
Demystifying Excess Immune Response in COVID-19 to Reposition an Orphan Drug for Down-Regulation of NF-κB: A Systematic Review


 ,
,  ,
,  and
and
Abstract
:1. Introduction
1.1. Normal Response of the Human Immune System to SARS-CoV-2 Infection
1.2. Need for Reviewing Excess Immune Response of Human Immune System to SARS-CoV-2 Infection in this Pandemic Situation
2. Materials and Methods
2.1. Search Strategy
2.2. Selection of Studies
2.2.1. Inclusion Criteria
- The articles describing immunology in COVID-19 patients especially in the context of excess immune response;
- Articles pertaining to adult population showing excess immune response in COVID-19.
2.2.2. Exclusion Criteria
- Articles related to drug repositioning, therapeutics, target drugs, therapies, treatments, and vaccines;
- Articles focusing on a sub-group of patients suffering from a particular co-morbidity;
- Articles with study design as clinical trial.
2.3. Data Extraction
2.4. Assessment of Risk of Bias and Quality of Studies
2.5. Strategy for Data Synthesis
2.6. Primary Outcome
3. Results
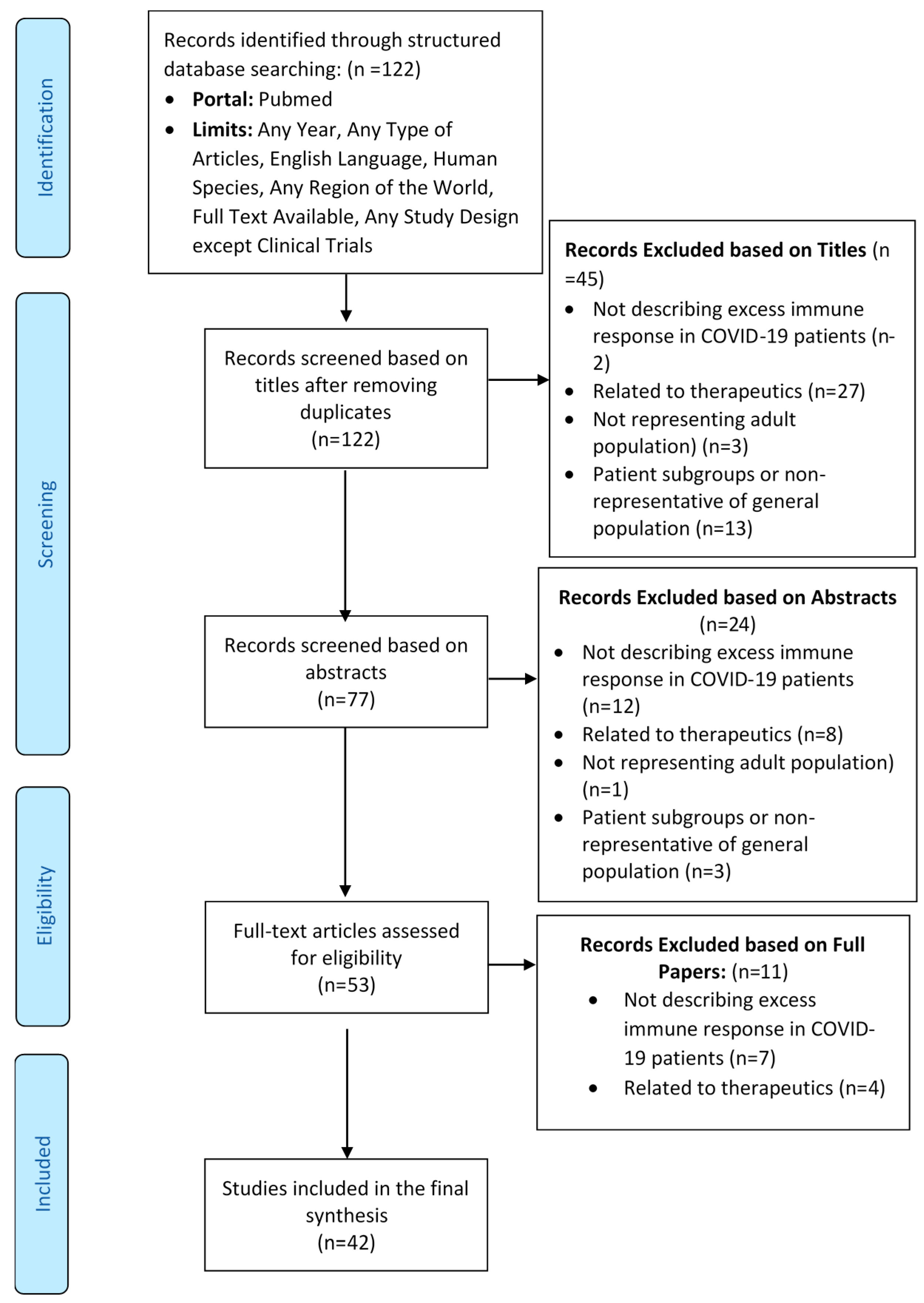
3.1. Flow of Studies through the Review Process
3.2. Mortality Rate due to Excess Immune Response in COVID-19
3.3. Reporting of Mechanisms of Excess Immune Response
4. Discussion
4.1. Mechanisms of Excess Immune Response of the Human Immune System due to SARS-CoV-2
4.1.1. Extracellular Mechanisms
Cytokine Storm and Related Mechanisms
Hyperferritinemia
MAS and sHLH
Neutrophil-Related Mechanisms
Immunothrombosis
Other Mechanisms
4.1.2. Intra-Cellular Mechanisms
4.1.3. Signaling Pathways in an Excess Immune Response
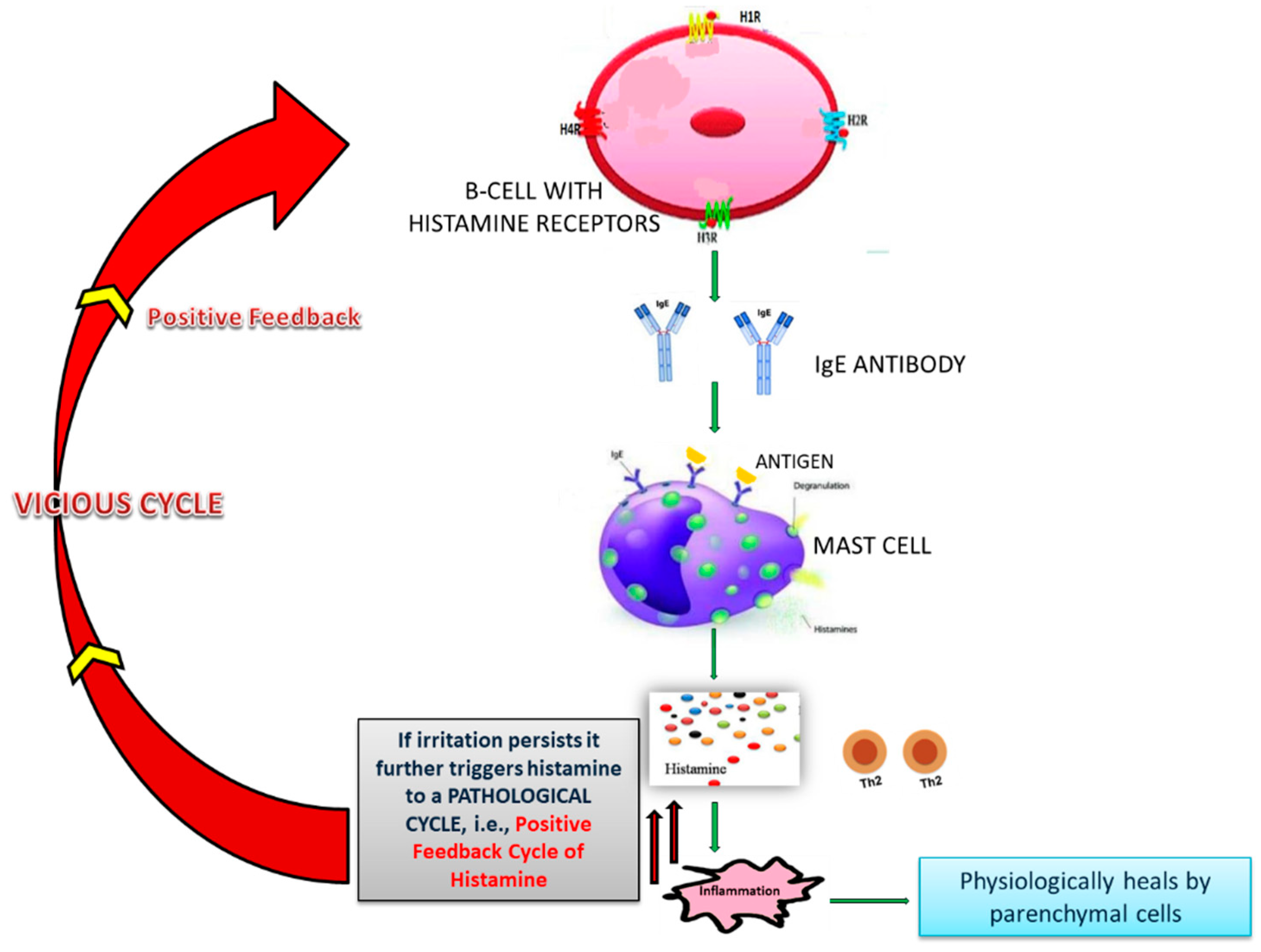
4.2. Role of Histamine and NF-κB in Excess Immune Response in COVID-19
4.3. Drugs Acting on NF-κB Signaling
4.4. Neutralization of Excess Histamine and Down-Regulation of NF-κB
5. Translational Value and Future Research Direction
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Pre-Registration
References and Note
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Moore, M.J.; Vasllieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greeneugh, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.; Najjar, A.; Chong, D.J.; Pramanik, B.K.; Kirsch, C.; Kuzniecky, R.I.; Pacia, S.V.; Azhar, S. Central nervous system complications associated with SARS-CoV-2 infection: Integrative concepts of pathophysiology and case reports. J. Neuroinflamm. 