



Identification of Genomic Variants of SARS-CoV-2 Using Nanopore Sequencing

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing/Ethics
2.2. RT-qPCR Confirmation
2.3. Library Preparation and Sequencing with MinION Mk1C
2.4. Ion Torrent Sequencing
3. Results
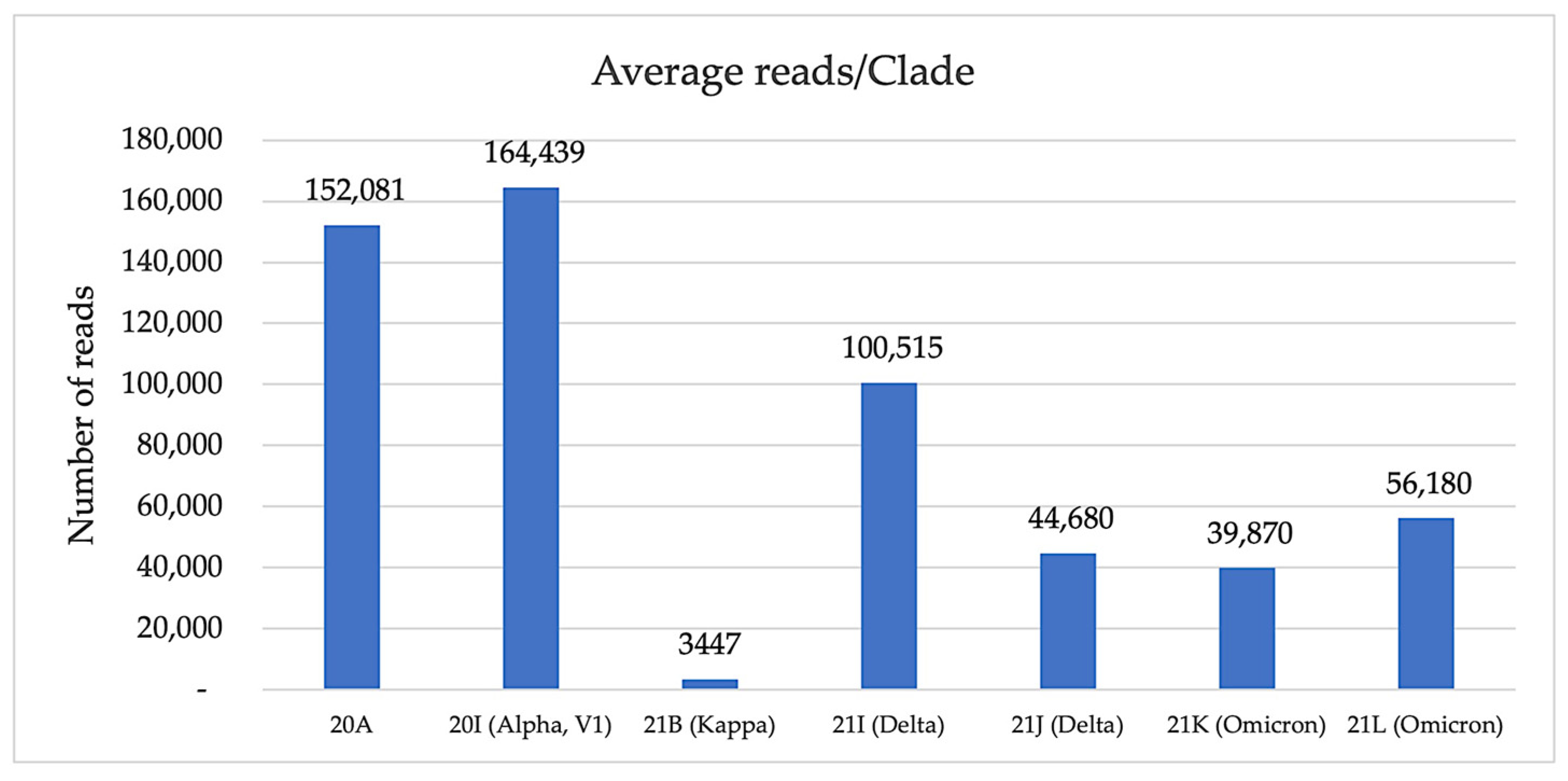
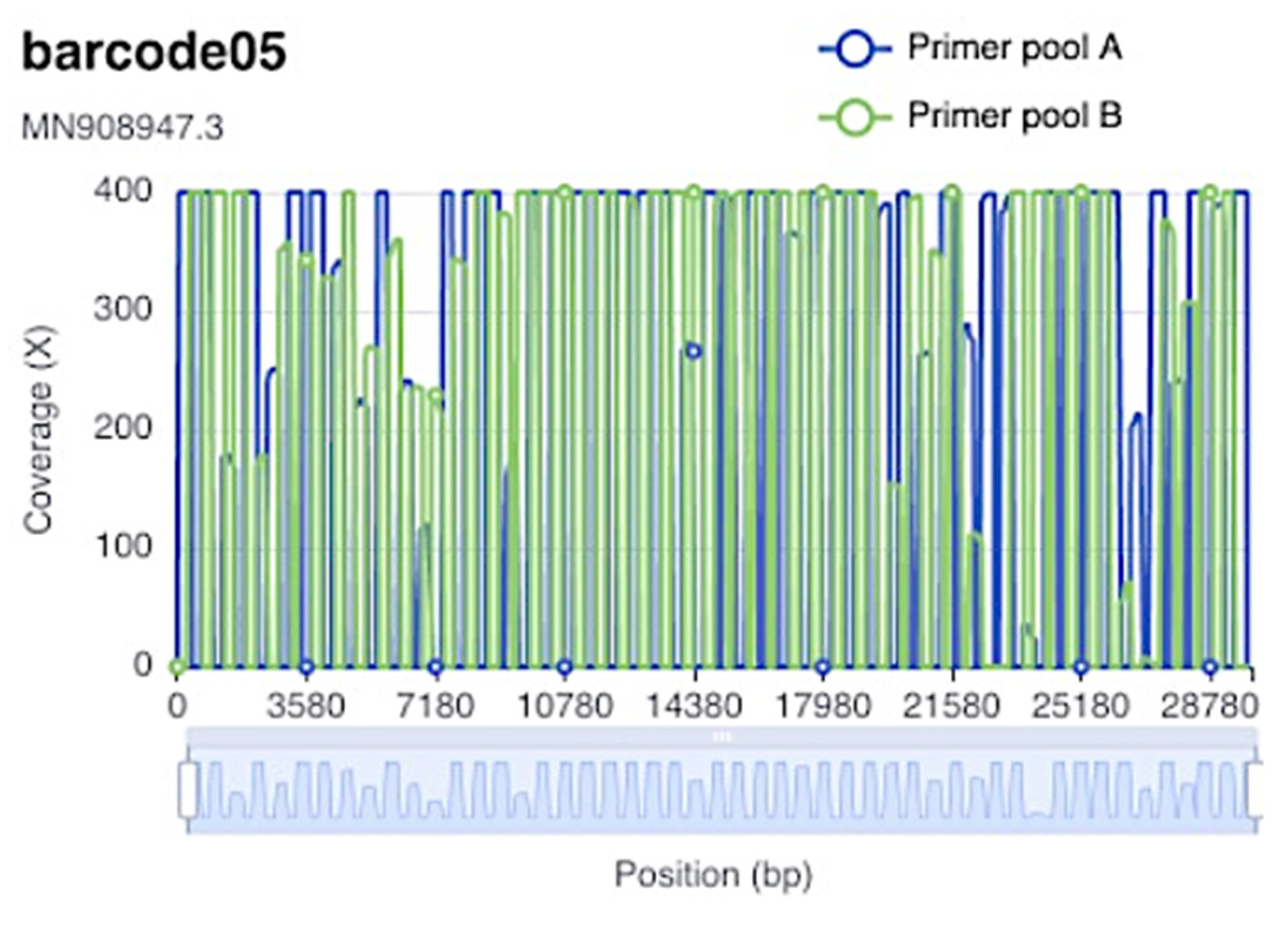
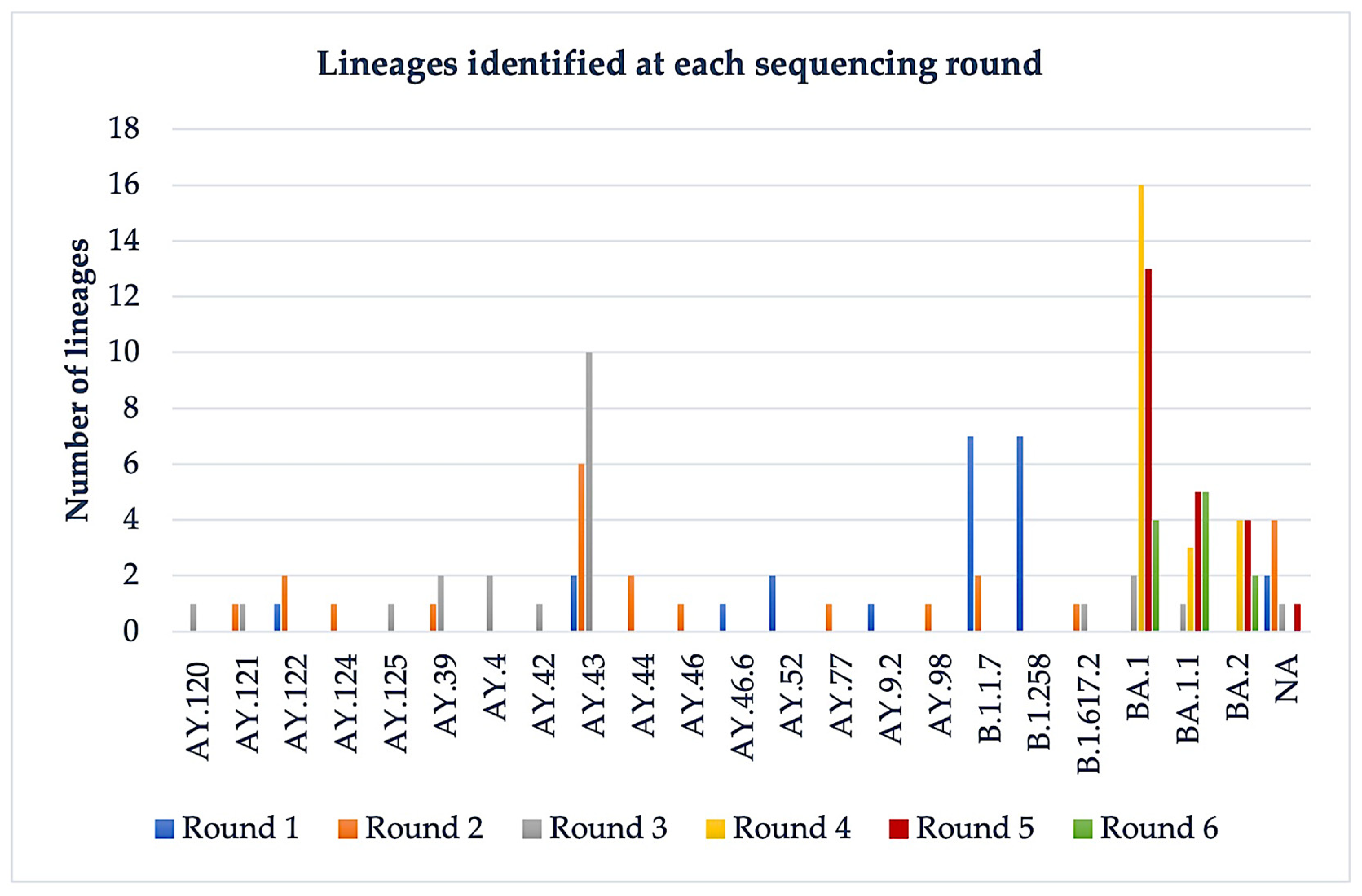
3.1. Nanopore Sequencing
3.2. Ion Torrent Sequencing
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baker, S.R.; Bloom, N.; Davis, S.J.; Terry, S.J. COVID-Induced Economic Uncertainty (No. w26983). National Bureau of Economic Research. Available online: https://www.policyuncertainty.com/media/COVID-Induced%20Economic%20Uncertainty.pdf (accessed on 13 April 2020).
