
Abstract
The novel Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV-2) has emerged to be the reason behind the COVID-19 pandemic. It was discovered in Wuhan, China and then began spreading around the world, impacting the health of millions. Efforts for treatment have been hampered as there are no antiviral drugs that are effective against this virus. In the present study, we have explored the phytochemical constituents of Salvia plebeia R. Br., in terms of its binding affinity by targeting COVID-19 main protease (Mpro) using computational analysis. Molecular docking analysis was performed using PyRx software. The ADMET and drug-likeness properties of the top 10 compounds showing binding affinity greater than or equal to − 8.0 kcal/mol were analysed using pkCSM and DruLiTo, respectively. Based on the docking studies, it was confirmed that Rutin and Plebeiosides B were the most potent inhibitors of the main protease of SARS-CoV-2 with the best binding affinities of − 9.1 kcal/mol and − 8.9 kcal/mol, respectively. Further, the two compounds were analysed by studying their biological activity using the PASS webserver. Molecular dynamics simulation analysis was performed for the selected protein–ligand complexes to confirm their stability at 300 ns. MM-PBSA provided the basis for analyzing the affinity of the phytochemicals towards Mpro by calculating the binding energy, and secondary structure analysis indicated the stability of protease structure when it is bound to Rutin and Plebeiosides B. Altogether, the study identifies Rutin and Plebeiosides B to be potent Mpro inhibitors of SARS-CoV-2.
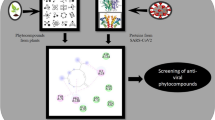
Graphic abstract

Similar content being viewed by others
Introduction
The current coronavirus pandemic, commonly known as COVID-19, is brought about by the Severe Acute Respiratory Syndrome coronavirus 2 (SARS-CoV-2) (Gil et al. 2020) and was first reported in Wuhan, China, in December 2019 (Das and Koner 2020). On February 12, 2020, the WHO proceeded to name the disease “coronavirus disease 2019” (COVID-19) and on March 11, 2020, they declared the outbreak a global pandemic. On conducting a whole viral genome evaluation, it was found that 88% of the viral sequence identity was shared with two SARS-like coronaviruses obtained from bats. This led to it being named “SARS coronavirus 2”, primarily based on the taxonomy and phylogeny (Fauci et al. 2020; Ranney et al. 2020). The Coronaviruses (CoVs) are a part of the subfamily Orthocoronavirinae, which belongs to the family Coronaviridae, coming under the order Nidovirales. Orthocoronavirinae has 4 genera within it, that is, Alphacoronavirus (α-CoV), Betacoronavirus (β-CoV), Gammacoronavirus (γ-CoV) and Deltacoronavirus (δ-CoV) (Li et al. 2020). The Coronaviruses are positively stranded RNA, enveloped viruses (Xiu et al. 2020). Mammals have been shown to be affected by α-CoV and β-CoV genera, and on the other hand, birds are known to be affected by γ-CoV and δ-CoV genera (Amin and Jha 2020).
The identification of biochemical events critical to the viral life cycle provides various significant targets to inhibit viral replication. An important process is the breakdown of the multidomain, viral polyprotein into 16 non-structural proteins (nsps). These individual proteins combine to form complexes that carry out the synthesis of viral RNA (Kandeel et al. 2020; Naidoo et al. 2020). Along with these non-structural proteins, the SARS-CoV-2 further possesses four structural proteins, namely the Spike surface glycoprotein, the membrane protein, the envelope protein and the nucleocapsid protein. These are all required for the infection and assembly of the virus (Li et al. 2020). When the virus is inhaled, the spike proteins help it interact with the epithelial cells by binding to the human receptor, ACE2. The virus then proceeds to proliferate and enter into the alveolar epithelial cells. This, in turn, triggers a vigorous immune response leading to cytokine storm syndromes as well as damage to the pulmonary tissue (Li et al. 2020; Mason 2020).
Main protease (Mpro) is an enzyme of great importance to the coronaviruses as it plays a crucial part in the replication of the virus and transcription. This functional significance of Mpro in the life cycle of the virus, along with the lack of any similarly related homologues in humans, has identified it as an attractive target for the development of medication against SARS-CoV-2. The structure of SARS-CoV-2 Mpro complexed with N3 is found in PDB (PDB ID: 6LU7) and it provides a basis to identify potential inhibitors against SARS-CoV-2 Mpro, with the help of virtual screening. From the literature, it can be seen that N3 is a Michael acceptor inhibitor that can inhibit Mpro from a range of coronaviruses like MERS-CoV and SARS-CoV and also displays potent antiviral activity. From the crystal structure of Mpro it was seen that it comprises protomers A and B which associate to form a dimer and each protomer consists of three domains. The residues of the active site of Mpro are conserved and make a catalytic Cys145-His41 dyad. N3 binds in the substrate-binding pocket situated in a cleft in the middle of domain I and domain II. The amino acid residues involved in the specific interactions of N3 with Mpro include Phe140, Cys145, Glu166, Leu4, Val3, Ala2, His172, Leu167, Asn142, His163, Met49, Leu141, His41, Met165, Tyr54, Gln192, Asp187, Phe185, Thr25, Gln189, Thr24 and Pro168 (Jin et al. 2020).