2020, 17, 231. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Rhee, J.W.; Cheng, P.; Waliany, S.; Chang, A.; Witteles, R.M.; Maecker, H.; Davis, M.M.; Nguyen, P.K.; Wu, S.M. Cardiovascular Complications in Patients with COVID-19: Consequences of Viral Toxicities and Host Immune Response. Curr. Cardiol. Rep. 2020, 22, 32. [Google Scholar] [CrossRef] [Green Version]
- Skevaki, C.; Fragkou, P.C.; Cheng, C.; Xie, M.; Renz, H. Laboratory characteristics of patients infected with the novel SARS-CoV-2 virus. J. Infect. 2020, 81, 205–212. [Google Scholar] [CrossRef]
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef]
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 533–535. [Google Scholar] [CrossRef] [Green Version]
- Maisch, B. SARS-CoV-2 as potential cause of cardiac inflammation and heart failure. Is it the virus, hyperinflammation, or MODS? Herz 2020, 45, 321–322. [Google Scholar] [CrossRef]
- Ling, Y.; Xu, S.B.; Lin, Y.X.; Tian, D.; Zhu, Z.Q.; Dai, F.H.; Wu, F.; Song, Z.G.; Huang, W.; Chen, J.; et al. Persistence and clearance of viral RNA in 2019 novel coronavirus disease rehabilitation patients. Chin. Med. J. 2020, 133, 1039–1043. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [Green Version]
- Turvey, S.E.; Broide, D.H. Innate immunity. J. Allergy Clin. Immunol. 2010, 125, S24. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, B.M.; Vikse, J.; Benoit, S.; Favaloro, E.J.; Lippi, G. Hyperinflammation and derangement of renin-angiotensin-aldosterone system in COVID-19: A novel hypothesis for clinically suspected hypercoagulopathy and microvascular immunothrombosis. Clin. Chim. Acta 2020, 507, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Du, S.Q.; Yuan, W. Mathematical Modeling of Interaction between Innate and Adaptive Immune Responses in COVID-19 and Implications for Viral Pathogenesis. J. Med. Virol. 2020, 92, 1615–1628. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The Adaptive Immune System. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Oberholzer, A.; Oberholzer, C.; Moldawer, L.L. Cytokine signaling—Regulation of the immune response in normal and critically ill states. Crit. Care Med. 2000, 28, N3–N12. [Google Scholar] [CrossRef]