- Bull, R.A.; Adikari, T.N.; Ferguson, J.M.; Hammond, J.M.; Stevanovski, I.; Beukers, A.G.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef] [PubMed]
- Hourdel, V.; Kwasiborski, A.; Balière, C.; Matheus, S.; Batéjat, C.F.; Manuguerra, J.C.; Vanhomwegen, J.; Caro, V. Rapid genomic characterization of SARS-CoV-2 by direct amplicon-based sequencing through comparison of MinION and Illumina iSeq100TM system. Front. Microbiol. 2020, 11, 571328. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Kasteren, P.B.; van Der Veer, B.; van den Brink, S.; Wijsman, L.; de Jonge, J.; van den Brandt, A.; Molenkamp, R.; Reusken, C.B.; Meijer, A. Comparison of seven commercial RT-PCR diagnostic kits for COVID-19. J. Clin. Virol. 2020, 128, 104412. [Google Scholar] [CrossRef]
- Martin, M.A.; VanInsberghe, D.; Koelle, K. Insights from SARS-CoV-2 sequences. Science 2021, 371, 466–467. [Google Scholar] [CrossRef]
- Pater, A.A.; Bosmeny, M.S.; White, A.A.; Sylvain, R.J.; Eddington, S.B.; Parasrampuria, M.; Ovington, K.N.; Metz, P.E.; Yinusa, A.O.; Barkau, C.L.; et al. High throughput nanopore sequencing of SARS-CoV-2 viral genomes from patient samples. J. Biol. Methods 2021, 8, e155. [Google Scholar] [CrossRef]
- Brejová, B.; Boršová, K.; Hodorová, V.; Čabanová, V.; Gafurov, A.; Fričová, D.; Neboháčová, M.; Vinař, T.; Klempa, B.; Nosek, J. Nanopore sequencing of SARS-CoV-2: Comparison of short and long PCR-tiling amplicon protocols. PLoS ONE 2021, 16, e0259277. [Google Scholar] [CrossRef]
- González-Recio, O.; Gutiérrez-Rivas, M.; Peiró-Pastor, R.; Aguilera-Sepúlveda, P.; Cano-Gómez, C.; Jiménez-Clavero, M.Á.; Fernández-Pinero, J. Sequencing of SARS-CoV-2 genome using different nanopore chemistries. Appl. Microbiol. Biotechnol. 2021, 105, 3225–3234. [Google Scholar] [CrossRef]
- Tillett, R.L.; Sevinsky, J.R.; Hartley, P.D.; Kerwin, H.; Crawford, N.; Gorzalski, A.; Laverdure, C.; Verma, S.C.; Rossetto, C.C.; Jackson, D.; et al. Genomic evidence for reinfection with SARS-CoV-2: A case study. Lancet Infect. Dis. 2021, 21, 52–58. [Google Scholar] [CrossRef]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomson, E.; Ip, C.L.; Badhan, A.; Christiansen, M.T.; Adamson, W.; Ansari, M.A.; Bibby, D.; Breuer, J.; Brown, A.; Bowden, R.; et al. Comparison of next-generation sequencing technologies for comprehensive assessment of full-length hepatitis C viral genomes. J. Clin. Microbiol. 2016, 54, 2470–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, H.; Mao, L.; Yu, H.; Yu, X.; Sun, Z.; Qian, X.; Cheng, S.; Chen, S.; Chen, J.; et al. Rapid genomic characterization of SARS-CoV-2 viruses from clinical specimens using nanopore sequencing. Sci. Rep. 2020, 10, 17492. [Google Scholar] [CrossRef]
