number of Lithuanian immigrants.
It is worth noticing that Denmark-related transmissions to Lithuania are more significant compared to what you could expect based on the size of Lithuanian diaspora. This could be related to the fact that Denmark was the first European country to remove COVID-19-related restrictions [
30]. On the other hand, countries such as India and Australia contributed to transmissions to Lithuania less than expected. This could be due to much higher distance and more restricted travel arrangements to these countries compared to other EU member.
We also hypothesized that higher evaluated R0 values or the characteristics of initial spread dynamics could be related to higher transmission intensities from abroad to Lithuania. However, the analysed data allowed us to detect only weak positive potential correlation between maximum infection growth rate and frequency of transmissions to Lithuania (
Figure 7). The relationship could be logical, as a more intensive transmission to Lithuania could result in a bigger number of initial spreaders.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