Presently, there is a lack of drugs targeted towards COVID-19 treatment, and effective therapeutic options are scarce (Jin et al. 2020). One of the potential methods of treatment that is currently being looked into by researchers is drug repurposing, which is also referred to as drug reprofiling or drug repositioning, where drugs that were intended as a treatment against other diseases or similar viruses (SARS and MERS) are used as a treatment against COVID-19. While this method substantially decreases the time it would have taken for the development of a new drug, drug repurposing does also pose certain challenges such as the absence of patent protection, establishment of the efficacy, demonstration of its safety and in certain cases the continuous need for risky and expensive trials (Nurton 2020; Pushpakom et al. 2018; Shineman et al. 2014; Talevi and Bellera 2020). Problems faced by synthetic or synthetically derived drugs such as harmful side effects and antibiotic resistance in the case of antibiotics, lead to the shift towards natural remedies. Herbal drug preparations, obtained from biologically active products, have been an important part of traditional medicine. The compounds obtained from natural products are known to be pharmacophores and possess certain properties, such as high stereochemistry and metabolite-likeness that make them ideal as alternative medicines (Marathe and Datey 2012; Harvey et al. 2015; Ngane et al. 2011; Veeresham 2012).
Salvia plebeia R. Br. belongs to the genus Salvia, which is a major genus of the family Lamiaceae (Wang et al. 2019). It is known to be an annual or biennial grass that is generally found in various countries, for example, China, India, Korea and Australia (Liang et al. 2020; Ren et al. 2014). In Traditional Chinese Medicine, it is believed to be used for various home remedies. A folk medicine, Badarangboya, made from S. plebeia, is used extensively in India (Ren et al. 2014). A majority of the traditional and folk medicines prepared from S. plebeia are known to be used for the treatment of the common cold, flu, cough, asthma, hepatitis, diarrhea, haemorrhoids and tumours (Bang et al. 2018; Liang et al. 2020; Ma et al. 2014; Nugroho et al. 2012; Ren et al. 2014; Wang et al. 2019).
The various phytochemical studies performed on S. plebeia unveiled the presence of diterpenoids, sesquiterpenoids, flavonoids, lignans, phenylpropanoids, aliphatic compounds and caffeic acid derivatives (Liang et al. 2020; Ren et al. 2014). All these compounds have shown various pharmacological properties such as anti-inflammatory activity, antioxidant activity, anti-tyrosinase, anti-viral activity against H1N1 and HSV-1, and anti-proliferative activity. Some of the other major activities shown by S. plebeia are inhibitory activity, antimicrobial activity, hypoglycemic activity, hepatoprotective activity, hemostatic activity, skin protective activity, antitumor activity, anti-obesity effects and anti-ageing effects (Liang et al. 2020).
In the present study, the computational screening of the biologically active compounds of Salvia plebeia R. Br. was performed to identify potential lead inhibitors of the Mpro enzyme of SARS-CoV-2. Molecular docking was initially carried out to screen the phytochemicals of S. plebeia and the pharmacokinetic or ADMET properties as well as the drug-likeness parameters of the phytochemicals were also studied to understand the disposition of the compounds and to ensure they possessed pharmacophoric features. The pharmacological activities of the selected compounds were analysed using the PASS webserver. To predict the dynamic behaviour as well as stability, molecular dynamics simulation and secondary structure analysis were executed for the protein–ligand complexes. MM-PBSA (Molecular Mechanics Poisson–Boltzmann Surface Area) supplied the grounds for analyzing the affinity of the phytochemicals towards Mpro by calculating the binding energy.
Materials and methods
Retrieval of ligands and ligand preparation
The structures of the biologically active compounds present in Salvia plebeia (Fig. S1) were identified from literature (Bang et al. 2018; Liang et al. 2020; Ma et al. 2014; Nugroho et al. 2012; Ren et al. 2014; Wang et al. 2019). The three-dimensional structures for each compound were downloaded from the NCBI PubChem Compound database (https://pubchem.ncbi.nlm.nih.gov/). The structures that were unavailable on the databases were drawn from the literature with the help of Marvin Sketch (https://chemaxon.com/products/marvin). The structures of all the compounds that were retrieved were then incorporated into a single file in the.sdf format with the help of Discovery Studio Client 2019 (Kumar et al. 2021).
Retrieval of SARS-CoV-2 drug targets and target preparation
Main protease complexed with N3 (PDB ID: 6LU7) (Fig. S2), was retrieved from the RCSB PDB database (https://www.rcsb.org/). Swiss PDB Viewer was then utilized to remove the recognized co-crystallized ligand along with any unwanted residues. This was followed by the energy minimization of the structure (https://spdbv.vital-it.ch/). The necessary changes such as the addition of Hydrogen atoms and the addition of Kollman charges were made.
Molecular docking studies
The molecular docking studies were executed to identify the binding affinity of each of the retrieved biologically active compounds of Salvia plebeia with the main protease of SARS-CoV-2. Molecular docking was carried out using AutoDock vina available in PyRx software (Kumar et al. 2021). The ligands in.sdf format and the energy minimized structure of the protein in.pdbqt format were uploaded here. The active sites of the protein were identified and selected for the docking procedure. The grid box was adjusted in such a manner to enclose the active sites within it. The remaining parameters were set as default. The molecular interactions between the protein and the ligands were visualised using Maestro 12.4 (Krishna et al. 2021). The top complexes which showed binding scores greater than − 8.0 kcal/mol were further selected for further studies.