- Branco, A.C.C.C.; Yoshikawa, F.S.Y.; Pietrobon, A.J.; Sato, M.N. Role of Histamine in Modulating the Immune Response and Inflammation. Mediat. Inflamm. 2018, 2018, 9524075. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Ananthanarayan, R.; Paniker, C.K.J. A Textbook of Microbiology; Orient Longman Limited: Hyderabad, India, 1992; ISBN 0-86311-194-7. [Google Scholar]
- Ennis, M.; Tiligada, K. Histamine receptors and COVID-19. Inflamm. Res. 2020, 1, 1. [Google Scholar]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: The hunt for new therapeutic targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, A.M.; Zhuo, J.; Mendelsohn, F.A.O. Localization and function of angiotensin AT1 receptors. Am. J. Hypertens. 2000, 13, S31–S38. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.D.; Karnik, S.S. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J. Cell Signal. 2016, 1. [Google Scholar] [CrossRef]
- Birnbaumer, M. Vasopressin receptors. Trends Endocrinol. Metab. 2000, 11, 406–410. [Google Scholar] [CrossRef]
- Barrett, O.; Wolak, T. Peripheral Adrenergic Blockers. In Hypertension: A Companion to Braunwald’s Heart Disease; Elsevier: Amsterdam, The Netherlands, 2018; pp. 222–229. ISBN 9780323429733. [Google Scholar]
- Ashina, K.; Tsubosaka, Y.; Nakamura, T.; Omori, K.; Kobayashi, K.; Hori, M.; Ozaki, H.; Murata, T. Histamine induces vascular hyperpermeability by increasing blood flow and endothelial barrier disruption in vivo. PLoS ONE 2015, 10, e0132367. [Google Scholar] [CrossRef]
- Katzung, B.G. Basic & Clinical Pharmacology; The McGraw-Hill Companies, Inc.: New York, NY, USA, 2012. [Google Scholar]
- Potempa, L.A.; Rajab, I.M.; Hart, P.C.; Bordon, J.; Fernandez-Botran, R. Insights into the use of C-reactive protein as a diagnostic index of disease severity in COVID-19 infections. Am. J. Trop. Med. Hyg. 2020, 103, 561–563. [Google Scholar] [CrossRef]
- Klok, F.A.; Kruip, M.J.H.A.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The experience of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Fernández, A.; Toledo-Pons, N.; Cosío, B.G.; Millán, A.; Calvo, N.; Ramón, L.; de Mendoza, S.H.; Morell-García, D.; Bauça-Rossello, J.M.; Núñez, B.; et al. Prevalence of pulmonary embolism in patients with COVID-19 pneumonia and high D-dimer values: A prospective study. PLoS ONE 2020, 15, e0238216. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Institute, J.B. Critical Appraisal Tools. Available online: https://joannabriggs.org/critical-appraisal-tools (accessed on 20 November 2020).
