
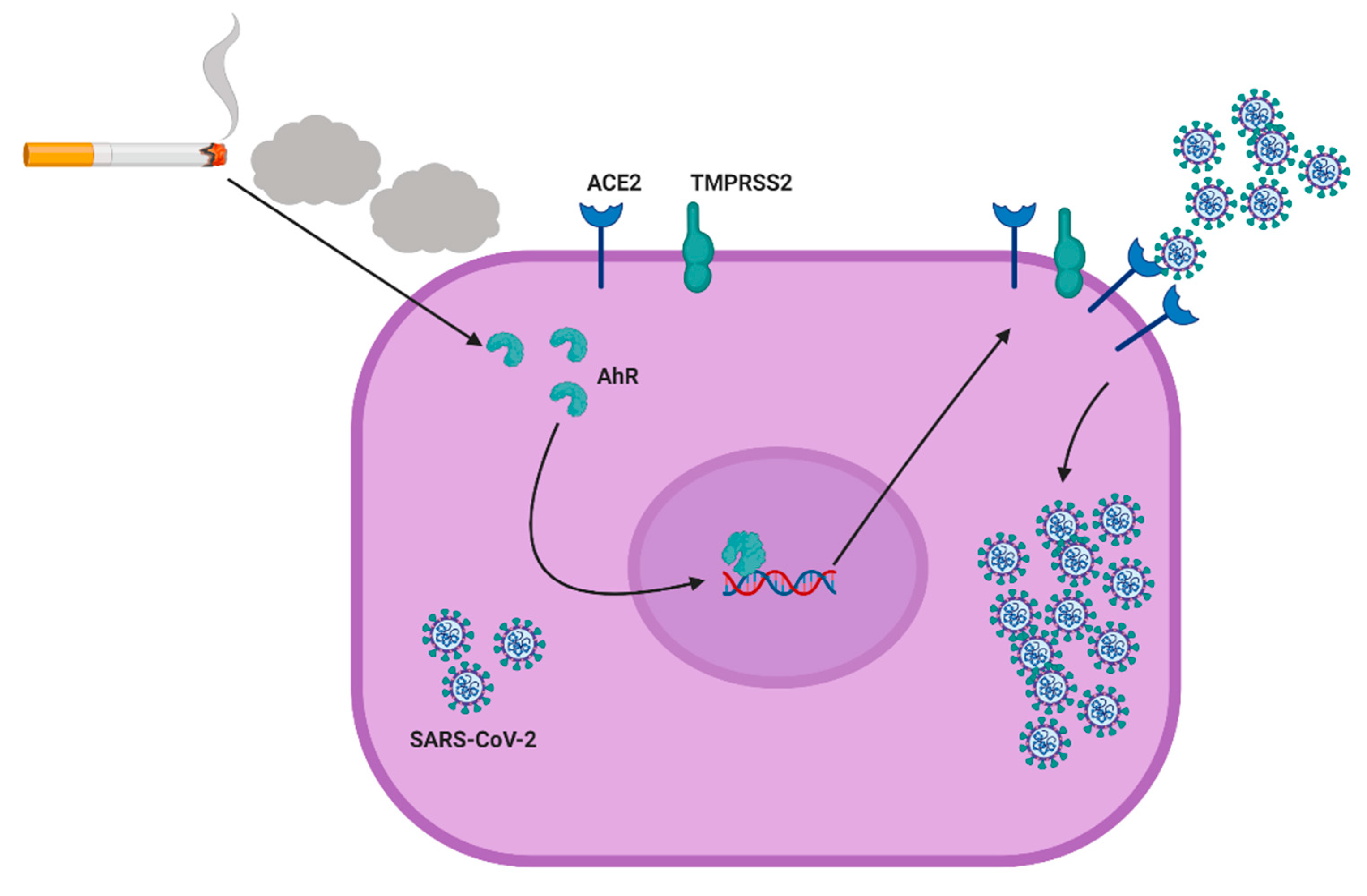
Cigarette Smoke Stimulates SARS-CoV-2 Internalization by Activating AhR and Increasing ACE2 Expression in Human Gingival Epithelial Cells

 ,
, 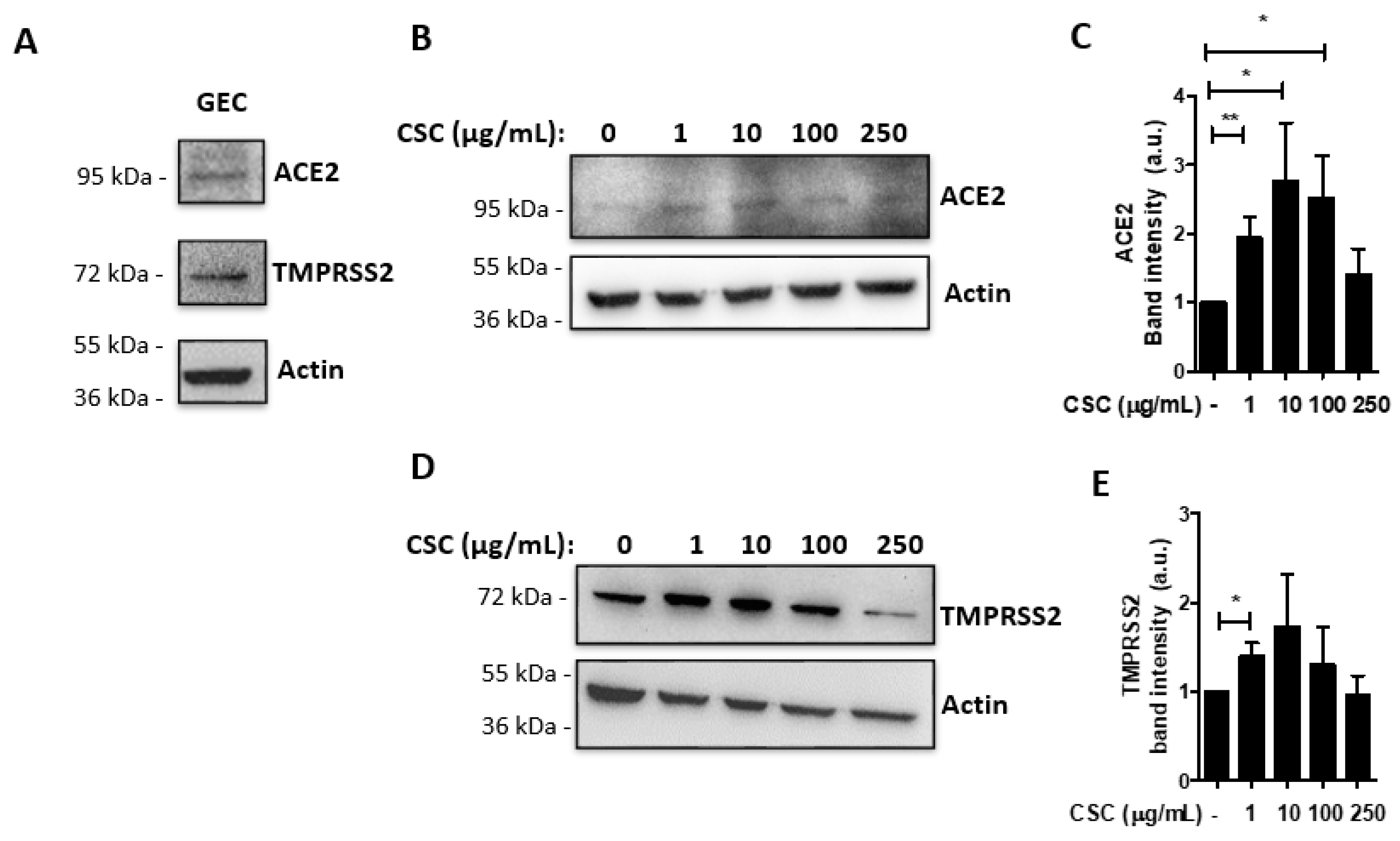{kind=link}
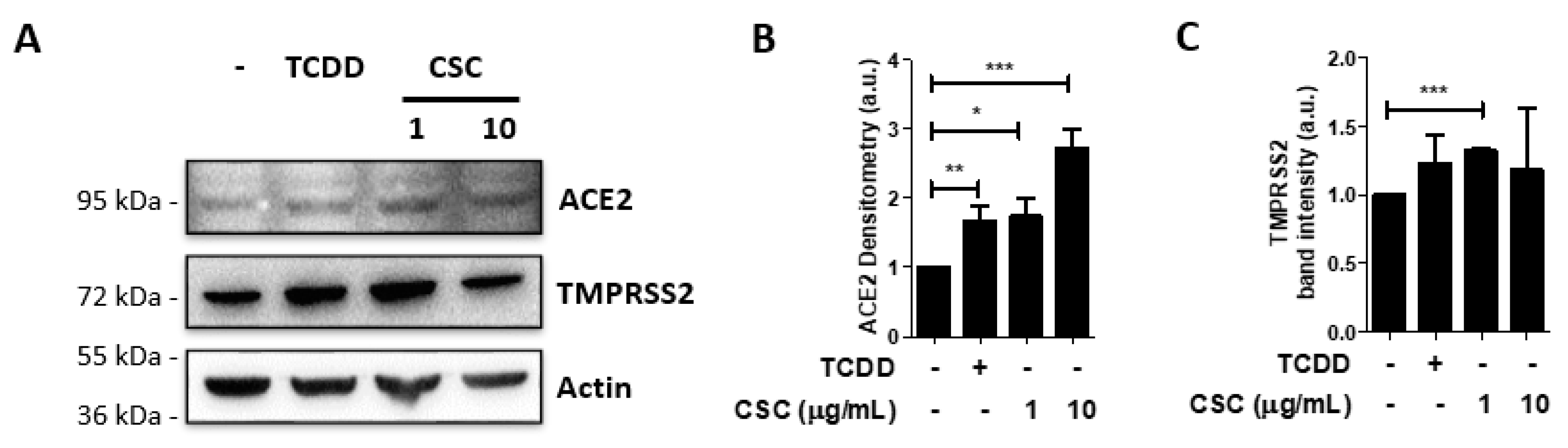{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Cigarette