Prediction of ADMET properties
The pharmacokinetic properties of the compounds which showed binding scores greater than − 8.0 kcal/mol with Mpro were determined with the help of pkCSM ADMET descriptors algorithm protocol (Han et al. 2019). Each of the five parameters has certain sub-parameters such as central nervous system permeability, CYP450 inhibitors, AMES toxicity, total clearance and so on. For each of the lead molecules that were selected, seven parameters for absorption, four for distribution, seven for metabolism, two for excretion and ten for toxicity were analysed.
Prediction of activity spectra of substances (PASS)
To predict pharmacological effects and biochemical processes of the hit compounds based on their structural formula, PASS (Prediction of activity spectra of substances) (http://www.pharmaexpert.ru/passonline/index.php) software was utilised (SB et al. 2021).
Prediction of drug likeness properties
The prediction of drug likeness properties involves a certain set of rules for the compound structural properties, used to identify the properties of a molecule that could make it an effective drug. To identify the drug-likeness of the selected compounds of S. plebeia an open-source virtual screening tool, DruLiTo was utilized (Fereidoonnezhad et al. 2018). Various descriptors for the drug-likeness were calculated such as the Molecular Weight (MW), H-Bond Acceptor and Donor (HBA and HBD), logP, the Total Polar Surface Area (TPSA), AlogP, the Atom Molar Refractivity (AMR), the number of Rotatable Bonds (nRB), the number of Atoms, the number of Acidic groups, the Rotatable bond Count (RC), the number of Rigid Bonds (nRigidB), nAtomRing, and nHB. It calculates the drug-likeness properties based on certain drug-likeness guidelines such as Veber rule, Lipinski’s rule, CmC-50 like rule, MDDR-like rule, BBB rule, Quantitative Estimate of Drug likeness (QED) and Ghose filter.
Molecular dynamics simulations
The movements and dynamics of the atoms present within the apo-protein as well as the protein complexed with the ligand were analysed by conducting molecular dynamics simulation. The GROMACS version 2019.4 was utilized to carry out the molecular dynamics simulation for the X-ray crystal structure of the main protease enzyme (6LU7) and its complexes with the top ligands as identified through molecular docking studies (Lindahl et al. 2019). The generation of the protein parameters was carried out with the help of the gromos54a7 force field. The Ligand topology was generated using the PRODRG webserver (Schüttelkopf and Van Aalten 2004) and the gmxeditconf tool was implemented to build the cubic simulation box. For the vacuum minimisation of the processed setup for 1500 steps, the steepest descent algorithm was employed. The SPC water model was used to carry out the solvation procedure by using the gmx solvate tool. The electro-neutralization of the system was carried out with the tool gmxgenion. To optimize the structure and remove the steric clashes, energy minimization was done. The system was then calibrated by implementing two steps. The NVT equilibration’s first step lasted for 100 picoseconds. The system’s temperature was stabilised by heating the system to 300 K. The NPT ensemble’s second step lasted for 100 picoseconds. The stabilization of pressure and density of the system was then carried out. Each of the final structures obtained from the NPT equilibration phase then underwent the final production run for 300 ns of simulation time (Gupta et al. 2020; Prasanth et al. 2020).
Trajectory analysis and free energy calculation
The calculations of the protein backbone’s Root Mean Square Deviation (RMSD) and the C–α atoms’ Root Mean Square Fluctuations (RMSF) were carried out using gmx rms and gmx rmsf tools, respectively (Gupta et al. 2020). Besides these, tools including, gmx hbond, gmx gyrate, and gmx sasa was also used to analyse the total number of hydrogen bonds that are formed between the protein and ligand, the radius of gyration of the backbone atoms, and the solvent-accessible surface area, respectively. Furthermore, the changes in the secondary structure for a simulation of every 1 ns for the main chain of the protein was studied with the help of the DSSP module. Additionally, the interpretation of the binding affinity of the selected protein with each of the inhibitors was done with the help of the Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) approach. The estimation of the binding free energy was performed using a GROMACS utility, g_mmpbsa (Kumari et al. 2014). ΔG binding was computed for the entire simulation time with dt 1000 frames for obtaining precise results (Prasanth et al. 2020).
Results and discussion
Screening of potential inhibitors
Virtual screening is a computational method to search a chemical compound database to identify molecules with desired biological activity. High throughput Virtual Screening (HTVS) programs through PyRx software with graphical user interfaces (GUIs) that employs AutoDock for predicting receptor–ligand interactions is beneficial for the comparison of ligands (Jacob et al. 2012). AutoDock Vina is a software that works on the premise of empirical scoring functions and also calculate the grid maps automatically (Karami et al. 2017).
AutoDock Vina implemented on PyRx 0.8 was used in this study to calculate the binding energies between the 208 compounds obtained from Salvia plebia R. Br. and Mpro (details in supplementary file, Table S1) (Bang et al. 2018; Liang et al. 2020; Ma et al. 2014; Nugroho et al. 2012; Ren et al. 2014; Wang et al. 2019). It is evident from the docking results that the binding scores range from − 9.1 to − 3.3 kcal/mol. The top 10 compounds with binding scores greater than − 8.0 kcal/mol were identified as potential and their interactions with the Mpro are summarized in Table 1. All the selected ligands had established associations with both the residues of the catalytic dyad (Cys145 and His41). The structures of the top 10 compounds are represented in Fig. S3. Maestro 12.4 was used to view the interactions between the amino acid residues of the ligands and the target. The top 2 compounds, Rutin and Plebeiosides B displayed the best binding scores of − 9.1 kcal/mol and − 8.9 kcal/mol, respectively, which suggests that these compounds have the potential to form strong and stable complexes with the target.