- Baethge, C.; Goldbeck-Wood, S.; Mertens, S. SANRA—a scale for the quality assessment of narrative review articles. Res. Integr. Peer Rev. 2019, 4, 5. [Google Scholar] [CrossRef] [Green Version]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- An, P.J.; Yi, Z.Z.; Yang, L.P. Biochemical indicators of coronavirus disease 2019 exacerbation and the clinical implications. Pharmacol. Res. 2020, 159, 104946. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Öztürk, R.; Taşova, Y.; Ayaz, A. Covid-19: Pathogenesis, genetic polymorphism, clinical features and laboratory findings. Turkish J. Med. Sci. 2020, 50, 638–657. [Google Scholar] [CrossRef]
- Girija, A.S.S.; Shankar, E.M.; Larsson, M. Could SARS-CoV-2-Induced Hyperinflammation Magnify the Severity of Coronavirus Disease (CoViD-19) Leading to Acute Respiratory Distress Syndrome? Front. Immunol. 2020, 11, 1206. [Google Scholar] [CrossRef]
- Wang, J.; Li, Q.; Yin, Y.; Zhang, Y.; Cao, Y.; Lin, X.; Huang, L.; Hoffmann, D.; Lu, M.; Qiu, Y. Excessive Neutrophils and Neutrophil Extracellular Traps in COVID-19. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- O’Brien, E.R.; Sandhu, J.K. Sex differences in COVID-19 mortality: Opportunity to develop HSP27 (HSPB1) immunotherapy to treat hyper-inflammation? Cell Stress Chaperones 2020, 25, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Silverman, G.J. Blood clots and TAM receptor signalling in COVID-19 pathogenesis. Nat. Rev. Immunol. 2020, 20, 395–396. [Google Scholar] [CrossRef]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mishra, K.P.; Singh, A.K.; Singh, S.B. Hyperinflammation and Immune Response Generation in COVID-19. Neuroimmunomodulation 2020, 27, 80–86. [Google Scholar] [CrossRef]
- Alunno, A.; Carubbi, F.; Rodríguez-Carrio, J. Storm, typhoon, cyclone or hurricane in patients with COVID-19? Beware of the same storm that has a different origin. RMD Open 2020, 6, e001295. [Google Scholar] [CrossRef]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef]
- Yazdanpanah, F.; Hamblin, M.R.; Rezaei, N. The immune system and COVID-19: Friend or foe? Life Sci. 2020, 256, 117900. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’’ in COVID-19’. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Alessandri, C.; Conti, F.; Priori, R. COVID-19 gone bad: A new character in the spectrum of the hyperferritinemic syndrome? Autoimmun. Rev. 2020, 19, 102573. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, G.; Dong, D.; Shang, P. Effects of iron overload and oxidative damage on the musculoskeletal system in the space environment: Data from spaceflights and ground-based simulation models. Int. J. Mol. Sci. 2018, 19, 2608. [Google Scholar] [CrossRef] [Green Version]
- Wenzhong, L.; Hualan, L. COVID-19:Attacks the 1-Beta Chain of Hemoglobin and Captures the Porphyrin to Inhibit Human Heme Metabolism. Am. Chem. Soc. 2020. [Google Scholar] [CrossRef]
- Ruscitti, P.; Berardicurti, O.; Di Benedetto, P.; Cipriani, P.; Iagnocco, A.; Shoenfeld, Y.; Giacomelli, R. Severe COVID-19, Another Piece in the Puzzle of the Hyperferritinemic Syndrome. An Immunomodulatory Perspective to Alleviate the Storm. Front. Immunol. 2020, 11, 1130. [Google Scholar] [CrossRef]
- Sukhbaatar, N.; Weichhart, T. Iron regulation: Macrophages in control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef] [Green Version]
- Paces, J.; Strizova, Z.; Smrz, D.; Cerny, J. COVID-19 and the immune system. Physiol. Res. 2020, 69, 379–388. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Garcia-Revilla, J.; Deierborg, T.; Venero, J.L.; Boza-Serrano, A. Hyperinflammation and Fibrosis in Severe COVID-19 Patients: Galectin-3, a Target Molecule to Consider. Front. Immunol. 2020, 11, 2069. [Google Scholar] [CrossRef]
- Mazzoni, A.; Salvati, L.; Maggi, L.; Capone, M.; Vanni, A.; Spinicci, M.; Mencarini, J.; Caporale, R.; Peruzzi, B.; Antonelli, A.; et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J. Clin. Invest. 2020, 130, 4694–4703. [Google Scholar] [CrossRef] [PubMed]
- Opoka-Winiarska, V.; Grywalska, E.; Roliński, J. Could hemophagocytic lymphohistiocytosis be the core issue of severe COVID-19 cases? BMC Med. 2020, 18. [Google Scholar] [CrossRef]
- Didangelos, A. COVID-19 Hyperinflammation: What about Neutrophils? mSphere 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Kondo, M.; Kelsoe, G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J. Exp. Med. 2005, 201, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Cicco, S.; Cicco, G.; Racanelli, V.; Vacca, A. Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs): Two Potential Targets for COVID-19 Treatment. Mediators Inflamm. 2020, 2020, 7527953. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef]
