
Abstract
Declared as a global public health emergency, coronavirus disease 2019 (COVID-19) is presented as a disease of the respiratory tract, although severe cases can affect the entire organism. Several studies have shown neurological symptoms, ranging from dizziness and loss of consciousness to cerebrovascular and neurodegenerative diseases. In this context, Guillain-Barré syndrome, an immune-mediated inflammatory neuropathy, has been closely associated with critical cases of infection with “severe acute respiratory syndrome of coronavirus 2” (SARS-CoV-2), the etiological agent of COVID-19. Its pathophysiology is related to a generalized inflammation that affects the nervous system, but neurotropism was also revealed by the new coronavirus, which may increase the risk of neurological sequel, as well as the mortality of the disease. Thus, considering the comorbidities that SARS-CoV-2 infection can promote, the modulation of purinergic signaling can be applied as a potential therapy. In this perspective, given the role of adenosine triphosphate (ATP) in neural intercommunication, the P2X7 receptor (P2X7R) acts on microglia cells and its inhibition may be able to reduce the inflammatory condition of neurodegenerative diseases. Finally, alternative measures to circumvent the reality of the COVID-19 pandemic need to be considered, given the severity of critical cases and the viral involvement of multiple organs.
Graphical Abstract

Similar content being viewed by others

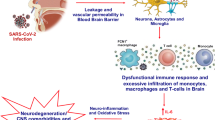

COVID-19 and Neurological Symptoms
Coronavirus acute respiratory syndrome (SARS-CoV-2) was the name chosen for the beta coronavirus of the Coronaviridae family responsible for coronavirus 2019 disease (COVID-19). Defined as a pandemic by the World Health Organization (WHO), its transmission occurs through hand-mouth-eye contact and infected droplets released by coughing and sneezing (Machhi et al. 2020). Thus, due to its high level of contagion, about 26,961,795 people were infected and the disease of the novel coronavirus had been responsible for the death of 880,955 people (Coronavirus disease 2019).
Among the symptoms, initial cases have shortness of breath, cough, and chest pain, which can progress to respiratory distress and acute respiratory distress syndrome, the main complication related to mortality (Chen et al. 2020a, b, c). Also, early signs of infection are dizziness, headache, obtundation, hypogeusia, ageusia, hyposmia, anosmia, and myalgia (Hartung 2020). This loss of consciousness indicates neurological effects and persistent sensory neuronal involvement, as these symptoms can persist for months after infection (Machhi et al. 2020).
Thus, the manifestation of COVID-19 has been related to neurological problems, including stroke (ischemic, hemorrhagic and secondary to coagulopathy), venous sinus thrombosis, cerebral hemorrhage, encephalopathy, encephalitis, and neurodegenerative diseases such as Guillain-Barré syndrome (GBS), or its variant Miller-Fisher syndrome (Hartung 2020).
GBS is a rare autoimmune disease characterized by bilateral weakness and neuromuscular paralysis (Willison et al. 2016) and classified into two subtypes: axonal and demyelinating. It has variants that include acute inflammatory demyelinating polyneuropathy (PDIA), acute motor axonal neuropathy (NAMA), acute motor axonal neuropathy (NAMSA), and Miller-Fisher syndrome (SMF) (Kaida 2019).
There are other phenotypes as SMF overlapping with GBS, acute ophthalmoparesis without ataxia, GBS with preserved reflexes variant regional pharyngeal-cervical-brachial GBS ataxia, GBS sensory ataxia, GBS pure sensory, encephalitis, Bickerstaff, Bickerstaff encephalitis overlapping with GBS (Kaida 2019). PDIA, the classic and most common variant of GBS develops after gastrointestinal or respiratory infections and has appeared after viral epidemics and pandemics (Zubair et al. 2020).
The relationship between infections and the development of GBS is explained by the mechanism of molecular mimicry, given the sharing of similar epitopes between peripheral nerve components and infecting viruses (Wijdicks and Klein, 2017). Thus, autoreactive T and/or B lymphocytes and the cross-reaction are stimulated, in which the antibodies that had the function of viral destruction bind to the peripheral nerve components. In the demyelinating subtype, like the classic variant (PDIA), the myelin sheath is compromised while, in axonal variants like NAMA, autoantibodies attack axonal membranes of peripheral nerves (Leonhard et al. 2019) (Fig. 1).

Representation of molecular mimicry, pathophysiological mechanism of GBS. The pathophysiology of GBS is evidenced by the mechanism of molecular mimicry, in which the virus shares epitopes common to neuron components, such as the myelin sheath and axonal lining. Thus, direct virus infection triggers the activation of the immune system and APC precursor cells, which differentiate into T and B lymphocytes. Also, the viral epitope is recognized by the B lymphocyte and the T lymphocyte releases isoleucine such as IL-6 and interleukin 4 (IL-4), in addition to cytokines such as TNF-α and interferon-gamma (IFN-γ) for macrophage activation, triggering local inflammation that can be exacerbated. Thus, there is the production of antibodies (ANTI-GM1) to attack the virus, however, by sharing epitopes, these antibodies also attack components of the neuron. The recruitment of more macrophages increases inflammation, as well as the performance of the microglia that release cytokines such as IFN-γ and interleukin 1 beta (IL-1β). Finally, the neuron is in hyperinflammation and operates with its reduced functionality or loses it definitively, being able to suffer cell death and causing irreversible damage to the patient with this syndrome
Thus, neurological parainfectious diseases, such as GBS, transverse myelitis, or acute disseminated encephalomyelitis, can be an associated disease (Ahmed et al. 2020). Neuronal dysfunction and clinical manifestations have also been described in Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS-CoV) infections, surprisingly two to three weeks after recovery from respiratory disease (Kim et al. 2017). In the period from 2015 to 2016, the Zika virus epidemic occurred and several studies reported the development of GBS in infected patients, which was related to a worse prognosis (Barbi et al. 2018; Dirlikov et al. 2018; Major et al. 2018; Arias et al. 2017; Araujo et al. 2016).
Studies claim that the manifestations of neurological diseases are a significant part of the manifestations of COVID-19 (Asadi-Pooya and Simani L 2020). Zhao et al. (2020) related GBS to SARS-CoV-2. Thus, in the same city as the outbreak, Wuhan, China, 214 hospitalized SARS-CoV-2 patients with neurological problems were reported (Mao et al. 2020). In France, among 58 patients, neural involvement was observed in almost 85% of this group (49 patients) (Helms et al. 2020).