The molecular interactions with the essential amino acid residues are considered to verify whether the ligand is docked in a favourable conformation. These associations play a vital role in maintaining a ligand energetically at the juncture of a macromolecule structure. The existence of a lead molecule close to the active site of the target will exhibit a better biological efficacy than a molecule that is not. Hydrophobic and hydrogen interactions predominantly contribute to protein stability (Patil et al. 2010). The relevance of drug–target interactions is related to polar interactions, as they play an important role in sustaining a stable 3D arrangement of the ligand in the binding pocket (Nisius et al. 2012).
The different amino acid interactions such as hydrogen, hydrophobic, electrostatic, polar and non-polar occurring between Mpro and the top 2 compounds, Rutin and Plebeiosides B is illustrated in Fig. 1. It was observed that Rutin interacts with the amino acid residues Thr26, Phe140 through Hydrogen bonds. Met49, Tyr54, Leu27, Cys145, Leu141, Phe140, Met165, Leu167, Pro168 are involved in hydrophobic interactions and Polar interaction involves Gln189, His41, Thr26, Ser144, His163, His164 and His172. Whereas, in the case of Plebeiosides B, it was observed that Hydrogen bonding involves Thr26, Cys145, Leu141, His163. Met49, Tyr54, Cys145, Leu141, Phe140 and Met165 were found to interact hydrophobically and polar interactions involved Thr26, Thr25, Ser144, Asn142, His163, His164, Gln189 and His41.

The molecular docking and interactions between main protease and a Rutin, b Plebeiosides B
Prediction of ADMET properties
The results of ADMET analysis for selected top 10 compounds obtained from pkCSM server is shown in Table 2. An ideal oral drug should be absorbed from the gastrointestinal tract, distributed to the target specifically, metabolized without eliminating its property and removed without any damage. The dynamic movements of the chemical structures during their passage through the body and their relation to physiological parameters helps us in understanding the pharmacokinetic properties (Gibaldi and Levy 1976). To understand these properties, the skin-permeability coefficient (logKp), apparent Caco-2 and MDCK were computed. The absorption level of the compounds can be anticipated using water solubility, Caco-2 permeability, skin permeability, intestinal absorption (human), and P-glycoprotein substrate or inhibitor. All ligands are found to be sparingly soluble in water. CaCo-2 permeability value greater than 0.9 indicates high permeability. All the compounds have shown a high permeability, except for rutin, cymaroside, 6 hydroxyluteolin 7 glucoside, plebeiosides B, plebeiosides A, salviaplebeiaside, rosmarinic acid and 6″-O-acetylhomoplantaginin. About skin permeability, log Kp > − 2.5 shows low skin permeability. Skin permeability is an important consideration for dermal drug delivery (Pires et al. 2015). All the compounds have shown good skin permeability. For intestinal absorption (humans), a value below 30% is associated with poor absorption. All the compounds have shown acceptable absorption, except rutin (23.446%). P-glycoprotein belongs to the ATP-binding transmembrane glycoprotein family [ATP-binding cassette (ABC)], which is found to be associated with the excretion of drugs or other exogenic chemicals from cells. The results suggest that all the compounds are substrates of P-Glycoprotein except Aflatoxin B2. Neocafhispidulin and salviaplebeiaside were found to be a P-glycoprotein I inhibitor. Neocafhispidulin was anticipated to be a P-glycoprotein II inhibitor.
Few necessary parameters for distribution include the distribution volume (VDss), blood–brain barrier membrane permeability (logBB), CNS permeability and Fraction unbound (human). Volume of distribution (Vd) is a pharmacokinetic parameter that indicates the tendency of the drug to exist in plasma or redistribute to other compartments of tissue. When VDss is lesser than 0.71 L kg−1 (log VDss < − 0.15), the volume of distribution is contemplated to be relatively low which indicates that the drug tends to remain in the plasma. When VDss is more than 2.81 L kg−1 (log VDss > 0.45), the volume of distribution is examined to be relatively more which indicates that the drug has the propensity to exit the plasma and pass into the body’s extravascular compartments (Mansoor and Mahabadi 2021). Neocafhispidulin, plebeiosides B, plebeiosides A, salviaplebeiaside, rosmarinic acid, 6″-O-acetylhomoplantaginin, Aflatoxin B2 showed relatively low distribution volumes. Rutin, cynaroside and 6 hydroxyluteolin 7 glucoside showed moderate values of distribution volume. In the case of blood–brain barrier membrane permeability, if the value of logBB is higher than 0.3, the compounds were thought to traverse the blood–brain barrier easily. LogBB value lower than − 1 suggests that the compounds do not easily traverse the blood–brain barrier. None of the compounds were found to traverse the blood–brain barrier easily indicating that the ligands express fairly minimal side effects and toxicity to the brain. For CNS permeability, logPS < − 3 shows that the compounds can easily penetrate the CNS. All the compounds were speculated to be able to penetrate the CNS except Aflatoxin B2.
Cytochrome P450s is a key enzyme system for the metabolism of drugs in the liver. CYP3A4 and CYP2D6 are known to be the two important subtypes of cytochrome P450. For metabolism parameters, the results suggested that only Aflatoxin B2 was a substrate for CYP3A4. None of the compounds were a substrate for CYP2D6. Aflatoxin B2 was anticipated to be a CYP1A2 and CYP2C19 inhibitor. Neocafhispidulin was found to be a CYP2C9 and CYP3A4 inhibitor. This indicated that these compounds may be metabolized in the liver.