- Kosmaczewska, A.; Frydecka, I. Dysregulation of the immune system as a driver of the critical course of novel coronavirus disease 2019. Polish Arch. Intern. Med. 2020, 130, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U. The cholinergic anti-inflammatory pathway alleviates acute lung injury. Mol. Med. 2020, 26. [Google Scholar] [CrossRef]
- Parackova, Z.; Zentsova, I.; Bloomfield, M.; Vrabcova, P.; Smetanova, J.; Klocperk, A.; Mesežnikov, G.; Casas Mendez, L.F.; Vymazal, T.; Sediva, A. Disharmonic Inflammatory Signatures in COVID-19: Augmented Neutrophils’ but Impaired Monocytes’ and Dendritic Cells’ Responsiveness. Cells 2020, 9, 2206. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Tang, N.; Liu, H.; Cao, W. Coagulation dysfunction: A hallmark in COVID-19. Arch. Pathol. Lab. Med. 2020, 144, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Smeda, M.; Chlopicki, S. Endothelial barrier integrity in covid-19-dependent hyperinflammation: Does the protective facet of platelet functionmatter? Cardiovasc. Res. 2020, 116, E118–E121. [Google Scholar] [CrossRef]
- Song, J.W.; Zhang, C.; Fan, X.; Meng, F.P.; Xu, Z.; Xia, P.; Cao, W.J.; Yang, T.; Dai, X.P.; Wang, S.Y.; et al. Immunological and inflammatory profiles in mild and severe cases of COVID-19. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Herrmann, M.; Schulte, S.; Wildner, N.H.; Wittner, M.; Brehm, T.T.; Ramharter, M.; Woost, R.; Lohse, A.W.; Jacobs, T.; Schulze zur Wiesch, J. Analysis of Co-inhibitory Receptor Expression in COVID-19 Infection Compared to Acute Plasmodium falciparum Malaria: LAG-3 and TIM-3 Correlate With T Cell Activation and Course of Disease. Front. Immunol. 2020, 11, 1870. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Fathi, N.; Rezaei, N. Lymphopenia in COVID-19: Therapeutic opportunities. Cell Biol. Int. 2020, 44, 1792–1797. [Google Scholar] [CrossRef]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 2020, 509, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Petralia, M.C.; Basile, M.S.; Bramanti, A.; Bramanti, P.; Nicoletti, F.; Spandidos, D.A.; Shoenfeld, Y.; Fagone, P. Transcriptomic analysis of covid-19 lungs and bronchoalveolar lavage fluid samples reveals predominant b cell activation responses to infection. Int. J. Mol. Med. 2020, 46, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Tetro, J.A. Is COVID-19 receiving ADE from other coronaviruses? Microbes Infect. 2020, 22, 72–73. [Google Scholar] [CrossRef]
- Mitsuhashi, M.; Tanaka, A.; Fujisawa, C.; Kawamoto, K.; Itakura, A.; Takaku, M.; Hironaka, T.; Sawada, S.; Matsuda, H. Necessity of Thromboxane A 2 for Initiation of Platelet-Mediated Contact Sensitivity: Dual Activation of Platelets and Vascular Endothelial Cells. J. Immunol. 2001, 166, 617–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheth, A.R.; Grewal, U.S.; Patel, H.P.; Thakkar, S.; Garikipati, S.; Gaddam, J.; Bawa, D. Possible mechanisms responsible for acute coronary events in COVID-19. Med. Hypotheses 2020, 143, 110125. [Google Scholar] [CrossRef]
- Patella, V.; Marinò, I.; Arbustini, E.; Lamparter-Schummert, B.; Verga, L.; Adt, M.; Marone, G. Stem Cell Factor in Mast Cells and Increased Mast Cell Density in Idiopathic and Ischemic Cardiomyopathy. Circulation 1998, 97, 971–978. [Google Scholar] [CrossRef] [Green Version]
- Vieira, L.M.F.; Emery, E.; Andriolo, A. Covid-19: Laboratory diagnosis for clinicians. An updating article. Sao Paulo Med. J. 2020, 138, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 Autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Schett, G.; Manger, B.; Simon, D.; Caporali, R. COVID-19 revisiting inflammatory pathways of arthritis. Nat. Rev. Rheumatol. 2020, 16, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E.; Feigin, V.; Caso, V.; Santalucia, P.; Logroscino, G. COVID-19 Infection and Neurological Complications: Present Findings and Future Predictions. Neuroepidemiology 2020, 54, 1–6. [Google Scholar] [CrossRef]
- Becker, R.C. COVID-19-associated vasculitis and vasculopathy. J. Thromb. Thrombolysis 2020, 50, 499–511. [Google Scholar] [CrossRef]
- Khoshdel-Rad, N.; Zahmatkesh, E.; Shpichka, A.; Timashev, P.; Vosough, M. Outbreak of chronic renal failure: Will this be a delayed heritage of COVID-19? J. Nephrol. 2020, 1, 3–5. [Google Scholar] [CrossRef]
- Komiyama, M.; Hasegawa, K.; Matsumori, A. Dilated Cardiomyopathy Risk in Patients with Coronavirus Disease 2019: How to Identify and Characterise it Early? Eur. Cardiol. Rev. 2020, 15, e49. [Google Scholar] [CrossRef]
- Cothran, T.P.; Kellman, S.; Singh, S.; Beck, J.S.; Powell, K.J.; Bolton, C.J.; Tam, J.W. A brewing storm: The neuropsychological sequelae of hyperinflammation due to COVID-19. J. Clean. Prod. 2020, 88, 957. [Google Scholar] [CrossRef]