Smoke Condensates Enhance ACE2 and TMPRSS2 Expression in Gingival Epithelial Cells
2.2. Activation of the Arzyl Hydrocarbon Receptor Upregulates ACE2 Expression in Gingival Epithelial Cells
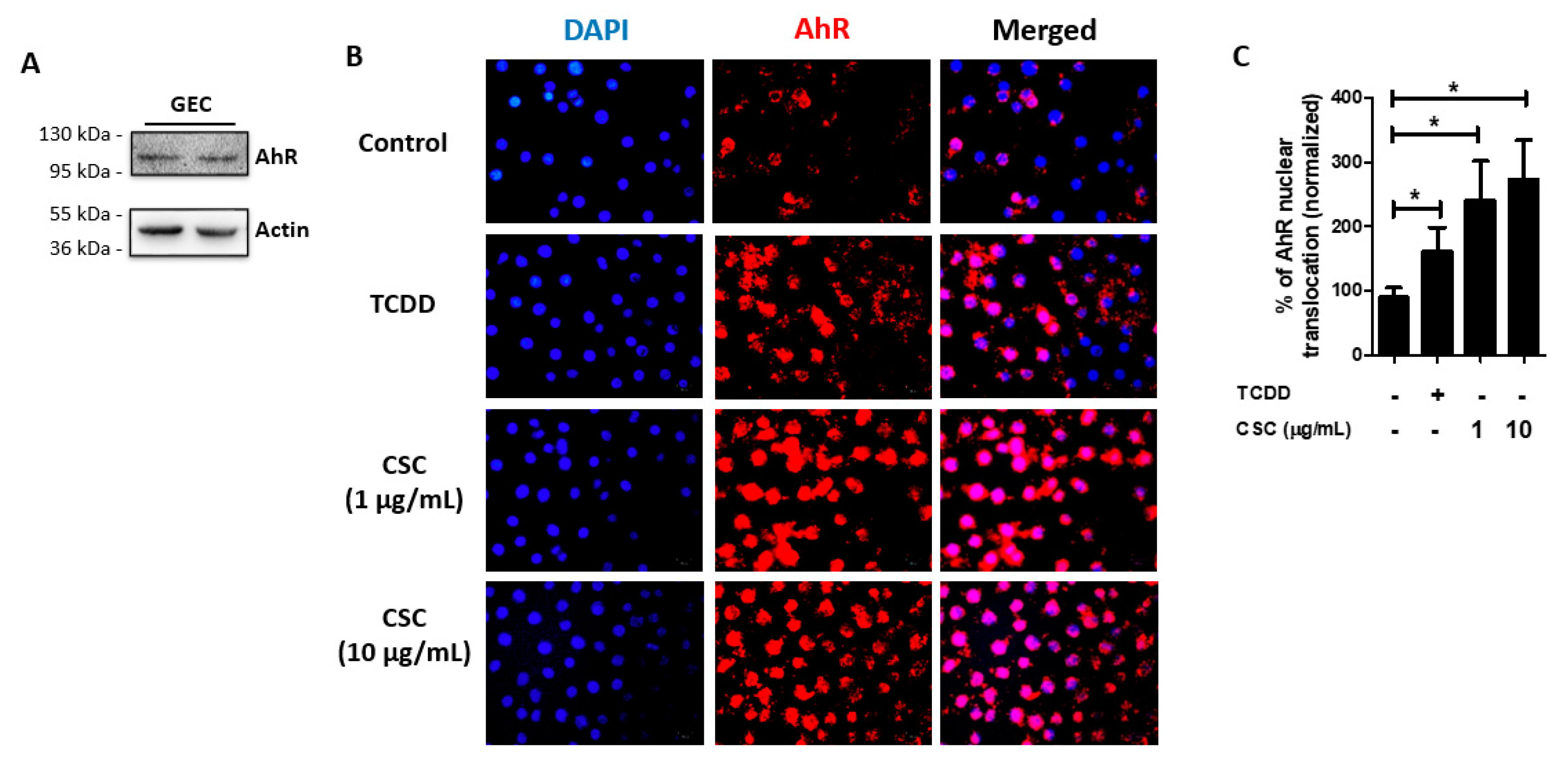
2.3. AhR Is Expressed and Activated in Gingival Epithelial Cells by Cigarette Smoke Condensate Treatment