Excretion parameters include renal OCT2 substrate and total clearance. The pkCSM results show that none of the compounds is an OCT2 substrate and almost all the compounds show high values of total clearance. Drug elimination is associated with the hydrophilicity of compounds and molecular weight. The results also suggest that none of the compounds is toxic in the AMES test except Plebeiosides B and Aflatoxin B2. MRTD is the maximum recommended tolerated dose. The value of MRTD ≤ 0.477 log (mg/kg/day) is regarded to be low and a value higher than 0.477 is considered to be high. Most of the ligands displayed low MRTD values. None of the compounds is hepatotoxic and might not possess skin sensitization or cardiotoxicity.
Prediction of activity spectra of substances (PASS)
The biological activities of the top compounds Rutin and Plebeiosides B were analysed using the online version of PASS software and the results indicate that the top compounds from Salvia plebeia possess different antioxidant and antiviral activities. The results are presented in Table 3.
Drug likeness
In silico drug-likeness prediction along with ADMET studies accelerates the discovery of lead compounds with desired biological activity (Ibrahim et al. 2020). Table 4 displays the drug-likeness properties of the top 10 compounds. Six compounds, i.e., plebeiosides B, plebeiosides A, salviaplebeiaside, neocafhispidulin, rosmarinic acid and Aflatoxin B2 are found to obey lipinski’s rule where the number of HBD was lower than or equal to 5 and number of HBA was lower than or equal to 10. The molecular weights of these compounds are found to be less than 500 suggesting that they can be easily transported, diffused and absorbed (Murugesan et al. 2020).
The value of AlogP was found to be less than 5. AlogP depicts the hydrophilicity of the compound. Greater value of AlogP suggests poor absorption and permeation due to low hydrophilicity. Neocafhispidulin and Aflatoxin B2 are found to obey Veber rule. According to this rule, the number of rotatable bonds should be lower than or equal to 10 and the value of topological polar surface area (TPSA) should be lower than or equal to 140A. TPSA is a good indicator of oral bioavailability (Murugesan et al. 2020).
Rutin was found to violate Lipinski’s rule and Veber rule. From literature, it is known that two of the main limitations of Lipinski’s rule of Five is that it excludes natural products and it overemphasizes the oral bioavailability (Zhang and Wilkinson 2007). Veber’s rule, which is a variation of Lipinski’s rule combines a descriptor for oral bioavailability with those of Lipinski’s rule (Pollastri 2010). Nearly 50% of the FDA approved small-molecule drugs are known to either violate the Rule of Five or are not used orally. This showed that following these rules strictly would lead to overlooking certain potential lead molecules (Zhang and Wilkinson 2007) and so they must be looked at as guidelines rather than strict rules (Pollastri 2010). Hence, even though these violations were seen, Rutin was still considered to be a prospective lead molecule against SARS-CoV-2.
Molecular dynamics simulations and MM-PBSA
The docking studies were followed by molecular dynamics simulations. The system stability, flexibility, and dynamic properties were studied using the apo-form of COVID-19 main protease (apo-Mpro) and the top 2 (Rutin, Plebeiosides B) docked ligand complexes. This was done by performing MD simulation for 300 ns and the results are as shown in Fig. 2.

The plots of molecular dynamics simulations for SARS-CoV-2 main protease (Mpro) in complex with the ligands, Rutin and Plebeiosides B, obtained from Salvia plebeia during 300 ns simulation. a The root mean square deviation (RMSD) plot for the complexes. b The root mean square fluctuation (RMSF) plot for the complexes. c Plot of number of hydrogen bond formation within the complexes. d Plot of radius of gyration (Rg) for the complexes. e Plot of solvent accessible surface area (SASA) for the complexes
Root mean square deviation (RMSD) analysis
The dynamic stability of the system and the conformational changes occurring in the backbone of the protein throughout the simulation period is signified by the RMSD. The calculation of the RMSD values for the apo-Mpro and the complexes of Mpro with Rutin and Plebeiosides B have been carried out as shown in Fig. 2a. It can be seen that apo-Mpro and the complexes show slight fluctuations up to 150 ns and have got the equilibration state thereafter. The average RMSD values for apo-Mpro, Mpro–Rutin and Mpro–Plebeiosides B is found to be 0.152 nm, 0.147 nm and 0.218 nm, respectively. The Mpro–Plebeiosides B complex has shown a comparatively greater RMSD value from 150 ns onwards. It has a constant RMSD of 0.35 nm for 150 ns and has suddenly risen to a value of 0.5 nm during 150–300 ns. The apo-Mpro and the Mpro–Rutin complexes have maintained a constant RMSD value of 0.3 nm.
Root mean square fluctuation (RMSF) analysis
The root mean square fluctuation (RMSF) plot shows fluctuations of the residues during the simulation (Fig. 2b). In both the complexes i.e., Mpro–Rutin and Mpro–Plebeiosides B, the fluctuations of Cα atoms of the protein were comparatively more in domain I and domain III and the amino acid residues which interacted with the ligands during docking exhibited minimal fluctuation values. Most importantly, the catalytic dyad residues (Cys145 and His41) which interacted with both the ligands during docking, fluctuated less. The average RMSF values of apo-Mpro, Mpro–Rutin and Mpro–Plebeiosides B complexes were calculated to be 0.81 nm, 0.436 nm and 0.653 nm, respectively. The analysis shows that the complexes were stable throughout the simulation with no major structural changes. MD trajectories corresponding to apo-Mpro, Mpro–Rutin and Mpro–Plebeiosides B systems were analysed considering the existence of the total number of intermolecular hydrogen bonds between the receptor and the ligand, radius of gyration (Rg) and solvent accessible surface area (SASA).