- Giaglis, S.; Stoikou, M.; Chowdhury, C.S.; Schaefer, G.; Grimolizzi, F.; Rossi, S.W.; Hoesli, I.M.; Lapaire, O.; Hasler, P.; Hahn, S. Multimodal regulation of NET formation in pregnancy: Progesterone antagonizes the pro-NETotic effect of estrogen and G-CSF. Front. Immunol. 2016, 7, 565. [Google Scholar] [CrossRef] [Green Version]
- Rossi, F.; Tortora, C.; Argenziano, M.; Di Paola, A.; Punzo, F. Cannabinoid receptor type 2: A possible target in SARS-CoV-2 (CoV-19) infection? Int. J. Mol. Sci. 2020, 21, 3809. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. From causes of aging to death from COVID-19. Aging 2020, 12, 10004–10021. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 84. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Rice, S.; Rosen, H.; Oldstone, M.B.A. Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3799–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeno, J.M.; Fernandez-Delgado, R.; Fett, C.; Castano-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF- B-Mediated Inflammation in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice Increases Survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Conti, P.; Caraffa, A.; Tetè, G.; Gallenga, C.E.; Ross, R.; Kritas, S.K.; Frydas, I.; Younes, A.; Di Emidio, P.; Ronconi, G. Mast cells activated by SARS-CoV-2 release histamine which increases IL-1 levels causing cytokine storm and inflammatory reaction in COVID-19. J. Biol. Regul. Homeost. Agents 2020, 34, 1629–1632. [Google Scholar]
- Coombs, R.; Gell, P. Classification of Allergic Reactions Responsible for Clinical Hypersensitity and Disease. In Clinical Aspect of Immunology, 3rd ed.; Gell, P.G.H., Coombs, R.R.A., Lachman, P.J., Eds.; Blackwell Scientific Publications: Oxford, UK, 1975. [Google Scholar]
- Holdsworth, S.R.; Kitching, A.R.; Tipping, P.G. Th1 and th2 T helper cell subsets affect patterns of injury and outcomes in glomerulonephritis. Kidney Int. 1999, 55, 1198–1216. [Google Scholar] [CrossRef] [Green Version]
- Deo, S.S.; Mistry, K.J.; Kakade, A.M.; Niphadkar, P.V. Role played by Th2 type cytokines in IgE mediated allergy and asthma. Lung India 2010, 27, 66–71. [Google Scholar] [CrossRef]
- Fierer, J.; Looney, D.; Pechère, J.-C. Nature and Pathogenicity of Micro-organisms. In Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2017; pp. 4–25. [Google Scholar]
- King, T.C. Elsevier’s Integrated Pathology; Mosby: Maryland Heights, MI, USA, 2007; ISBN 9780323043281. [Google Scholar]
- Justiz Vaillant, A.A.; Ramphul, K. Hypersensitivity Reactions, Delayed; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Justiz Vaillant, A.A.; Zito, P.M. Hypersensitivity Reactions, Immediate; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Berger, A. Science commentary: Th1 and Th2 responses: What are they? Br. Med. J. 2000, 321, 424. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, S. Researchers discover “feedback loop” in allergic reactions. BMJ Br. Med. J. 1999, 318, 1306. [Google Scholar] [CrossRef]
- Bax, H.J.; Keeble, A.H.; Gould, H.J. Cytokinergic IgE action in mast cell activation. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Takatsu, K.; Kouro, T.; Nagai, Y. Interleukin 5 in the Link Between the Innate and Acquired Immune Response—PubMed. Adv Immunol 2009, 101, 191–236. [Google Scholar] [CrossRef]
- Sehra, S.; Yao, Y.; Howell, M.D.; Nguyen, E.T.; Kansas, G.S.; Leung, D.Y.M.; Travers, J.B.; Kaplan, M.H. IL-4 Regulates Skin Homeostasis and the Predisposition toward Allergic Skin Inflammation. J. Immunol. 2010, 184, 3186–3190. [Google Scholar] [CrossRef] [Green Version]
- Luzina, I.G.; Keegan, A.D.; Heller, N.M.; Rook, G.A.W.; Shea-Donohue, T.; Atamas, S.P. Regulation of inflammation by interleukin-4: A review of “alternatives”. J. Leukoc. Biol. 2012, 92, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Müller, U.; Stenzel, W.; Köhler, G.; Werner, C.; Polte, T.; Hansen, G.; Schütze, N.; Straubinger, R.K.; Blessing, M.; McKenzie, A.N.J.; et al. IL-13 Induces Disease-Promoting Type 2 Cytokines, Alternatively Activated Macrophages and Allergic Inflammation during Pulmonary Infection of Mice with Cryptococcus neoformans. J. Immunol. 2007, 179, 5367–5377. [Google Scholar] [CrossRef] [Green Version]
- De Vries, J.E. The role of IL-13 and its receptor in allergy and inflammatory responses. J. Allergy Clin. Immunol. 1998, 102, 165–169. [Google Scholar] [CrossRef]
- Klimpel, G.R. Immune Defense. In Encyclopedic Reference of Molecular Pharmacology; Springer: Berlin/Heidelberg, Germany, 2006; pp. 476–481. ISBN 0963117211. [Google Scholar]
- Rock, K.L.; Kono, H. The Inflammatory Response to Cell Death. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 99–126. [Google Scholar] [CrossRef]