2.4. Cigarette Smoke Condensates Enhance SARS-CoV-2 Pseudovirus Internalization in Gingival Epithelial Cells
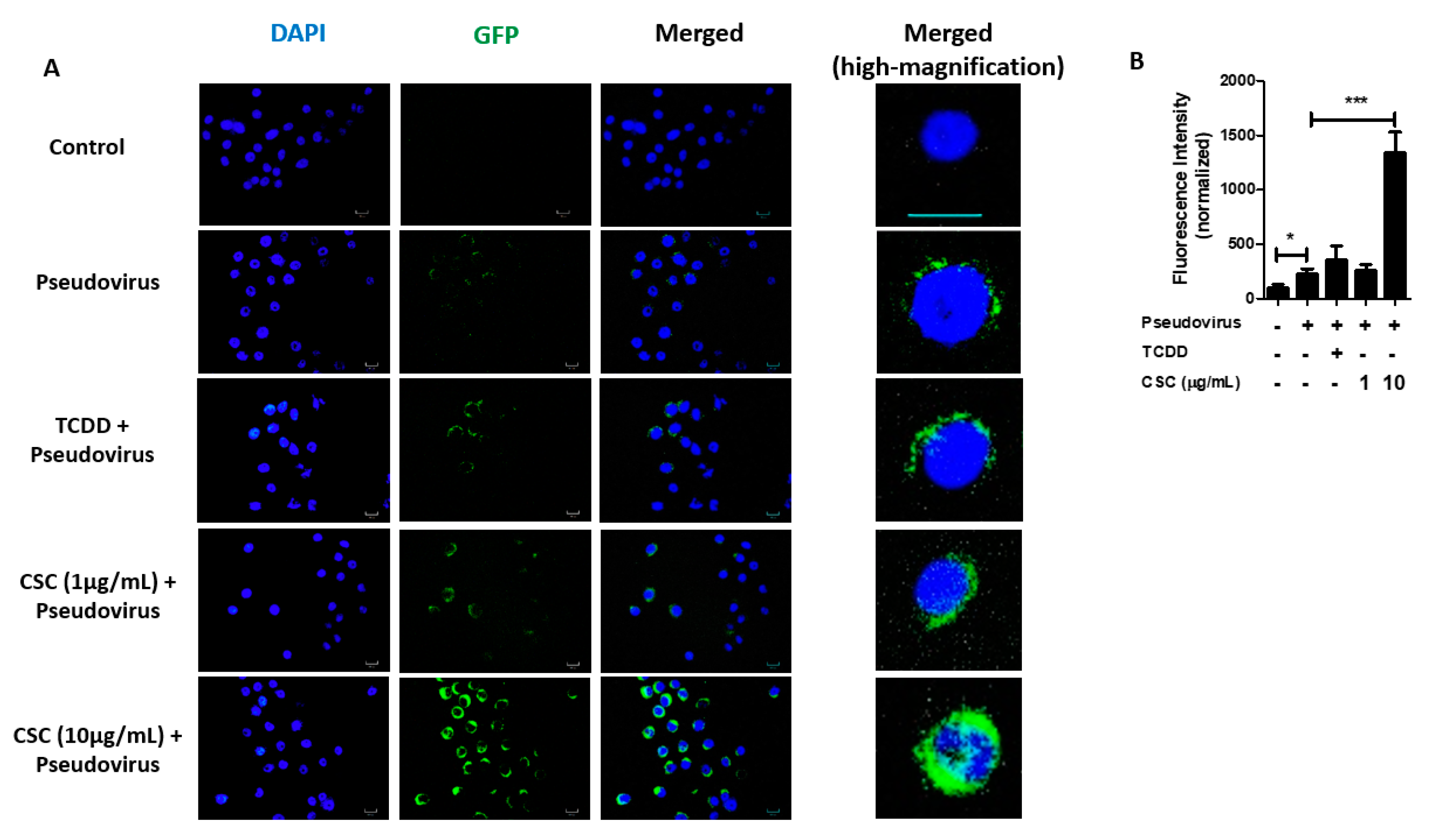
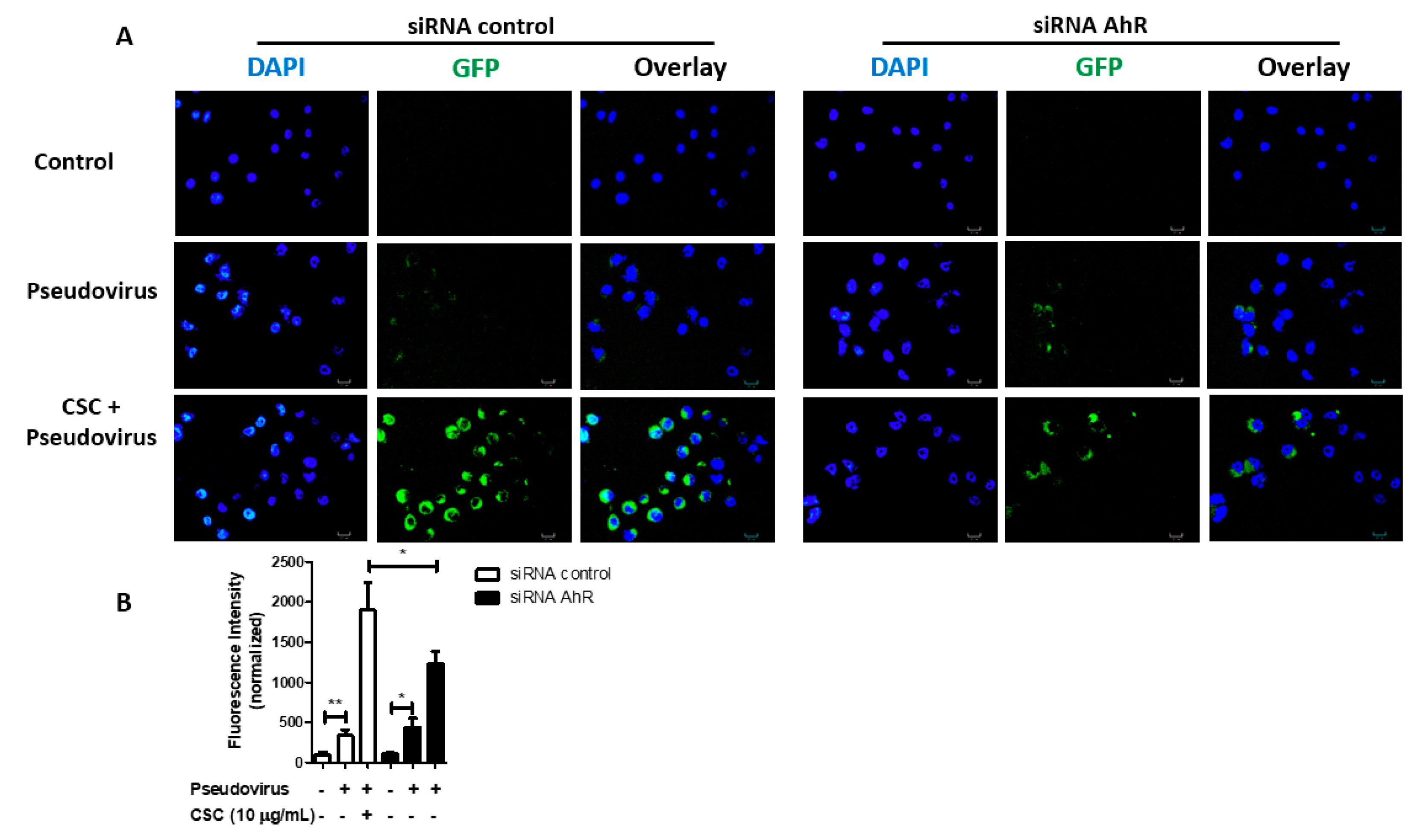
2.5. Cigarette Smoke Condensates Promote SARS-CoV-2 Pseudovirus Internalization via AhR Activation in Gingival Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Human Gingival Epithelial Cells and Cell Culture
4.2. Reagents
4.3. SARS-CoV-2 Spike Protein Pseudotyped GFP Lentivirus
4.4. Transient RNA Depletion Using siRNA
4.5. Werstern Blot Analysis
4.6. AhR Immunofluorescence
4.7. Lactate Dehydrogenase Detection Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Almeida-da-Silva, C.L.C.; Matshik Dakafay, H.; O’Brien, K.; Montierth, D.; Xiao, N.; Ojcius, D.M. Effects of electronic cigarette aerosol exposure on oral and systemic health. Biomed. J. 2020. [Google Scholar] [CrossRef]
- The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General; Centers for Disease Control and Prevention: Atlanta, GA, USA, 2014.