As for GBS reports, cases have been reported in China, Iran, and Europe - most notably Italy (Alberti et al. 2020; El Otmani et al. 2020; Gutiérrez-Ortiz et al. 2020; Padroni et al. 2020; Scheidl et al. 2020; Toscano et al. 2020; Sedaghat and Karimi 2020; Zhao et al. 2020). The first group (Toscano et al. 2020) consists of five patients, who developed symptoms between five and ten days after the viral infection, and four patients had weakness in the lower limbs and paresthesia. Through electromyography, two patients with GBS and three with acute motor axonal neuropathy (Toscano et al. 2020) were confirmed. In another Italian patient, as well as an Iranian (Sedaghat and Karimi 2020) patient, the Miller-Fisher GBS variant was found (Gutiérrez-Ortiz et al. 2020).
Mechanism of Neural Infection by SARS-CoV-2
The angiotensin-converting enzyme (ACE2) has the function of enzymatic modification of the vasoconstrictor peptides angiotensin II and angiotensin I in the vasodilating peptides angiotensin [1–7] (Ang [1–7]) and angiotensin [1–9], respectively. Thus, Ang [1–7] is the predominant form in several regions of the brain, including the hypothalamus and amygdala, as well as in the oblong medulla, which inhibits hypothalamic noradrenergic neurotransmission, reducing inflammation, oxidative stress, and neuronal apoptosis (Rocha et al. 2018).
Also, coronaviruses use the ACE2 protein as a receptor associated with serine transmembrane protease 2 (TMPRSS2) for cell invasion and replication in human cells (Yan et al. 2020). Thus, organs and tissues that express the ACE2 gene can be considered targets for infection. The SARS-CoV-2 neurovirulence is related to the degree of expression of the ACE2 receptor in the central nervous system (CNS), which is highly expressed in the substantia nigra, choroid plexus, ventricles, middle temporal gyrus, posterior cingulate cortex, olfactory bulb (Chen et al. 2020a, b, c), in the sympathetic tract of the brain stem, and the motor cortex (Xia and Lazartigues 2008).
ACE2 is considered a key element in the anti-inflammatory and hypotensive arm of the renin-angiotensin-aldosterone system (RAAS), which is operating endogenously in the CNS (Garcia-Garrote et al. 2019; Xu et al. 2011; Xia and Lazartigues 2008). This protein is present both in the superficial membrane and in the cytoplasm of the neuronal and glial cells of the human brain (Xiao et al. 2013). Furthermore, using transcriptomic analysis, the expression of ACE2 has been reported in astrocytes, oligodendrocytes, endothelial cells, excitatory and inhibitory neurons (Chen et al. 2020a, b, c), increasing the possibility of viral attack.
Other evidence found this receptor in the cardiorespiratory nuclei of the brain stem, which may indicate direct viral contact and justify the atypical form of acute respiratory distress syndrome (ARDS) (Marini.2020; Marini and Gattinoni 2020). Likewise, being expressed in the capillary endothelium, the viral S protein can allow the virus to interact in the cerebral microcirculation, triggering endothelial damage, and spreading to neurons (Baig et al. 2020).
Autopsies have demonstrated SARS-CoV-2 ribonucleic acid (RNA) in the respiratory and cardiovascular regulatory centers in the medulla (Meinhardt et al. 2020). The way SARS-CoV-2 infects nerve tissue remains uncertain though. Thus, five different ways are being hypothesized for the virus to enter the nervous system and to bind ACE2 expressed in this organ, including the neuronal, hematogenous, Trojan, pericellular, and lymphatic routes (Briguglio et al. 2020) (Fig. 2).

Different hypothetical routes of viral infiltration of the new coronavirus to the CNS. Representation of virus entry and involvement of nervous tissue by different pathways. A neuronal pathway, which can be through the olfactory nerve (a) or transsynaptic axonal reverse pathway (b), occurs when the virus comes into contact with the nasal cavity (a) or with peripheral nerves (b) and is transported through the synapses. In the first situation (a), the virus has contact with the olfactory nerves that allowed the olfactory bulb to enter and spread through the brain. As for the retrograde transsynaptic pathway, the virus that is being transported by the peripheral nerves follows the opposite pathway by endocytosis of the neurotransmitters by the presynaptic neuron. Thus, this virus can follow up to the central nervous system and infect the brain. Another route may be the hematogenic (c), in which the virus injures and infects blood-brain barrier endothelial vascular cells and this infected cell has contact with neural cells such as the astrocyte, which can spread SARS-CoV-2 through neuronal tissue until infection direct to the brain. Another possible route is the lymphatic route (d), which occurs through the infection of the coronavirus and the lymphatic vessels through contact with infected capillaries, thus, these infected lymphatic capillaries may have contact with the brain and enable viral dissemination in this organ. The Trojan route, on the other hand, occurs through the movement of leukocytes infected by the blood-brain barrier and the contamination of these cells. Finally, a pericellular route can occur, for example, in contact between alveoli and blood capillaries, thus, SARS-CoV-2 can cross the alveolar membrane and spread through the blood and infect blood cells
The mechanism of involvement of multiple organs is shared by other coronaviruses, so understanding how they infect the brain can be a way of understanding SARS-CoV-2. Previous studies have analyzed the nervous system of SARS patients and reported the presence of viral particles in the brain, exclusively in neurons (Xu et al. 2005). In vivo experiments with transgenic mice reproduced the infection by SARS and MERS intranasally and perceived the entrance through the olfactory nerve and the dissemination through brain regions such as the brainstem and thalamus (Li et al. 2016).
Occurring at different stages, the invasion of the CNS in the COVID-19 scenario can be reported both early and late and the manifestations of CNS disease can appear directly after infection, at the end of the disease course, or after recovery (Conde Cardona et al. 2020). Baig et al. (2020) found the presence of RNA in the cerebrospinal fluid as evidence, which supports neurovirulence and the route through the SARS-CoV-2 cribriform plate to the brain (Fig. 2a). Also, the likelihood of virus binding is increased in the nasal olfactory epithelium, as Bilinska et al. (2020) demonstrated the expression of two host receptors, ACE2 and TMPRSS2, which facilitate the binding, replication, and accumulation of SARS-CoV-2.
Thus, the new coronavirus can spread via the transcervical pathway from the olfactory epithelium along the olfactory nerve to the olfactory bulb within the CNS, in addition to using the trigeminal nerve branch through the transsynaptic route (de Freitas Ferreira et al. 2020) (Fig. 2a). Besides, another form of dissemination would be the vesicular transport of the virus through microtubules and neuronal cell bodies (Zubair et al. 2020), through transsynaptic transfer using an axonal transport mechanism and endocytosis or exocytosis (Fig. 2b).