- Hwa, K.Y.; Lin, W.M.; Hou, Y.I.; Yeh, T.M. Peptide Mimicrying Between SARS Coronavirus Spike Protein and Human Proteins Reacts with SARS Patient Serum. J. Biomed. Biotechnol. 2008, 2008, 326464. [Google Scholar] [CrossRef]
- Zicari, S.; Sessa, L.; Cotugno, N.; Ruggiero, A.; Morrocchi, E.; Concato, C.; Rocca, S.; Zangari, P.; Manno, E.; Palma, P. Immune Activation, Inflammation, and Non-AIDS Co-Morbidities in HIV-Infected Patients under Long-Term ART. Viruses 2019, 11, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Packard, K.A.; Khan, M.M. Effects of histamine on Th1/Th2 cytokine balance. Int. Immunopharmacol. 2003, 3, 909–920. [Google Scholar] [CrossRef]
- Kofler, L.; Ulmer, H.; Kofler, H. Histamine 50-Skin-Prick Test: A Tool to Diagnose Histamine Intolerance. ISRN Allergy 2011, 2011, 353045. [Google Scholar] [CrossRef] [Green Version]
- Majno, G.; Palade, G. The Effect of Histamine and Serotonin on Vascular Permeability: An Electron Microscopic Study. J. Biophys. Biochem. Cytol. 1961, 11, 586. [Google Scholar]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tan, M.; Chen, X.; Liu, Y.; Huang, J.; Ou, J.; Deng, X. Immunopathological characteristics of coronavirus disease 2019 cases in Guangzhou, China. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Ghebrehiwet, B.; Randazzo, B.P.; Dunn, J.T.; Silverberg, M.; Kaplan, A.P. Mechanisms of activation of the classical pathway of complement by Hageman factor fragment. J. Clin. Invest. 1983, 71, 1450–1456. [Google Scholar] [CrossRef] [Green Version]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Duchene, J. Kallikrein-Kinin Kystem in Inflammatory Diseases; De Gruyter: Berlin, Germany, 2011; ISBN 978-3-11-025235-4. [Google Scholar]
- Chaudhry, R.; Babiker, H.M. Physiology, Coagulation Pathways; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Nayak, A.; Dodagatta-Marri, E.; Tsolaki, A.G.; Kishore, U. An insight into the diverse roles of surfactant proteins, SP-A and SP-D in innate and adaptive immunity. Front. Immunol. 2012, 3, 131. [Google Scholar] [CrossRef] [Green Version]
- Hardin, C.C.; Chivukula, R. Surfactant Worth Studying as Treatment for COVID-19–Related ARDS—Mass General Advances in Motion. Available online: https://advances.massgeneral.org/research-and-innovation/article.aspx?id=1196 (accessed on 1 January 2021).
- Holden, N.S.; Gong, W.; King, E.M.; Kaur, M.; Giembycz, M.A.; Newton, R. Potentiation of NF-κB-dependent transcription and inflammatory mediator release by histamine in human airway epithelial cells. Br. J. Pharmacol. 2007, 152, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Ayoub, M.; Mittenbühler, K.; Sütterlin, B.W.; Bessler, W.G. The anti-allergic drug histaglobin inhibits NF-κB nuclear translocation and down-regulates proinflammatory cytokines. Int. J. Immunopharmacol. 2000, 22, 755–763. [Google Scholar] [CrossRef]
- Hiscott, J.; Nguyen, T.L.A.; Arguello, M.; Nakhaei, P.; Paz, S. Manipulation of the nuclear factor-κB pathway and the innate immune response by viruses. Oncogene 2006, 25, 6844–6867. [Google Scholar] [CrossRef] [Green Version]
- ChiCTR2000030089, I.T.I. A clinical Study for the Efficacy and Safety of Adalimumab Injection in the Treatment of Patients A Randomized, Open-Label, Controlled Trial for the Efficacy and Safety of Adalimumab Injection in the Treatment of Patients with Severe Novel Coronavirus pne. Available online: http://www.chictr.org.cn/showprojen.aspx?proj=49889 (accessed on 1 January 2021).
- Galloway, J.B.; Hyrich, K.L.; Mercer, L.K.; Dixon, W.G.; Fu, B.; Ustianowski, A.P.; Watson, K.D.; Lunt, M.; Symmons, D.P.M. Anti-TNF therapy is associated with an increased risk of serious infections in patients with rheumatoid arthritis especially in the first 6 months of treatment: Updated results from the British Society for Rheumatology Biologics Register with special emphasis on risks in the elderly. Rheumatology 2011, 50, 124–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Comas-Basté, O.; Sánchez-Pérez, S.; Veciana-Nogués, M.T.; Latorre-Moratalla, M.; Vidal-Carou, M.D.C. Histamine intolerance: The current state of the art. Biomolecules 2020, 10, 1181. [Google Scholar] [CrossRef] [PubMed]
- Laborde, C.; Parrot, J.L. The Histamine-Fixing Power of Blood Serum; Its Modification After an Injection of Normal Human Serum or of an Azoprotein of Histamine. J. Physiol. 1954, 46, 492–495. [Google Scholar]
- Laborde, C.; Parrot, J.L.; Urquia, D. Le pouir histaminopexique du serum sanguin. Thechinique mesre. Pre Med 1953, 61, 1151–1153. [Google Scholar]
- Yoshii, H.; Fukata, Y. US Patent Application for Immunomodulating and antiinflammatory agent Patent Application (Application #20020004058 issued 10 January 2002).