- Creamer, M.R.; Wang, T.W.; Babb, S.; Cullen, K.A.; Day, H.; Willis, G.; Jamal, A.; Neff, L. Tobacco Product Use and Cessation Indicators Among Adults—United States, 2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 1013–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omboni, S. Smoking and hypertension: What is behind the mask? J. Hypertens 2020, 38, 1029–1030. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.J.; Stewart, C.P.; Davis, S.K.; Harvey, D.J.; Leistikow, B.N. The risk and burden of smoking related heart disease mortality among young people in the United States. Tob. Induc. Dis. 2015, 13, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimatani, K.; Ito, H.; Matsuo, K.; Tajima, K.; Takezaki, T. Cumulative cigarette tar exposure and lung cancer risk among Japanese smokers. Jpn. J. Clin. Oncol. 2020, 50, 1009–1017. [Google Scholar] [CrossRef]
- Kispert, S.; McHowat, J. Recent insights into cigarette smoking as a lifestyle risk factor for breast cancer. Breast Cancer 2017, 9, 127–132. [Google Scholar] [CrossRef] [Green Version]
- Nocini, R.; Lippi, G.; Mattiuzzi, C. The worldwide burden of smoking-related oral cancer deaths. Clin. Exp. Dent. Res. 2020, 6, 161–164. [Google Scholar] [CrossRef]
- Szwarcbard, N.; Villani, M.; Earnest, A.; Flack, J.; Andrikopoulos, S.; Wischer, N.; Soldatos, G.; Gasevic, D.; Zoungas, S. The association of smoking status with glycemic control, metabolic profile and diabetic complications- Results of the Australian National Diabetes Audit (ANDA). J. Diabetes Complicat. 2020, 34, 107626. [Google Scholar] [CrossRef]
- Calfee, C.S.; Matthay, M.A.; Kangelaris, K.N.; Siew, E.D.; Janz, D.R.; Bernard, G.R.; May, A.K.; Jacob, P.; Havel, C.; Benowitz, N.L.; et al. Cigarette Smoke Exposure and the Acute Respiratory Distress Syndrome. Crit. Care. Med. 2015, 43, 1790–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Gottlieb, E.; Rounds, S. Effects of cigarette smoke on pulmonary endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L743–L756. [Google Scholar] [CrossRef] [PubMed]
- Rachmawati, E.; Listiowati, E.; Kurniawan, D.W.; Suraya, I.; Ahsan, A.; Nurmansyah, M.I. Significance of Chronic Diseases and Smoking Behavior in the Development of Acute Respiratory Distress Syndrome Among Hospitalized COVID-19 Patients in Indonesia. Asia Pac. J. Public Health 2021. [Google Scholar] [CrossRef]
- Gulsen, A.; Yigitbas, B.A.; Uslu, B.; Dromann, D.; Kilinc, O. The Effect of Smoking on COVID-19 Symptom Severity: Systematic Review and Meta-Analysis. Pulm. Med. 2020, 2020, 7590207. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Carvalho, T.; Krammer, F.; Iwasaki, A. The first 12 months of COVID-19: A timeline of immunological insights. Nat. Rev. Immunol. 2021, 21, 245–256. [Google Scholar] [CrossRef]
- World Health Organization. Available online: https://covid19.who.int/ (accessed on 22 June 2021).
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Wu, L.; O’Kane, A.M.; Peng, H.; Bi, Y.; Motriuk-Smith, D.; Ren, J. SARS-CoV-2 and cardiovascular complications: From molecular mechanisms to pharmaceutical management. Biochem. Pharm. 2020, 178, 114114. [Google Scholar] [CrossRef] [PubMed]
- Pascarella, G.; Strumia, A.; Piliego, C.; Bruno, F.; Del Buono, R.; Costa, F.; Scarlata, S.; Agro, F.E. COVID-19 diagnosis and management: A comprehensive review. J. Intern. Med. 2020, 288, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Kamal, M.; Abo Omirah, M.; Hussein, A.; Saeed, H. Assessment and characterisation of post-COVID-19 manifestations. Int. J. Clin. Pract 2021, 75, e13746. [Google Scholar] [CrossRef] [PubMed]
- Vardavas, C.I.; Nikitara, K. COVID-19 and smoking: A systematic review of the evidence. Tob. Induc. Dis. 2020, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Bosse, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco Smoking Increases the Lung Gene Expression of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 201, 1557–1559. [Google Scholar] [CrossRef]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV-2 Receptor ACE2 in the Respiratory Tract. Dev. Cell. 2020, 53, 514–529.e3. [Google Scholar] [CrossRef]
- Li, G.; He, X.; Zhang, L.; Ran, Q.; Wang, J.; Xiong, A.; Wu, D.; Chen, F.; Sun, J.; Chang, C. Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J. Autoimmun 2020, 112, 102463. [Google Scholar] [CrossRef]