The biological evidence supporting the retrograde transsynaptic pathway of peripheral nerve endings is a finding that demonstrates that coronaviruses can penetrate the CNS through the cribriform plate of the ethmoid bone (Li et al. 2020). Gosztonyi (1986) demonstrated the axonal transport of viral nucleic acid between some neurotropic viruses, as well as Taylor and Enquist (2015) profoundly presented this route in several viruses in a review. Also, Li et al. (2013a, b) demonstrated that other viruses spread in the CNS through a transsynaptic route. Thus, joining these observations, we can base the direct infection of SARS-CoV-2 as a possibility in transsynaptic transport (Fig. 2a and b).
Another mechanism of CNS involvement can be by hematogenous propagation (Fig. 2c), or by the lymphatic pathway (Fig. 2d). Thus, damage to the capillary epithelial barrier allows the virus to reach the bloodstream and lymphatic capillaries, spreading through the body and reaching the brain. SARS-CoV-2 particles were found in the capillary endothelium and neurons in a frontal lobe sample from an autopsy case study (Paniz-Mondolfi; et al. 2020), infecting vascular and neural cells. ACE2 interaction of these cells can occur, increasing their replication and damage to vascular and neuronal tissue (Baig et al. 2020) (Fig. 2c and d).
Furthermore, there is the possibility of the Trojan pathway, also called the “Trojan horse mechanism”, in which infected leukocytes pass through the barrier (BBB) and spread the virus (Fig. 2e). Generalized systemic inflammation can increase BBB permeability and as lymphocytes, monocytes and granulocytes express ACE2. Thus, the invasion of the CNS by infected immune cells is facilitated (Sankowski et al. 2015). Finally, contamination of adjacent cells via the pericellular route can occur, in which the virus passes through the gap between the efferent cells of the olfactory sensory neurons and infects their surrounding cells, as well as diffusion through the mucous layer of the respiratory tract or the cerebrospinal fluid (Briguglio et al. 2020) (Fig. 2f).
SARS-CoV-2 and the SGB
SARS-CoV-2 infiltrated in neural tissue is capable to generate the pathophysiology of GBS through the mechanism of molecular mimicry, however, it is not the only way to develop this pathology when the patient presents COVID-19. The clinical picture of this disease is closely related to a “cytokine storm” and the subsequent hyperinflammation may also be responsible for the development of the pathogenesis of GBS, as they can result in the breakdown of the blood-brain barrier without direct intracranial viral invasion (Ahmed et al. 2020; Zhou et al. 2020).
Thus, cytokines such as interleukin-6 (IL-6) act on multiple organ dysfunction, with fatal consequences (Ahmed et al. 2020; McCrayet al. 2007). The same is true for tumor necrosis factor alpha (TNF-α), which was identified as pro-inflammatory and related to the increased severity of GBS (Li et al. 2013a, b) (Fig. 3a). Also, the “macrophage activation syndrome”, associated with the coagulopathy observed in the disease, causes cerebrovascular events, both thrombotic and hemorrhagic (Ahmed et al. 2020) (Fig. 3b).

Cytokine storm and hypoxia in COVID-19 as a possible trigger of GBS. In addition to direct viral infection, pathology of GBS and other neurological disorders may occur due to the cytokine storm and hypoxia caused by COVID-19. Thus, a cytokine storm (a) can break through a blood-brain barrier and contribute to pathogenesis, in addition to activating the “macrophage activation syndrome” (b) that triggers both thrombotic and hemorrhagic cerebrovascular events. Furthermore, hypoxia (c) forces the anaerobic metabolic function of neural cells, by reducing the concentration of oxygen, causing interstitial edema and cerebral vasodilation, which compromises its functioning and can progress to stroke, syncope, and anoxic crisis
Considering that coronaviruses can infect immune cells, the host’s immune-mediated response may play a role in brain failure, given that the hyperinflammatory syndrome has been linked to increased mortality due to COVID-19 (Mehta et al. 2020). Studies have shown a significant increase in the levels of pro-inflammatory cytokines such as IL-6, TNF-α, interleukin 1 (IL-1) (Yesilkaya and Balcioglu 2020), and the chemokine-1 monocyte protein after infiltration of infected T lymphocytes (Jacomy et al. 2010) (Fig. 3b).
Hypoxia, caused by infection and respiratory tract dysfunction, is closely associated with indirect neuronal damage. With the reduction of oxygen levels, CNS cells increase anaerobic metabolism, in addition to the induction of ischemia, interstitial edema and vasodilation in the cerebral circulation, which ends up causing stroke, syncope and anoxic crisis (Wu et al. 2020) (Fig. 3c).
Regardless of the etiology, GBS generates neurological problems that persist in up to 20% of patients with the disease. Half of these patients are severely deficient (Ahmed et al. 2020), affecting several components of the nervous system and changing the dynamics of functioning. Adenosine triphosphate (ATP) is seen as a neuronal modulating agent, as it can involve several important intracellular pathways, such as calcium, adenosine 3’, 5’-cyclic monophosphate (cAMP), inositol-1,4,5-triphosphate, phospholipase C, and others (Fields and Burnstock 2006). Also, both microglia and immune cells respond to a wide range of ATP receptor agonists, secreting cytokines (Hide et al. 2000), plasminogen (Inoue et al. 1998), increasing intracellular calcium (Toescu et al. 1998), and triggering potassium currents (Boucsein et al. 2003).
In GBS, cell damage can result in the release of large amounts of ATP in the extracellular environment, which stimulates the proliferation of microglia - immune cells active in the CNS and act as a powerful chemoattractant at the brain injury site (Ahmed et al. 2020; Li et al. 2013a, b). Responses to ATP released during brain injury are not always neuroprotective though and can contribute to pathophysiology (Hide et al. 2000). Thus, purinergic signaling is evidenced as therapy, due to the regulation of ATP release.
Purinergic System and Coronavirus‐induced GBS
Proposed in 1972, purinergic signaling is characterized by the relationship between signaling components, receptors, and specific enzymes (Burnstock 2020a). The signaling molecules include adenosine (Ado) and ATP, which are co-transmitters and act in the process of synaptic transmission. Ado plays an important role in presynaptic neuromodulation and ATP has been identified as a short-term signaling molecule in neuromodulation, neurosecretion, and neurotransmission, in addition to playing an important role in cell proliferation, differentiation, and death (Burnstock 2020b).