Hydrogen bond analysis
The conformational stability of the complexes was analysed by the number of intermolecular hydrogen bonds formed. Analysis of hydrogen bonds is very important as they are highly specific (Das et al. 2020). An adequate number of H-bonds were developed between the receptor and the ligand over the course of the simulation (Fig. 2c). The average number of intermolecular hydrogen bonds in the Mpro–Rutin complex was 6 and the maximum bonds formed were 17 and in case of Mpro–Plebeiosides B complex, the average and maximum number of bonds were 4 and 11, respectively.
Radius of gyration (Rg) analysis
Radius of gyration (Rg) of the backbone atoms is examined to analyse the compactness of the protein. The Mpro in the 6LU7-RUT complex didn’t exhibit major structural changes while the Mpro in 6LU7-PLE and the apo-Mpro was more compact at the end of the simulation (Fig. 2d). The obtained average values of Rg for the complexes Mpro–Rutin, Mpro–Plebeiosides B and apo-Mpro were found to be 2.19 nm, 2.18 nm and 2.17 nm, respectively. A stable Rg of about 2.2 nm was exhibited by the ligands chosen from the natural source throughout the 300 ns simulation time and suggested that these complexes were rigid.
Solvent accessible surface area
SASA is examined to determine the degree of expansion of protein volume. Average SASA values corresponding to the complexes Mpro–Rutin, Mpro–Plebeiosides B and apo-Mpro (Fig. 2e) were found to be 154.34 nm2,155.02 nm2 and 149.89 nm2, respectively. The complete analysis revealed the favourable binding of both the ligands to Mpro.
MM-PBSA binding free energy calculations
The binding free energies of complexes were predicted using the MM-PBSA scheme. MM-PBSA can be used to perceive the contribution of amino acid residues within the active site to the structural stability of ligands and binding free energy throughout the simulation period (Kumar and Pandey 2013). MM-PBSA calculates absolute binding free energies that are the sum of the electrostatic interaction, polar solvation free energy, van der Waals interaction and nonpolar solvation free energy. The predicted energy components for the binding of Rutin and Plebeiosides B with main protease are summarised in Table 5. From the results, it is observed that electrostatic, SASA and van der Waals energy has a negative contribution to the total interaction energy and the polar solvation energy has a positive contribution to the total binding energy. The van der Waals interaction’s contribution between the protein and ligands is larger in magnitude and the SASA’s contribution is comparatively less. A high negative value of van der Waals energy indicates the massive hydrophobic interactions between the ligands and the main protease (Das et al. 2020). The total binding energies of Rutin and Plebeiosides B are found to be − 75.100 ± 21.854 kJ/mol and − 121.182 ± 20.640 kJ/mol, respectively. The MM-PBSA results confirm the stable binding of the ligands to the protein.
Secondary structure analysis
Structural changes during the simulation were studied to observe the evolution in the 3D structure of the complex by comparing 4 snapshots of the protein–ligand complexes (Fig. 3). From the overall secondary structure analysis, it is observed that the protease structure remains conserved and shows minimal changes throughout the 300 ns simulation period, which shows the stability of the protease-ligand complexes. Figure 4 and Table 6 display the secondary structural analysis of the apo-protein (Mpro) as well as that of the Mpro–ligand complexes on the whole. The percentage of A-Helices/B-Sheets in the secondary structure determine its structural rigidity and the percentage of coils/turns found within the structure determine its flexibility. When Mpro binds to Rutin and Plebeiosides B, marginal structural changes were observed. Rutin showed a slightly greater percentage of coils and α-helix in comparison to the Plebeiosides B docked complex and the apo protein. The Plebeiosides B docked complex showed the same percentage of secondary structure elements as the apo-protein.

Snapshot of a the apo form of SARS-CoV-2 main protease and the SARS-CoV-2 main protease complexes docked with b Rutin and c Plebeiosides B over the 300 ns MD simulation trajectory

The secondary structure analysis of the a apo form of main protease and the Mpro complexes docked with b Rutin, and c Plebeiosides B
Based on previous studies, rutin has shown anticancer/antineoplastic effects, antimicrobial activity and antiviral activity (Araruna et al. 2012; Lin et al. 2009). Sodium rutin sulfate which is a sulfated rutin analogue, was previously studied for its anti-HIV activity against HIV-1 X 4 viruses IIIB, HIV-1 R5 isolates Ada-M and Ba-L strains (Tao et al. 2007). Plebeiosides B is a relatively newly isolated benzoylated monoterpene glycoside. This compound has been found to be structurally similar to salviaplebeiaside, isolated from S. plebeia and (1R,2S,4R,7S)-vicodiol 9-O-β-d-glucopyranoside that was isolated from Amomum xanthioides. Such camphane monoterpenes are generally found in essential oils such as citronella oil, valerian, turpentine and so on (Bang et al. 2016). Monoterpenoids are widely found in the aerial parts of the plant like leaves and flowers, and are the most important components of naturally occurring volatile products (Ghasemnezhad et al. 2020). However, as Plebeiosides B is a relatively novel compound and has not been studied extensively, further studies would be required to confirm the activities identified through in silico studies.