- Liu, A.; Zhang, X.; Li, R.; Zheng, M.; Yang, S.; Dai, L.; Wu, A.; Hu, C.; Huang, Y.; Xie, M.; et al. Overexpression of the SARS-CoV-2 receptor ACE2 is induced by cigarette smoke in bronchial and alveolar epithelia. J. Pathol. 2021, 253, 17–30. [Google Scholar] [CrossRef]
- Amorim Dos Santos, J.; Normando, A.G.C.; Carvalho da Silva, R.L.; Acevedo, A.C.; De Luca Canto, G.; Sugaya, N.; Santos-Silva, A.R.; Guerra, E.N.S. Oral Manifestations in Patients with COVID-19: A Living Systematic Review. J. Dent. Res. 2021, 100, 141–154. [Google Scholar] [CrossRef]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Lin, B.; Pathak, J.L.; Gao, H.; Young, A.J.; Wang, X.; Liu, C.; Wu, K.; Liu, M.; Chen, J.M.; et al. ACE2 and Furin Expressions in Oral Epithelial Cells Possibly Facilitate COVID-19 Infection via Respiratory and Fecal-Oral Routes. Front. Med. (Lausanne) 2020, 7, 580796. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, W.; Kubota, N.; Shimizu, T.; Saruta, J.; Fuchida, S.; Kawata, A.; Yamamoto, Y.; Sugimoto, M.; Yakeishi, M.; Tsukinoki, K. Existence of SARS-CoV-2 Entry Molecules in the Oral Cavity. Int. J. Mol. Sci. 2020, 21, 6000. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.C.; Fachin, S.; da Fonseca, J.G.; Angst, P.D.M.; Lamers, M.L.; da Silva, I.S.B.; Nunes, L.N. Dental biofilm of symptomatic COVID-19 patients harbours SARS-CoV-2. J. Clin. Periodontol. 2021, 48, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Perez, P.; Kato, T.; Mikami, Y.; Okuda, K.; Gilmore, R.C.; Conde, C.D.; Gasmi, B.; Stein, S.; Beach, M.; et al. SARS-CoV-2 infection of the oral cavity and saliva. Nat. Med. 2021, 27, 892–903. [Google Scholar] [CrossRef]
- Imamura, K.; Kokubu, E.; Kita, D.; Ota, K.; Ishihara, K.; Saito, A. Cigarette smoke condensate modulates migration of human gingival epithelial cells and their interactions with Porphyromonas gingivalis. J. Periodontal Res. 2015, 50, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Guerrina, N.; Traboulsi, H.; Eidelman, D.H.; Baglole, C.J. The Aryl Hydrocarbon Receptor and the Maintenance of Lung Health. Int. J. Mol. Sci. 2018, 19, 3882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Engen, S.A.; Rorvik, G.H.; Schreurs, O.; Blix, I.J.; Schenck, K. The oral commensal Streptococcus mitis activates the aryl hydrocarbon receptor in human oral epithelial cells. Int. J. Oral Sci. 2017, 9, 145–150. [Google Scholar] [CrossRef]
- Martelli, D.R.; Coletta, R.D.; Oliveira, E.A.; Swerts, M.S.; Rodrigues, L.A.; Oliveira, M.C.; Martelli Junior, H. Association between maternal smoking, gender, and cleft lip and palate. Braz. J. Otorhinolaryngol. 2015, 81, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidrich, J.; Wellmann, J.; Heuschmann, P.U.; Kraywinkel, K.; Keil, U. Mortality and morbidity from coronary heart disease attributable to passive smoking. Eur. Heart J. 2007, 28, 2498–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, M.D.; Balmes, J.; Katz, P.P.; Trupin, L.; Yelin, E.H.; Blanc, P.D. Lifetime environmental tobacco smoke exposure and the risk of chronic obstructive pulmonary disease. Environ. Health 2005, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Buscetta, M.; Di Vincenzo, S.; Miele, M.; Badami, E.; Pace, E.; Cipollina, C. Cigarette smoke inhibits the NLRP3 inflammasome and leads to caspase-1 activation via the TLR4-TRIF-caspase-8 axis in human macrophages. FASEB J. 2020, 34, 1819–1832. [Google Scholar] [CrossRef] [Green Version]
- White, P.C.; Hirschfeld, J.; Milward, M.R.; Cooper, P.R.; Wright, H.J.; Matthews, J.B.; Chapple, I.L.C. Cigarette smoke modifies neutrophil chemotaxis, neutrophil extracellular trap formation and inflammatory response-related gene expression. J. Periodontal Res. 2018, 53, 525–535. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Fang, M.; Song, F.; Windsor, L.J. Effects of cigarette smoke condensate and nicotine on human gingival fibroblast-mediated collagen degradation. J. Periodontol. 2011, 82, 1071–1079. [Google Scholar] [CrossRef]
- Robson, N.; Bond, A.; Wolff, K. Salivary nicotine and cotinine concentrations in unstimulated and stimulated saliva. Afr. J. Pharm. Pharmacol. 2010, 4, 61–65. [Google Scholar] [CrossRef]