Purinergic receptors are widely distributed in different cell types, both non-neuronal and neuronal and are divided into two groups: Ado receptors (P1) and ATP receptors (P2). The former play an important role in presynaptic neuromodulation and are divided into four subtypes. The second group is divided into seven P2X ionotropic nucleotide receptors, which are involved in rapid synaptic transmission and synaptic plasticity; and eight P2Y metabotropic nucleotide receptors widely involved in presynaptic activities, as well as in mediating long-term (trophic) signaling of cellular activity (Burnstock 2020b). Also, there are the ectonucleotidases: the ectoenzyme adenosine deaminase (ADA) responsible for the deamination of Ado in inosine and the ectonucleotidases E-NPP, E-NTPDases, ecto-5’-nucleotidase, and alkaline phosphatase, which regulate the nucleotides (Leal et al. 2005).
Acting in conjunction with glutamate, norepinephrine, GABA, acetylcholine, and dopamine in the brain, ATP has receptors in both neurons and glial cells (Illes 2020). Within the receptor-channel family of this nucleotide, the P2X7R is widely distributed in the CNS, especially in microglia, but also in neurons and neuroglia (astrocytes, oligodendrocytes) (Illes et al. 2020). It plays a role in transmembrane flows of Na+, Ca 2+, and K+, in addition to the slow permeation of larger organic molecules, acting on cellular functions such as, for example, the excess influx of Ca2+, is capable of promoting necrosis (Illes 2020).
Furthermore, stimulation of this receptor leads to the release of pro-inflammatory cytokines, proteases, chemokines, reactive oxygen species (ROS), and the excitotoxic glutamate and ATP (Illes 2020). These bioactive molecules inducing neurodegeneration when released in excess, by signaling effect, mechanical deformation or injury, are closely associated with a variety of neurological problems, including stroke, thrombosis, Alzheimer’s disease (AD), Parkinson’s (Burnstock 2020b), trauma, ischemia, epilepsy, neurodegenerative diseases, and cancer, including gliomas (Illes 2020).
Thus, after these events, a massive release of ATP occurs through the permeable plasma membrane of neural tissue, causing cell damage, in addition to the original consequences of neurodegeneration, increasing comorbidities (Illes 2020). Thus, the modulation of purinergic signaling can act as a possible therapy for Guillain-Barré syndrome triggered by the SARS-CoV-2 infection, with emphasis on P2X7R antagonists permeable to the blood-brain barrier with excellent bioavailability.
As the main objective of this therapy, the use of P2X7R antagonists may be able to reduce the release of pro-inflammatory molecules and thereby reduce the damage from the cytokine storm. However, an obstacle to its application may be a less significant reduction of these biomarkers in relation to their uncontrolled production. Thus, it would be necessary to use an adjuvant therapy or analyze these markers to enhance the results.
Thus, including both neurological pathologies and other diseases that plague the world as well as cancer, recent studies have worked and shown good results with the modulation of the P2X7R. These include infections (Di Virgilio et al., 2017), inflammation (Freire 2019), glioma (Kan et al. 2020), tumors (De Marchi et al. 2019), sclerosis (Cieślak, Roszek and Wujak, 2019), depression and anxiety (Hong et al. 2020). Thus, it is understood that the association made between the purinergic system, with emphasis on P2X7R, in Guillain Barré Syndrome is unprecedented, but strongly supported by the literature, although there are no clinical experiments.
As confirmed several times in the literature, P2X7R acts directly on neuroinflammation in neurological diseases and the performance of its antagonists is related to a reduction in inflammatory markers, which must be analyzed to assess the effectiveness of the therapy. As an example, the study by Lu et al. (2013), demonstrated the role of the low toxicity and selective P2X7R antagonist brilliant blue G (BBG) in reducing microglial activity and its consequent inflammation by reducing levels of cyclooxygenase-2 and IL-6.
Thus, the use of antagonists becomes a universally used therapy against inflammatory scenarios in the nervous environment. In the study by Wang et al. (2020) the anti-inflammatory effect of the BBG was able to attenuate the neuroinflammatory response, being perceived by the reduction in the levels of pro-inflammatory cytokines as interleukins and TNF-a, as well as levels of mRNA, inducible oxide synthase protein and cyclooxygenase-2. In addition, these antagonists were used to protect against brain ischemia and reperfusion injury, reducing the levels of IL-1ß, TNF-α and IL-6 and the overexpression of these cytokines in the hippocampus (Chu 2012).
In addition, the role of purinergic signaling in epilepsy enables treatment with modulation of both Ado and ATP receptors. Thus, Ado acts to inhibit seizures, which can induce inflammatory processes and vice versa, as well as activation of P2X7R. Therefore, the use of its antagonists reduces the uncontrolled release of pro-inflammatory cytokines such as IL-1ß and TNF-α and act in the way of inhibiting seizures (Ciéslak et al. 2017). Likewise, in cases of alcohol abuse, treatment with the antagonist P2X7R A804598 was able to reverse changes in microglia and astrocytes, in addition to significantly reducing levels of mRNA and inflammatory markers such as IL-1ß in the brain (Freire et al. 2019).
This therapeutic hypothesis becomes stronger when analyzing the gene expression of this receptor in pathology. In a resting situation, microglia act in different physiological functions, such as protection of the blood-brain barrier, conformation of synaptic structures, and elimination of excess synapses during development (Illes et al. 2020). When in a scenario of neurodegenerative disease, however, its activation can aggravate the disease and trigger an uncontrolled neuroinflammatory reaction (Fig. 4a).

Representation of quantitative changes in P2X7R in microglia in neuropathologies. In a resting state and basal cell action, the microglia acts in neuronal protection and the structural action of the synapse. In the neuropathies scenario, there is greater activation of these cells and the development of neuroinflammation. As for the disposition of the receptor, in the case of neurodegenerative diseases, there is a greater expression of P2X7R in glial cells, which release pro-inflammatory cytokines and a smaller one in neurons. Thus, with greater microglial activation and migration, this release becomes exacerbated and ends up worsening the disease. Thus, considering that there is an increase in the production of ATP in GBS by cells, the inhibition of receptors would be able to reduce inflammation and assist in the treatment of both GSB and COVID-19
Whereas the microglia is regulated by a set of purinergic receptors, namely P2Y12, P2Y6, P2Y4, P2X4, P2X7R, A2A, and A3 (Illes et al. 2020), modulation P2X7R was chosen by previous studies to demonstrate its role in microglia associated with neurodegenerative diseases. Thus, there is therapeutic potential in GBS, as P2X7R regulates different microglial functions, including migratory capacity (Davalos et al. 2005).