Conclusion
In conclusion, this study is intended to discover lead compounds that would show high efficiency in the treatment of COVID-19 by screening all the biologically active compounds obtained from Salvia plebeia against the main protease enzyme of SARS-CoV-2. A library of the 208 compounds identified was scrutinized by molecular docking and molecular dynamics simulation studies which led to the identification of Rutin and Plebeiosides B which showed maximum inhibitory activity against Mpro. The protein–ligand interactions displayed that hydrogen bonding, hydrophobic, electrostatic, polar and non-polar interactions played key roles in the contribution of the interactions at the active pocket of the protein. The compounds showed strong affinity towards the target and the analysis of the ADMET properties, biological activities, as well as drug-likeness properties, showed that they could be effective potential drug molecules against SARS-CoV-2. Furthermore, the secondary structure analyses showed the stability of the complexes formed. However, further in vitro assays can confirm if the lead molecules could be used in the treatment of COVID-19.
References
Amin SA, Jha T (2020) Fight against novel coronavirus: a perspective of medicinal chemists. Eur J Med Chem 201:112559
Araruna MKA et al (2012) Evaluation of antibiotic & antibiotic modifying activity of pilocarpine & rutin. Indian J Med Res 135(2):252
Bang S et al (2016) Antiviral activities of compounds from aerial parts of Salvia plebeia R. Br. J Ethnopharmacol 192:398–405
Bang S et al (2018) Anti-influenza effect of the major flavonoids from Salvia plebeia R.Br. via inhibition of influenza H1N1 virus neuraminidase. Nat Prod Res 32(10):1224–1228
Das S, Koner BC (2020) Pre-analytical, analytical, and post-analytical considerations while processing samples of COVID-19 patients: perspective from a clinical chemistry laboratory in India. Asian J Med Sci 11(5):112–115
Das P, Majumder R, Mandal M, Basak P (2020) In-silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid based phytochemical constituents of calendula officinalis. J Biomol Struct Dyn 39:1–16
Fauci AS, Clifford Lane H, Redfield RR (2020) Covid-19—navigating the uncharted. N Engl J Med 382(13):1268–1269
Fereidoonnezhad M et al (2018) Multitarget drug design, molecular docking and PLIF studies of novel tacrine−coumarin hybrids for the treatment of Alzheimer’s disease. Iran J Pharm Res 17(4):1217–1228
Ghasemnezhad A, Ghorbanzadeh A, Sarmast MK, Ghorbanpour M (2020) A review on botanical, phytochemical, and pharmacological characteristics of Iranian Junipers (Juniperus Spp.). Plant-derived bioactives: production, properties and therapeutic applications. Springer, Singapore
Gibaldi M, Levy G (1976) Pharmacokinetics in clinical practice: I. Concepts. JAMA J Am Med Assoc 235(17):1864–1867
Gil C et al (2020) COVID-19: drug targets and potential treatments. J Med Chem. https://doi.org/10.1021/acs.jmedchem.0c00606
Gupta S et al (2020) Identification of potential natural inhibitors of SARS-CoV2 main protease by molecular docking and simulation studies. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1776157
Han Y et al (2019) In silico ADME and toxicity prediction of ceftazidime and its impurities. Front Pharmacol 10(APR):434
Harvey AL, Edrada-Ebel R, Quinn RJ (2015) The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discovery 14(2):111–129
Ibrahim MAA, Abdeljawaad KAA, Abdelrahman AHM, Hegazy MEF (2020) Natural-like products as potential SARS-CoV-2 Mpro inhibitors: in-silico drug discovery. J Biomol Struct Dyn 39:1–13
Jacob RB, Andersen T, Mcdougal OM (2012) “Accessible high-throughput virtual screening molecular docking software for students and educators” ed Fran Lewitter. PLoS Comput Biol 8(5):e1002499
Jin Z et al (2020) Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 582(7811):289–293. https://doi.org/10.1038/s41586-020-2223-y
JP, Lin, et al (2009) Rutin inhibits the proliferation of murine leukemia WEHI-3 cells in vivo and promotes immune response in vivo. Leuk Res 33(6):823–828
Kandeel M, Ibrahim A, Fayez M, Al-Nazawi M (2020) From SARS and MERS CoVs to SARS-CoV-2: moving toward more biased codon usage in viral structural and nonstructural genes. J Med Virol 92(6):660–666
Karami M, Jalali C, Mirzaie S (2017) Combined virtual screening, MMPBSA, molecular docking and dynamics studies against deadly anthrax: an in silico effort to inhibit bacillus anthracis nucleoside hydrolase. J Theor Biol 420:180–189
Krishna S, Kumar SB, Krishna Murthy TP, Murahari M (2021) Structure-based design approach of potential BCL-2 inhibitors for cancer chemotherapy. Comput Biol Med 134:104455
Kumar S, Pandey AK (2013) Chemistry and biological activities of flavonoids: an overview. Sci World J 2013:1–16
Kumar SB et al (2021) Screening of natural compounds from Cyperus Rotundus Linn against SARS-CoV-2 main protease (Mpro): an integrated computational approach. Comput Biol Med 134:104524–104524