- Gkogkou, E.; Barnasas, G.; Vougas, K.; Trougakos, I.P. Expression profiling meta-analysis of ACE2 and TMPRSS2, the putative anti-inflammatory receptor and priming protease of SARS-CoV-2 in human cells, and identification of putative modulators. Redox. Biol. 2020, 36, 101615. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.Y.; Qiu, H.; Cao, Q.Q.; Duan, Z.L.; Liu, F.L.; Song, T.Z.; Liu, Y.; Fang, Y.Q.; Wu, G.M.; Zheng, Y.T.; et al. Particulate matter exposure exacerbates susceptibility to SARS-CoV-2 infection in humanized ACE2 mice. Zool. Res. 2021, 42, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Lv, J.; Yu, P.; Wang, Z.; Deng, W.; Bao, L.; Liu, J.; Li, F.; Zhu, Q.; Zhou, N.; Lv, Q.; et al. ACE2 expression is regulated by AhR in SARS-CoV-2-infected macaques. Cell Mol. Immunol. 2021, 18, 1308–1310. [Google Scholar] [CrossRef]
- Cochran, C.J.; Gallicchio, L.; Miller, S.R.; Zacur, H.; Flaws, J.A. Cigarette smoking, androgen levels, and hot flushes in midlife women. Obstet. Gynecol. 2008, 112, 1037–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stopsack, K.H.; Mucci, L.A.; Antonarakis, E.S.; Nelson, P.S.; Kantoff, P.W. TMPRSS2 and COVID-19: Serendipity or Opportunity for Intervention? Cancer Discov. 2020, 10, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Chakladar, J.; Shende, N.; Li, W.T.; Rajasekaran, M.; Chang, E.Y.; Ongkeko, W.M. Smoking-Mediated Upregulation of the Androgen Pathway Leads to Increased SARS-CoV-2 Susceptibility. Int. J. Mol. Sci. 2020, 21, 3627. [Google Scholar] [CrossRef]
- Gao, Y.D.; Ding, M.; Dong, X.; Zhang, J.J.; Kursat Azkur, A.; Azkur, D.; Gan, H.; Sun, Y.L.; Fu, W.; Li, W.; et al. Risk factors for severe and critically ill COVID-19 patients: A review. Allergy 2021, 76, 428–455. [Google Scholar] [CrossRef] [PubMed]
- Babb, S.; Malarcher, A.; Schauer, G.; Asman, K.; Jamal, A. Quitting Smoking Among Adults—United States, 2000–2015. MMWR Morb. Mortal. Wkly. Rep. 2017, 65, 1457–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doll, R.; Peto, R.; Boreham, J.; Sutherland, I. Mortality in relation to smoking: 50 years’ observations on male British doctors. BMJ 2004, 328, 1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention. Quitting smoking among adults—United States, 2001–2010. MMWR Morb. Mortal. Wkly. Rep. 2011, 60, 1513–1519. [Google Scholar]
- Hamner, L.; Dubbel, P.; Capron, I.; Ross, A.; Jordan, A.; Lee, J.; Lynn, J.; Ball, A.; Narwal, S.; Russell, S.; et al. High SARS-CoV-2 Attack Rate Following Exposure at a Choir Practice—Skagit County, Washington, March 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Ghinai, I.; McPherson, T.D.; Hunter, J.C.; Kirking, H.L.; Christiansen, D.; Joshi, K.; Rubin, R.; Morales-Estrada, S.; Black, S.R.; Pacilli, M.; et al. First known person-to-person transmission of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in the USA. Lancet 2020, 395, 1137–1144. [Google Scholar] [CrossRef]
- Almeida-da-Silva, C.L.C.; Alpagot, T.; Zhu, Y.; Lee, S.S.; Roberts, B.P.; Hung, S.C.; Tang, N.; Ojcius, D.M. Chlamydia pneumoniae is present in the dental plaque of periodontitis patients and stimulates an inflammatory response in gingival epithelial cells. Microb. Cell 2019, 6, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Roztocil, E.; Hammond, C.L.; Gonzalez, M.O.; Feldon, S.E.; Woeller, C.F. The aryl hydrocarbon receptor pathway controls matrix metalloproteinase-1 and collagen levels in human orbital fibroblasts. Sci. Rep. 2020, 10, 8477. [Google Scholar] [CrossRef]
- Wessel, A.W.; Hanson, E.P. A method for the quantitative analysis of stimulation-induced nuclear translocation of the p65 subunit of NF-kappaB from patient-derived dermal fibroblasts. Methods Mol. Biol. 2015, 1280, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida-da-Silva, C.L.C.; Matshik Dakafay, H.; Liu, K.; Ojcius, D.M. Cigarette Smoke Stimulates SARS-CoV-2 Internalization by Activating AhR and Increasing ACE2 Expression in Human Gingival Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 7669. https://doi.org/10.3390/ijms22147669
Almeida-da-Silva CLC, Matshik Dakafay H, Liu K, Ojcius DM. Cigarette Smoke Stimulates SARS-CoV-2 Internalization by Activating AhR and Increasing ACE2 Expression in Human Gingival Epithelial Cells. International Journal of Molecular Sciences. 2021; 22(14):7669. https://doi.org/10.3390/ijms22147669
Chicago/Turabian StyleAlmeida-da-Silva, Cassio Luiz Coutinho, Harmony Matshik Dakafay, Kaitlyn Liu, and David M. Ojcius. 2021. "Cigarette Smoke Stimulates SARS-CoV-2 Internalization by Activating AhR and Increasing ACE2 Expression in Human Gingival Epithelial Cells" International Journal of Molecular Sciences 22, no. 14: 7669. https://doi.org/10.3390/ijms22147669