Martínez-Frailes et al. (2019) confirmed in vitro studies the action of ATP in promoting microglial mobility by activating P2X7R. Thus, there is an interdependent relationship between the increase in ATP and microglial activity, however, it is also related to a release of cytokines by the microglia (Di Virgilio et al. 2017). It has also been reported in changes in the cytoskeleton, reducing phagocytic capacity (Fang et al. 2009) (Fig. 4b).
From this perspective, studies have found P2X7R in neurons (Miras-Portugal et al. 2017) and others have detected it mainly in glial cells (Illes et al. 2017) (Fig. 4a), although the distribution pattern is altered in pathological conditions (Fig. 4b). Neuroinflammation causes microglial proliferation and activation and reduces the transcription of neuronal P2X7R and increases in microglial cells, demonstrating how the pathology regulates the expression of this receptor (Martínez-frailes et al. 2019).
Thus, the increase in pro-inflammatory cytokines released by P2X7R can promote its overload, characterizing a positive feedback cycle that promotes an exacerbated microglial response and contributes to the adverse effects of neuroinflammation (Mosher and Wyss-Coray 2014) (Fig. 4b). Therefore, P2X7R blockade is ratified by the reduction of microglial activity and subsequent inflammation, as well as its ability to attenuate LPS-induced neuroinflammation in the study by Lu et al. (2013), also reducing microglial proliferation (Bianco et al. 2006) (Fig. 4c).
Lee et al. (2011) applied this therapy in AD and was able to create a close relationship between the positive regulation of P2X7R in the microglia and the increase in synaptic toxicity in this pathology. The compression of brain tissue by extracellular β-amyloid deposits is observed in the final stages of the disease and can cause a mechanical release of ATP (Burnstock et al. 2011), which favors microglial migration, which also expresses the receptor towards these deposits. Also, it can promote sustained activation of neuronal P2X7R, compromising cell viability, and increasing neuronal loss associated with neurodegenerative diseases (Martínez-Frailes et al. 2019).
The Ado is also present in the nerve intercom, as Schwann cells respond to nerve stimulation and are intimately involved in signal Ado and ATP the release of transmitters. Thus, the nucleoside may be neuroprotective, activating A1R in a situation of depression of synaptic transmission in the hippocampus during hypoxia, although the activation of A2B receptors in astroglioma cells increases the synthesis of IL-6 messenger RNA and IL-6 protein. Hence, in addition to the discussion about the P2X7R and other ATP receptors, the P1 subtype can also be implemented as therapy, hypoxia being closely related to neuropathologies, with Ado modulation acting as a protector of this system (Fields and Burnstock 2006).
Finally, inhibition of P2X7R in patients with GBS would be able to reduce the existing neuroinflammation and reduce damage to the pathology, given the exacerbated action of the microglia. Thus, as discussed above, antagonists of this receptor can negatively modulate the inflammatory cycle and bring more comfort to the neural environment. Therefore, both in GBS and in its variants triggered by the coronavirus, which is closely associated with generalized and uncontrolled systemic inflammation, purinergic signaling can be used as adjuvant therapy in the treatment of COVID-19.
Conclusions
In a COVID-19 pandemic scenario, the development of immunotherapies becomes a priority. The fact that SARS-CoV-2 affects the nervous system increases this concern even more, as the involvement of the brain increases the level of fatality and can trigger comorbidities. Thus, neurological symptoms, ranging from sensory losses to cerebrovascular diseases, are highlighted in the literature as associated with the action of the neural tissue coronavirus. Therefore, the study and application of the modulation of purinergic signaling in neurodegenerative diseases, with emphasis on GBS and its variants, may be able to work together with the treatment of COVID-19 and reduce the level of mortality. Finally, the use of P2X7R antagonists may indicate a usual therapy, as this block reduces neuronal inflammation and microglial activity.
References
Ahmed MU, Hanif M, Ali MJ, Haider MA, Kherani D, Memon GM, Karim AH, Sattar A (2020) Neurological manifestations of COVID-19 (SARS-CoV-2): A review. Front Neurol 11. https://doi.org/10.3389/fneur.2020.00518
Alberti P, Beretta S, Piatti M, Karantzoulis A, Piatti ML, Santoro P, Viganò M, Giovannelli G, Pirro F, Montisano DA, Appollonio I, Ferrarese C (2020) Guillain-Barré syndrome related to COVID-19 infection. Neurol Neurophysiol Neurosci 7(4). https://doi.org/10.1212/NXI.0000000000000741
Araujo LM, Ferreira MLB, Nascimento OJ (2016) Guillain-Barré syndrome associated with the Zika virus outbreak in Brazil. Arquivos De Neuro-Psiquiatria 74(3):253–255. https://doi.org/10.1590/0004-282X20160035
Arias A, Torres-Tobar L, Hernández G, Paipilla D, Palacios E, Torres Y, Duran J, Ugarte U, Ardila-Sierra S, A., & Castellanos G (2017) Guillain-Barré syndrome in patients with a recent history of Zika in Cúcuta, Colombia: A descriptive case series of 19 patients from December 2015 to March 2016. J Crit Care 37:19–23. https://doi.org/10.1016/j.jcrc.2016.08.016
Asadi-Pooya AA, Simani L (2020) Central nervous system manifestations of COVID-19: A systematic review. J Neurol Sci 413:116832. https://doi.org/10.1016/j.jns.2020.116832
Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host–virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11(7):995–998. https://doi.org/10.1021/acschemneuro.0c00122
Barbi L, Coelho AVC, Alencar LCA de, Crovella S (2018) Prevalence of Guillain-Barré syndrome among Zika virus infected cases: A systematic review and meta-analysis. The Brazilian Journal of Infectious Diseases: An Official Publication of the Brazilian Society of Infectious Diseases 22(2):137–141. https://doi.org/10.1016/j.bjid.2018.02.005
Bianco F, Ceruti S, Colombo A, Fumagalli M, Ferrari D, Pizzirani C et al (2006) A role for P2 × 7 in microglial proliferation. J Neurochem 99:745–758. https://doi.org/10.1111/j.1471-4159.2006.04101.x
Bilinska K, Jakubowska P, Von Bartheld CS, Butowt R (2020) Expression of the SARS-CoV-2 entry proteins, ACE2 and TMPRSS2, in cells of the olfactory epithelium: identification of cell types and trends with age. ACS Chem Neurosci. https://doi.org/10.1021/acschemneuro.0c00210
Boucsein C, Zacharias R, Färber K, Pavlovic S, Hanisch U-K, Kettenmann H (2003) Purinergic receptors on microglial cells: Functional expression in acute brain slices and modulation of microglial activation in vitro. Eur J Neurosci 17(11):2267–2276. https://doi.org/10.1046/j.1460-9568.2003.02663.x
Briguglio M, Bona A, Porta M, Dell’Osso B, Pregliasco FE, Banfi G (2020) Disentangling the hypothesis of host dysosmia and SARS-CoV-2: the bait symptom that hides neglected neurophysiological routes. Front Physiol 11. https://doi.org/10.3389/fphys.2020.00671
Burnstock G (2020a) Introduction to Purinergic Signaling. Methods Mol Biol 2041:1–15. https://doi.org/10.1007/978-1-4939-9717-6_1
Burnstock G (2020b) Introduction to Purinergic Signalling in the Brain. Adv Exp Med Biol 1202:1–12. https://doi.org/10.1007/978-3-030-30651-9_1
Burnstock G, Fredholm BB, Verkhratsky A (2011) Adenosine and ATP receptors in the brain. Curr Top Med Chem 11:973–1011. https://doi.org/10.2174/156802611795347627
Chen R, Wang K, Yu J et al. (2020a) The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in human and mouse brain. Neuroscience. https://www.biorxiv.org/content/biorxiv/early/2020/05/19/2020.04.07.030650.full.pdf. Accessed 6 Sep 2020
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Xia J, Yu T, Zhang X, Zhang L (2020b) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 395(10223):507–513. https://doi.org/10.1016/S0140-6736(20)30211-7
Chen R, Wang K, Yu J, Howard D, French L, Chen Z, Wen C, Xu Z (2020c) The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in human and mouse brain [Preprint]. Neuroscience. https://doi.org/10.1101/2020.04.07.030650
Chu K, Yin B, Wang J, Peng G, Liang H, Xu Z, Du Y, Fang M, Xia Q, Luo B (2012) Inhibition of P2 × 7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J Neuroinflammation 18:9:69. https://doi.org/10.1186/1742-2094-9-69
Cieślak M, Wojtczak A, Komoszyński M (2017) Role of the purinergic signaling in epilepsy. Pharmacol Rep 69(1):130–138. https://doi.org/10.1016/j.pharep.2016.09.018
Cieślak M, Roszek K, Wujak M (2019) Purinergic implication in amyotrophic lateral sclerosis-from pathological mechanisms to therapeutic perspectives. Purinergic Signal 15(1):1–15. https://doi.org/10.1007/s11302-018-9633-4
Conde Cardona G, Quintana Pájaro LD, Marzola Q, Villegas ID,R, Y., & Moscote Salazar LR (2020) Neurotropism of SARS-CoV 2: Mechanisms and manifestations. J Neurol Sci 412:116824. https://doi.org/10.1016/j.jns.2020.116824
Coronavirus disease 2019 [Internet]. Available from: https://www.who.int/emergencies/diseases/novel-coronavirus-2019. Accessed 10 Sep 2020
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758. https://doi.org/10.1038/nn1472
de Freitas Ferreira ACA, Romão TT, SIlva Macedo Y, Pupe C, Nascimento OJ (2020) COVID-19 and herpes zoster co‐infection presenting with trigeminal neuropathy. Eur J Neurol. https://doi.org/10.1111/ene.14361
De Marchi E, Orioli E, Pegoraro A, Sangaletti S, Portararo P, Curti A, Colombo MP, Di Virgilio F, Adinolfi E (2019) O receptor P2 × 7 modula a infiltração de células do sistema imunológico, a expressão de ectonucleotidases e os níveis extracelulares de ATP no microambiente tumoral. Oncogene 38(19):3636–3650. https://doi.org/10.1038/s41388-019-0684-y
Di Virgilio F, Ben D, Sarti D, Giuliani AC, A. L., and Falzoni S (2017) The P2 × 7 receptor in infection and inflammation. Immunity 47:15–31. https://doi.org/10.1016/j.immuni.2017.06.020
Dirlikov E, Major CG, Medina NA, Lugo-Robles R, Matos D, Muñoz-Jordan JL, Colon-Sanchez C, Garcia M, Olivero-Segarra M, Malave G, Rodríguez-Vega GM, Thomas DL, Waterman SH, Sejvar JJ, Luciano CA, Sharp TM, Rivera-García B (2018) Clinical features of Guillain-Barré Syndrome with vs without zika virus infection, Puerto Rico, 2016. JAMA Neurol 75(9):1089–1097. https://doi.org/10.1001/jamaneurol.2018.1058
El Otmani H, El Moutawakil B, Rafai M-A, El Benna N, Kettani E, Soussi C, El Mdaghri M, Barrou N, Afif H (2020) Covid-19 and Guillain-Barré syndrome: More than a coincidence! Rev Neurol 176(6):518–519. https://doi.org/10.1016/j.neurol.2020.04.007
Fang KM, Yang CS, Sun SH, Tzeng SF (2009) Microglial phagocytosis attenuated by short-term exposure to exogenous ATP through P2X receptor action. J Neurochem 111:1225–1237. https://doi.org/10.1111/j.1471-4159.2009.06409.x
Fields RD, Burnstock G (2006) Purinergic signalling in neuron–glia interactions. Nat Rev Neurosci 7(6):423–436. https://doi.org/10.1038/nrn1928