Kumar B et al (2021) In silico screening of therapeutic potentials from strychnos nux-vomica against the dimeric main protease (Mpro) structure of SARS-CoV-2. J Biolmol Struct Dyn. https://doi.org/10.1080/07391102.2021.1902394
Kumari R, Kumar R, Lynn A (2014) G-Mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54(7):1951–1962. https://doi.org/10.1021/ci500020m
Li H et al (2020) Coronavirus Disease 2019 (COVID-19): current status and future perspectives. Int J Antimicrobial Agents 55(5):105951
Liang Y et al (2020) Salvia plebeia R. Br.: an overview about its traditional uses, chemical constituents, pharmacology and modern applications. Biomed Pharmacother 121(219):109589
Lindahl, Abraham, Hess, van der Spoel (2019) GROMACS 2019.4 manual
Ma Q et al (2014) Chemistry and Pharmacology of Salvia Plebeia R. Brown (Lamiaceae ). J Chem Pharm Res 6(10):777–783
Mansoor A, Mahabadi N (2021) Volume of Distribution. In: StatPearls [Internet]. StatPearls Publishing, Treasure Island (FL). https://www.ncbi.nlm.nih.gov/books/NBK545280/
Marathe SA, Datey AA (2012) Herbal cocktail as anti-infective: promising therapeutic for the treatment of viral diseases. Recent Pat Anti-Infective Drug Discov 7(2):123–132
Mason RJ (2020) Pathogenesis of COVID-19 from a cell biology perspective. Eur Respir J 55(4):2000607
Murugesan S, Venkateswaran MR, Jayabal S, Periyasamy S (2020) Evaluation of the antioxidant and anti-arthritic potential of zingiber officinale Rosc. by in vitro and in silico analysis. S Afr J Bot 130:45–53
Naidoo D et al (2020) Cyanobacterial metabolites as promising drug leads against the Mpro and PLpro of SARS-CoV-2: an in silico analysis. J Biomol Struct Dyn 39:1–13
Ngane A, Ngono R et al (2011) Ethnobotanical survey of some cameroonian plants used for treatment of viral diseases. Afr J Plant Sci 5(1):15–21
Nisius B, Sha F, Gohlke H (2012) Structure-based computational analysis of protein binding sites for function and druggability prediction. J Biotechnol 159(3):123–134
Nugroho A et al (2012) In vivo sedative and gastroprotective activities of Salvia plebeia extract and its composition of polyphenols. Arch Pharmacal Res 35(8):1403–1411
Nurton J (2020) Drug repurposing and the COVID-19 pandemic, WIPO Magazine. https://www.wipo.int/wipo_magazine/en/2020/02/article_0004.html
Patil R et al (2010) “Optimized hydrophobic interactions and hydrogen bonding at the target-ligand interface leads the pathways of drug-designing” ed Sridhar Hannenhalli. PLoS ONE 5(8):e12029
Pires DEV, Blundell TL, Ascher DB (2015) PkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med cinal Chem 58(9):4066–72. http://structure.bioc.cam.ac.uk/. Accessed 28 Jul 2020
Pollastri MP (2010) Overview on the rule of five. Curr Protoc Pharmacol 49(1):9.12.1-9.12.8
Prasanth DSNBK et al (2020) In silico identification of potential inhibitors from cinnamon against main protease and spike glycoprotein of SARS CoV-2. J Biomol Struct Dyn. https://doi.org/10.1080/07391102.2020.1779129
Pushpakom S et al (2018) Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discovery 18(1):41–58
Ranney ML, Griffeth V, Jha AK (2020) Critical supply shortages—the need for ventilators and personal protective equipment during the Covid-19 pandemic. N Engl J Med 382(18):E41
Ren DB et al (2014) Separation of nine compounds from Salvia plebeia R.Br. using two-step high-speed counter-current chromatography with different elution modes. J Sep Sci 37(16):2118–2125
Schüttelkopf AW, Van Aalten DMF (2004) “PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr Sect D Biol Crystallogr 60(8):1355–1363. http://scripts.iucr.org/cgi-bin/paper?S0907444904011679. Accessed 29 Jul 2020
Shineman DW et al (2014) Overcoming obstacles to repurposing for neurodegenerative disease. Ann Clin Transl Neurol 1(7):512–518
Talevi A, Bellera CL (2020) Challenges and opportunities with drug repurposing: finding strategies to find alternative uses of therapeutics. Expert Opin Drug Discov 15(4):397–401
Tao J et al (2007) In vitro anti-HIV and -HSV activity and safety of sodium rutin sulfate as a microbicide candidate. Antiviral Res 75(3):227–233
Veeresham C (2012) Natural products derived from plants as a source of drugs. J Adv Pharm Technol Res 3(4):200–201
Wang J et al (2019) Biosynthesis, chemistry, and pharmacology of polyphenols from Chinese salvia species: a review. Molecules 24(1):1–23
Xiu S et al (2020) Inhibitors of SARS-CoV-2 entry: current and future opportunities. J Med Chem. https://doi.org/10.1021/acs.jmedchem.0c00502
Zhang MQ, Wilkinson B (2007) Drug discovery beyond the ‘rule-of-five.’ Curr Opin Biotechnol 18(6):478–488
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zackria, A.A., Pattabiraman, R., Murthy, T.P.K. et al. Computational screening of natural compounds from Salvia plebeia R. Br. for inhibition of SARS-CoV-2 main protease. Vegetos 35, 345–359 (2022). https://doi.org/10.1007/s42535-021-00304-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42535-021-00304-z