- Yoshii, H.; Fukata, Y. US Patent for Activated Immunoglobulin Patent. U.S. Patent 6,627,194, 30 September 2003. [Google Scholar]
- Yoshii, H.; Fukata, Y.; Yamamoto, K.; Yago, H.; Suehiro, S.; Yanagihara, Y.; Okudaira, H. Inhibitory effect of histamine-added mouse gamma-globulin on eosinophil accumulation induced by allergen in BALB/c mice. Arerugi 1995, 44, 567–570. [Google Scholar] [PubMed]
- Yoshii, H.; Fukata, Y.; Yamamoto, K.; Yago, H.; Suehiro, S.; Yanagihara, Y.; Okudaira, H. A new assay system detecting antibody production and delayed-type hypersensitivity responses to trinitrophenyl hapten in an individual mouse. Int. J. Immunopharmacol. 1996, 18, 31–36. [Google Scholar] [CrossRef]
- Mangadevi Thota, S. Efficacy of Injection Histoglob in Treatment of Chronic Urticaria—A Prospective Study. IOSR J. Dent. Med. Sci. e-ISSN 2019, 18, 57–62. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar]
- Zeshan, H.; Muhammad Muneeb, S.; Muhammad Ansar, F.; Maryum, I.; Maryam, K.; Rao Sohail Ahmad, K.; Adnan Khan, N. In Silico Discovery of Novel Inhibitors Against Main Protease (Mpro) of SARS-CoV-2 Using Pharmacophore and Molecular Docking Based Virtual Screening from ZINC Database. Preprints 2020. [Google Scholar] [CrossRef]
- Hu, F.; Jiang, J.; Yin, P. Prediction of potential commercially inhibitors against SARS-CoV-2 by multi-task deep model. arXiv 2020, arXiv:2003.00728. [Google Scholar]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Duan, Y.; et al. Structure-based drug design, virtual screening and high-throughput screening rapidly identify antiviral leads targeting COVID-19. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kong, R.; Yang, G.; Xue, R.; Liu, M.; Wang, F.; Hu, J.; Guo, X.; Chang, S. COVID-19 Docking Server: An interactive server for docking small molecules, peptides and antibodies against potential targets of COVID-19. Bioinformatics 2020, 36, 5109–5111. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Arya, R.; Das, A.; Prashar, V.; Kumar, M. Potential inhibitors against papain-like protease of novel coronavirus (SARS-CoV-2) from FDA approved drugs. Chemrxiv.Org 2020, 1–8. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Tung, Y.-A.; Lee, K.-H.; Chen, T.-F.; Hsiao, Y.-C.; Chang, H.-C.; Hsieh, T.-T.; Su, C.-H.; Wang, S.-S.; Yu, J.-Y.; et al. Potential Therapeutic Agents for COVID-19 Based on the Analysis of Protease and RNA Polymerase Docking. Preprints 2020. [Google Scholar] [CrossRef] [Green Version]
- Gehani, M.; Peddapalli, A.; Kalle, A.; Peddapalli, S.; Peter, A.; Sharad, S. Demystifying Excess Immune Response in COVID-19 to Reposition an Orphan Drug for Down-Regulation of NF-κB: A Systematic Review. Available online: https://doi.org/10.7910/DVN/EQXE0E (accessed on 1 January 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic 1 | Number of Studies |
|---|---|
| Month of publication | N = 42 |
| April 2020 | 6 |
| May 2020 | 8 |
| June 2020 | 12 |
| July 2020 | 8 |
| August 2020 | 3 |
| September 2020 | 3 |
| October 2020 | 2 |
| Article type | N = 42 |
| Research Paper | 8 |
| Systematic Review | 1 |
| Narrative Review | 22 |
| Editorial | 1 |
| Commentary | 2 |
| Comment | 3 |
| Hypothesis | 2 |
| Viewpoint/perspective | 2 |
| Updating article | 1 |
| Outcome (Mechanism of excess immune response) | N = 42 |
| Extracellular mechanisms 2 | |
| Cytokine storm | 12 |
| Neutrophil-related mechanism (NETosis, neutrophilia, and HMGB1 induced inflammation) | 6 |
| Lymphocyte-related mechanisms | 4 |
| Secondary haemophagocytic lymphohistocytosis | 3 |
| Direct injury | 3 |
| Immunothrombosis-related mechanisms | 3 |
| Hyperferritinemia | 2 |
| Macrophage activation syndrome (including Galactin-3 up-regulation) | 2 |
| Hypoxia-induced dysregulated immune response | 1 |
| Cannabinoid receptor-mediated immune suppression | 1 |
| Quasi-programmed aging | 1 |
| IL-6 attenuated HLA-DR expression | 1 |
| Intracellular mechanisms | |
| NLRP3 inflammasome activation | 2 |
| Dysfunction of platelet mitochondria | 1 |
| PROS1 signalling | 1 |
| NF-κB pathway | 1 |
| ACE2/bradykinin B1R/DABK axis involvement | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peddapalli, A.; Gehani, M.; Kalle, A.M.; Peddapalli, S.R.; Peter, A.E.; Sharad, S. Demystifying Excess Immune Response in COVID-19 to Reposition an Orphan Drug for Down-Regulation of NF-κB: A Systematic Review. Viruses 2021, 13, 378. https://doi.org/10.3390/v13030378
Peddapalli A, Gehani M, Kalle AM, Peddapalli SR, Peter AE, Sharad S. Demystifying Excess Immune Response in COVID-19 to Reposition an Orphan Drug for Down-Regulation of NF-κB: A Systematic Review. Viruses. 2021; 13(3):378. https://doi.org/10.3390/v13030378
Chicago/Turabian StylePeddapalli, Apparao, Manish Gehani, Arunasree M. Kalle, Siva R. Peddapalli, Angela E. Peter, and Shashwat Sharad. 2021. "Demystifying Excess Immune Response in COVID-19 to Reposition an Orphan Drug for Down-Regulation of NF-κB: A Systematic Review" Viruses 13, no. 3: 378. https://doi.org/10.3390/v13030378