Freire D, Reyes RE, Baghram A, Davies DL, Asatryan L (2019) P2 × 7 receptor antagonist A804598 inhibits inflammation in brain and liver in C57BL/6J mice exposed to chronic ethanol and high fat diet. J Neuroimmune Pharmacol 14(2):263–277. https://doi.org/10.1007/s11481-018-9816-3
Garcia-Garrote M, Perez-Villalba A, Garrido-Gil P, Belenguer G, Parga JA, Perez-Sanchez F, Labandeira-Garcia JL, Fariñas I, Rodriguez-Pallares J (2019) Interaction between Angiotensin Type 1, Type 2, and mas receptors to regulate adult neurogenesis in the brain ventricular–subventricular zone. Cells 8(12). https://doi.org/10.3390/cells8121551
Gosztonyi G (1986) Propagação de vírus ao longo de redes de neurônios por passagem transsináptica - uma contribuição para a patogênese da raiva. Tierarztl Prax 14(2):199–204
Gutiérrez-Ortiz C, Méndez-Guerrero A, Rodrigo-Rey S, Pedro-Murillo S, Bermejo-Guerrero E, Gordo-Mañas L, de Aragón-Gómez R, F., & Benito-León J (2020) Miller Fisher syndrome and polyneuritis cranialis in COVID-19. Neurology 95(5):e601–e605. https://doi.org/10.1212/WNL.0000000000009619
Hartung H-P, Aktas O (2020) COVID-19 and management of neuroimmunological disorders. Nat Rev Neurol 16(7):347–348. https://doi.org/10.1038/s41582-020-0368-9
Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, Collange O, Boulay C, Fafi-Kremer S, Ohana M, Anheim M, Meziani F (2020) Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. https://doi.org/10.1056/NEJMc2008597
Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, Nakata Y (2000) Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem 75(3):965–972. https://doi.org/10.1046/j.1471-4159.2000.0750965.x
Hong S, Xin Y, JiaWen W, ShuQin Z, GuiLian Z, HaiQin W, Zhen G, HongWei R, YongNan L (2020) The P2 × 7 receptor in activated microglia promotes depression- and anxiety-like behaviors in lithium -pilocarpine induced epileptic rats. Neurochem Int. 138:104773. https://doi.org/10.1016/j.neuint.2020.104773
Illes P (2020) P2X7 Receptors Amplify CNS Damage in Neurodegenerative Diseases. Int J Mol Sci 21(17):5996. https://doi.org/10.3390/ijms21175996
Illes P, Khan TM, Rubini P (2017) Neuronal P2 × 7 receptors revisited: do they really exist? J Neurosci 37:7049–7062. https://doi.org/10.1523/JNEUROSCI.3103-16.2017
Illes P, Rubini P, Ulrich H, Zhao Y, Tang Y (2020) Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 9(5):1108. https://doi.org/10.3390/cells9051108
Inoue K, Nakajima K, Morimoto T, Kikuchi Y, Koizumi S, Illes P, Kohsaka S (1998) ATP stimulation of Ca2+ -dependent plasminogen release from cultured microglia. Br J Pharmacol 123(7):1304–1310. https://doi.org/10.1038/sj.bjp.0701732
Jacomy H, St-Jean JR, Brison E, Marceau G, Desforges M, Talbot PJ (2010) Mutations in the spike glycoprotein of human coronavirus OC43 modulate disease in BALB/c mice from encephalitis to flaccid paralysis and demyelination. J Neurovirol 16(4):279–293. https://doi.org/10.3109/13550284.2010.497806
Kaida K (2019) Guillain-Barré Syndrome. Adv Exp Med Biol 1190:323–331. https://doi.org/10.1007/978-981-32-9636-7_20
Kan LK, Seneviratne S, Drummond KJ, Williams DA, O’Brien TJ, Monif M (2020) P2 × 7 receptor antagonism inhibits tumour growth in human high-grade gliomas. Purinergic Signal 16(3):327–336. https://doi.org/10.1007/s11302-020-09705-2
Kim J-E, Heo J-H, Kim H, Song S, Park S-S, Park T-H, Ahn J-Y, Kim M-K, Choi J-P (2017) Neurological complications during treatment of Middle East respiratory syndrome. J Clin Neurol (Seoul Korea) 13(3):227–233. https://doi.org/10.3988/jcn.2017.13.3.227
Leal DBR, Streher CA, Neu TN, Bittencourt FP, Leal CAM, da Silva JEP, Morsch VM, Schetinger MRC (2005) Characterization of NTPDase (NTPDase1; ecto-apyrase; ecto-diphosphohydrolase; CD39; EC 3.6.1.5) activity in human lymphocytes. Biochim Biophys Acta 1721(1–3):9–15. https://doi.org/10.1016/j.bbagen.2004.09.006
Lee HG, Won SM, Gwag BJ, Lee YB (2011) Microglial P2 × (7) receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Exp Mol Med 43:7–14. https://doi.org/10.3858/emm.2011.43.1.001
Leonhard SE, Mandarakas MR, Gondim FAA, Bateman K, Ferreira MLB, Cornblath DR, van Doorn PA, Dourado ME, Hughes RAC, Islam B, Kusunoki S, Pardo CA, Reisin R, Sejvar JJ, Shahrizaila N, Soares C, Umapathi T, Wang Y, Yiu EM, Jacobs BC (2019) Diagnosis and management of Guillain-Barré syndrome in ten steps. Nat Rev Neurol 15(11):671–683. https://doi.org/10.1038/s41582-019-0250-9
Li C, Zhao P, Sun X, Che Y, Jiang Y (2013a). Elevated levels of cerebrospinal fluid and plasma interleukin-37 in patients with Guillain-Barré syndrome [Clinical Study]. Mediat Inflamm. https://doi.org/10.1155/2013/639712
Li Y-C, Bai W-Z, Hirano N, Hayashida T, Taniguchi T, Sugita Y, Tohyama K, Hashikawa T (2013b) Neurotropic virus tracing suggests a membranous-coating-mediated mechanism for transsynaptic communication. J Comp Neurol 521(1):203–212. https://doi.org/10.1002/cne.23171
Li Z, He W, Lan Y, Zhao K, Lv X, Lu H, Ding N, Zhang J, Shi J, Shan C, Gao F (2016) The evidence of porcine hemagglutinating encephalomyelitis virus induced nonsuppurative encephalitis as the cause of death in piglets. PeerJ 4:e2443. https://doi.org/10.7717/peerj.2443
Li Y-C, Bai W-Z, Hashikawa T (2020) The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J Med Virol 92(6):552–555. https://doi.org/10.1002/jmv.25728
Lu K, Wang J, Hu B, Shi X, Zhou J, Tang Y et al (2013) Brilliant blue G attenuates lipopolysaccharide-mediated microglial activation and inflammation. Neural Regen Res 8:599–608. https://doi.org/10.3969/j.issn.1673-5374.2013.07.003