- Gohl, D.M.; Garbe, J.; Grady, P.; Daniel, J.; Watson, R.H.; Auch, B.; Nelson, A.; Yohe, S.; Beckman, K.B. A rapid, cost-effective tailed amplicon method for sequencing SARS-CoV-2. BMC Genom. 2020, 21, 863. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faria, N.R.; Quick, J.; Claro, I.M.; Theze, J.; de Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.; Hill, S.C.; Black, A.; da Costa, A.C.; et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Kafetzopoulou, L.E.; Pullan, S.T.; Lemey, P.; Suchard, M.A.; Ehichioya, D.U.; Pahlmann, M.; Thielebein, A.; Hinzmann, J.; Oestereich, L.; Wozniak, D.M.; et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science 2019, 363, 74–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovanetti, M.; de Mendonça, M.C.L.; Fonseca, V.; Mares-Guia, M.A.; Fabri, A.; Xavier, J.; de Jesus, J.G.; Gräf, T.; dos Santos Rodrigues, C.D.; Dos Santos, C.C.; et al. Yellow fever virus reemergence and spread in Southeast Brazil, 2016–2019. J. Virol. 2019, 94, e01623-19. [Google Scholar] [CrossRef]
- Wang, J.; Moore, N.E.; Deng, Y.M.; Eccles, D.A.; Hall, R.J. MinION nanopore sequencing of an influenza genome. Front. Microbiol. 2015, 6, 766. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.J.; Long, S.W.; Christensen, P.A.; Olsen, R.J.; Olson, R.; Shukla, M.; Subedi, S.; Stevens, R.; Musser, J.M. Analysis of the ARTIC version 3 and version 4 SARS-CoV-2 primers and their impact on the detection of the G142D amino acid substitution in the spike protein. Microbiol. Spectr. 2021, 9, e01803-21. [Google Scholar] [CrossRef]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Plitnick, J.; Griesemer, S.; Lasek-Nesselquist, E.; Singh, N.; Lamson, D.M.; George, K.S. Whole-genome sequencing of SARS-CoV-2: Assessment of the Ion Torrent AmpliSeq panel and comparison with the Illumina MiSeq ARTIC Protocol. J. Clin. Microbiol. 2021, 59, e00649-21. [Google Scholar] [CrossRef] [PubMed]
- Andeweg, S.P.; Vennema, H.; Veldhuijzen, I.; Smorenburg, N.; Schmitz, D.; Zwagemaker, F.; van Gageldonk-Lafeber, A.B.; Hahné, S.J.; Reusken, C.; Knol, M.J.; et al. Elevated risk of infection with SARS-CoV-2 Beta, Gamma, and Delta variant compared to Alpha variant in vaccinated individuals. Sci. Transl. Med. 2022, eabn4338. [Google Scholar] [CrossRef]
- Birnie, E.; Biemond, J.J.; Appelman, B.; de Bree, G.J.; Jonges, M.; Welkers, M.R.; Wiersinga, W.J. Development of resistance-associated mutations after sotrovimab administration in high-risk individuals infected with the SARS-CoV-2 omicron variant. JAMA 2022, 328, 1104–1107. [Google Scholar] [CrossRef]
- Dächert, C.; Muenchhoff, M.; Graf, A.; Autenrieth, H.; Bender, S.; Mairhofer, H.; Wratil, P.R.; Thieme, S.; Krebs, S.; Grzimek-Koschewa, N.; et al. Rapid and sensitive identification of omicron by variant-specific PCR and nanopore sequencing: Paradigm for diagnostics of emerging SARS-CoV-2 variants. Med. Microbiol. Immunol. 2022, 211, 71–77. [Google Scholar] [CrossRef]
- Tshiabuila, D.; Giandhari, J.; Pillay, S.; Ramphal, U.; Ramphal, Y.; Maharaj, A.; Anyaneji, U.J.; Naidoo, Y.; Tegally, H.; San, E.J.; et al. Comparison of SARS-CoV-2 sequencing using the ONT GridION and the Illumina MiSeq. BMC Genom. 2022, 23, 319. [Google Scholar] [CrossRef]
- Freed, N.E.; Vlková, M.; Faisal, M.B.; Silander, O.K. Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 2020, 5, bpaa014. [Google Scholar] [CrossRef]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. BioRxiv. 2020. [Google Scholar] [CrossRef]
- Laver, T.; Harrison, J.; O’neill, P.A.; Moore, K.; Farbos, A.; Paszkiewicz, K.; Studholme, D.J. Assessing the performance of the oxford nanopore technologies minion. Biomol. Detect. Quantif. 2015, 3, 1–8. [Google Scholar] [CrossRef]
- Lambisia, A.W.; Mohammed, K.S.; Makori, T.O.; Ndwiga, L.; Mburu, M.W.; Morobe, J.M.; Moraa, E.O.; Musyoki, J.; Murunga, N.; Mwangi, J.N.; et al. Optimization of the SARS-CoV-2 ARTIC Network V4 Primers and Whole Genome Sequencing Protocol. Front. Med. 2022, 9, 836728. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, L.D.; Richter, S.R.; Midgley, S.E.; Franck, K.T. Detecting SARS-CoV-2 Omicron B.1.1.529 variant in wastewater samples by using nanopore sequencing. Emerg. Infect. Dis. 2022, 28, 1296. [Google Scholar] [CrossRef] [PubMed]
- Dharmadhikari, T.; Rajput, V.; Yadav, R.; Boargaonkar, R.; Patil, D.; Kale, S.; Kamble, S.P.; Dastager, S.G.; Dharne, M.S. High throughput sequencing based direct detection of SARS-CoV-2 fragments in wastewater of Pune, West India. Sci. Total Environ. 2022, 807, 151038. [Google Scholar] [CrossRef] [PubMed]
- Cheng, V.C.C.; Ip, J.D.; Chu, A.W.H.; Tam, A.R.; Chan, W.M.; Abdullah, S.M.U.; Chan, B.P.C.; Wong, S.C.; Kwan, M.Y.W.; Chua, G.T.; et al. Rapid spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Omicron subvariant BA. 2 in a single-source community outbreak. Clin. Infect. Dis. 2022, 75, e44–e49. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, D.; Zhu, C.; Zhao, Z.; Gao, S.; Gou, J.; Guo, Y.; Kong, X. Genetic Surveillance of Five SARS-CoV-2 Clinical Samples in Henan Province Using Nanopore Sequencing. Front. Immunol. 2022, 13, 814806. [Google Scholar] [CrossRef]
- Chen, Z.; Azman, A.S.; Chen, X.; Zou, J.; Tian, Y.; Sun, R.; Xu, X.; Wu, Y.; Lu, W.; Ge, S.; et al. Global landscape of SARS-CoV-2 genomic surveillance and data sharing. Nat. Genet. 2022, 54, 499–507. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Round 1 | Round 2 | Round 3 | Round 4 | Round 5 | Round 6 | |
|---|---|---|---|---|---|---|
| Reads Analyzed | 4,465,541 | 1,765,872 | 998,468 | 1,095,194 | 705,499 | 849,462 |
| Unclassified Reads | 890,927 | 1,214,457 | 62,643 | 74,064 | 52,421 | 47,970 |
| Total Yield | 2.3 Gbases | 920.8 Mbases | 513.9 Mbases | 533.9 Mbases | 393.9 Mbases | 434.4 Mbases |
| Average Quality Score | 14.1 | 11.84 | 11.73 | 11.32 | 11.6 | 11.56 |
| Average Sequence Length (Bp) | 524 | 521 | 514 | 487 | 558 | 511 |
| Ion Torrent | Nanopore | No. of Common Mutations | Concordance (%) | ||||
|---|---|---|---|---|---|---|---|
| Clade | Lineage | Total No. of Mutations | Clade | Lineage | Total No. of Mutations | ||
| 20I (Alpha, V1) | B.1.1.7 | 35 | 20I (Alpha, V1) | B.1.1.7 | 35 | 35 | 100.0 |
| 20A | B.1.258.3 | 32 | 20A | B.1.258 | 28 | 27 | 84.4 |
| 20A | B.1.258 | 25 | 20A | B.1.258 | 20 | 19 | 76.0 |
| 20A | B.1.258 | 30 | 20A | B.1.258 | 22 | 21 | 70.0 |
| 21I (Delta) | B.1.617.2 | 38 | 21I (Delta) | AY.52 | 35 | 33 | 86.8 |
| 20I (Alpha, V1) | B.1.1.7 | 36 | 20I (Alpha, V1) | B.1.1.7 | 33 | 32 | 88.9 |
| 20A | B.1.258 | 29 | 20A | B.1.258 | 29 | 28 | 96.6 |
| 20I (Alpha, V1) | B.1.1.7 | 30 | 20I (Alpha, V1) | B.1.1.7 | 30 | 30 | 100.0 |
| 20I (Alpha, V1) | B.1.1.7 | 32 | 20I (Alpha, V1) | B.1.1.7 | 31 | 31 | 96.9 |
| 20I (Alpha, V1) | B.1.1.7 | 34 | 20I (Alpha, V1) | B.1.1.7 | 33 | 33 | 97.1 |
| 21J (Delta) | AY.122 | 48 | 21J (Delta) | AY.122 | 41 | 41 | 85.4 |
| 20I (Alpha, V1) | B.1.1.7 | 36 | 20I (Alpha, V1) | B.1.1.7 | 34 | 34 | 94.4 |
| 20A | B.1.258 | 25 | 20A | B.1.258 | 21 | 21 | 84.0 |
| 20A | B.1.258 | 21 | 20A | B.1.258 | 19 | 19 | 90.5 |
| 21I (Delta) | B.1.617.2 | 39 | 21J (Delta) | AY.46.6 | 36 | 16 | 41.0 |
| 21J (Delta) | AY.43 | 37 | 21J (Delta) | AY.43 | 34 | 34 | 91.9 |
| 21J (Delta) | AY.43 | 38 | 21J (Delta) | AY.43 | 32 | 32 | 84.2 |
| 20A | B.1.258 | 33 | 20A | B.1.258 | 30 | 30 | 90.9 |
| 21I (Delta) | AY.9.2 | 33 | 21I (Delta) | AY.9.2 | 27 | 26 | 78.8 |
| 21J (Delta) | AY.125 | 45 | 21J (Delta) | None | 21 | 21 | 46.7 |
| 21J (Delta) | AY.46.6 | 40 | 21I (Delta) | AY.52 | 34 | 17 | 42.5 |
| 21I (Delta) | B.1.617.2 | 40 | 21I (Delta) | None | 12 | 12 | 30.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capraru, I.D.; Romanescu, M.; Anghel, F.M.; Oancea, C.; Marian, C.; Sirbu, I.O.; Chis, A.R.; Ciordas, P.D. Identification of Genomic Variants of SARS-CoV-2 Using Nanopore Sequencing. Medicina 2022, 58, 1841. https://doi.org/10.3390/medicina58121841
Capraru ID, Romanescu M, Anghel FM, Oancea C, Marian C, Sirbu IO, Chis AR, Ciordas PD. Identification of Genomic Variants of SARS-CoV-2 Using Nanopore Sequencing. Medicina. 2022; 58(12):1841. https://doi.org/10.3390/medicina58121841
Chicago/Turabian StyleCapraru, Ionut Dragos, Mirabela Romanescu, Flavia Medana Anghel, Cristian Oancea, Catalin Marian, Ioan Ovidiu Sirbu, Aimee Rodica Chis, and Paula Diana Ciordas. 2022. "Identification of Genomic Variants of SARS-CoV-2 Using Nanopore Sequencing" Medicina 58, no. 12: 1841. https://doi.org/10.3390/medicina58121841