Machhi J, Herskovitz J, Senan AM, Dutta D, Nath B, Oleynikov MD, Blomberg WR, Meigs DD, Hasan M, Patel M, Kline P, Chang RC-C, Chang L, Gendelman HE, Kevadiya BD (2020) The natural history, pathobiology, and clinical manifestations of SARS-CoV-2 infections. J Neuroimmune Pharmacol. https://doi.org/10.1007/s11481-020-09944-5
Major CG, Dirlikov E, Medina NA, Lugo-Robles R, Matos D, Muñoz-Jordán J, Colón-Sánchez C, García-Negrón M, Olivero-Segarra M, Malavé-González G, Thomas DL, Luciano CA, Waterman SH, Sejvar J, Sharp TM, Rivera-García B (2018) Implementation and Evaluation of Guillain-Barré Syndrome Surveillance in Puerto Rico during the 2016 Zika Virus Epidemic. P R Health Sci J 37(Spec Issue):S85–S92
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Miao X, Li Y, Hu B (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol 77(6):1–9. https://doi.org/10.1001/jamaneurol.2020.1127
Marini JJ (2020) Dealing with the CARDS of COVID-19. Crit Care Med 48(8):1239–1241. https://doi.org/10.1097/CCM.0000000000004427
Marini JJ, Gattinoni L (2020) Management of COVID-19 respiratory distress. JAMA. https://doi.org/10.1001/jama.2020.6825
Martínez-Frailes C, Di Lauro C, Bianchi C, de Diego-García L, Sebastián-Serrano Á, Boscá L, Díaz-Hernández M (2019) Amyloid peptide induced neuroinflammation increases the P2 × 7 receptor expression in microglial cells, impacting on its functionality. Front Cell Neurosci 13. https://doi.org/10.3389/fncel.2019.00143
McCray PB, Pewe L, Wohlford-Lenane C, Hickey M, Manzel L, Shi L, Netland J, Jia HP, Halabi C, Sigmund CD, Meyerholz DK, Kirby P, Look DC, Perlman S (2007) Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J Virol 81(2):813–821. https://doi.org/10.1128/JVI.02012-06
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, HLH Across Speciality Collaboration, UK (2020) COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 395(10229):1033–1034. https://doi.org/10.1016/S0140-6736(20)30628-0
Meinhardt J, Radke J, Dittmayer C, Mothes R, Franz J, Laue M, Schneider J, Brünink S, Hassan O, Stenzel W, Windgassen M, Rößler L, Goebel H-H, Martin H, Nitsche A, Schulz-Schaeffer WJ, Hakroush S, Winkler MS, Tampe B, … Heppner FL (2020) Olfactory transmucosal SARS-CoV-2 invasion as port of Central Nervous System entry in COVID-19 patients. BioRxiv 2020. https://doi.org/10.1101/2020.06.04.135012
Miras-Portugal MT, Sebastian-Serrano A, De Diego Garcia L, Diaz-Hernandez M (2017) Neuronal P2 × 7 receptor: involvement in neuronal physiology and pathology. J Neurosci 37:7063–7072. https://doi.org/10.1523/JNEUROSCI.3104-16.2017
Mosher KI, Wyss-Coray T (2014) Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol 88:594–604. https://doi.org/10.1016/j.bcp.2014.01.008
Padroni M, Mastrangelo V, Asioli GM, Pavolucci L, Abu-Rumeileh S, Piscaglia MG, Querzani P, Callegarini C, Foschi M (2020) Guillain-Barré syndrome following COVID-19: New infection, old complication? J Neurol 1–3. https://doi.org/10.1007/s00415-020-09849-6
Paniz-Mondolfi A, Bryce C, Grimes Z, Gordon RE, Reidy J, Lednicky J, Sordillo EM, Fowkes M (2020) Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J Med Virol 92(7):699–702. https://doi.org/10.1002/jmv.25915
Rocha NP, Simoes E, Silva AC, Prestes TRR, Feracin V, Machado CA, Ferreira RN, Teixeira AL, de Miranda AS (2018) RAS in the central nervous system: potential role in neuropsychiatric disorders. Curr Med Chem 25(28):3333–3352. https://doi.org/10.2174/0929867325666180226102358
Sankowski R, Mader S, Valdés-Ferrer SI (2015) Systemic Inflammation and the Brain: Novel Roles of Genetic, Molecular, and Environmental Cues as Drivers ofNeurodegeneration. Frente Célula Neurosci 9:28. https://doi.org/10.3389/fncel.2015.00028
Scheidl E, Canseco DD, Hadji-Naumov A, Bereznai B (2020) Guillain‐Barré syndrome during SARS‐CoV‐2 pandemic: A case report and review of recent literature. J Peripher Nerv Syst. https://doi.org/10.1111/jns.12382
Sedaghat Z, Karimi N (2020) Guillain Barre syndrome associated with COVID-19 infection: A case report. J Clin Neurosci 76:233–235. https://doi.org/10.1016/j.jocn.2020.04.062
Taylor MP, Enquist LW (2015) Axonal spread of neuroinvasive viral infections. Trends Microbiol 23(5):283–288. https://doi.org/10.1016/j.tim.2015.01.002
Toescu EC, Möller T, Kettenmann H, Verkhratsky A (1998) Long-term activation of capacitative Ca2 + entry in mouse microglial cells. Neuroscience 86(3):925–935. https://doi.org/10.1016/s0306-4522(98)00123-7
Toscano G, Palmerini F, Ravaglia S, Ruiz L, Invernizzi P, Cuzzoni MG, Franciotta D, Baldanti F, Daturi R, Postorino P, Cavallini A, Micieli G (2020) Guillain–Barré syndrome associated with SARS-CoV-2. N Engl J Med. https://doi.org/10.1056/NEJMc2009191
Wang W, Huang F, Jiang W, Wang W, Xiang J (2020) Brilliant blue G attenuates neuro-inflammation via regulating MAPKs and NF-κB signaling pathways in lipopolysaccharide-induced BV2 microglia cells. Exp Ther Med 20(5):116. https://doi.org/10.3892/etm.2020.9244
Wijdicks EF, Klein CJ (2017) Guillain-Barré syndrome. Mayo Clin Proc 92(3):467–479
Willison HJ, Jacobs BC, van Doorn PA (2016) Guillain-Barré syndrome. Lancet 388(10045):717–727. https://doi.org/10.1016/S0140-6736(16)00339-1
Wu Y, Xu X, Chen Z, Duan J, Hashimoto K, Yang L, Liu C, Yang C (2020) Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav Immun 87:18–22. https://doi.org/10.1016/j.bbi.2020.03.031
Xia H, Lazartigues E (2008) Angiotensin-converting enzyme 2 in the brain: Properties and future directions. J Neurochem 107(6):1482–1494. https://doi.org/10.1111/j.1471-4159.2008.05723.x
Xiao L, Haack KKV, Zucker IH (2013) Angiotensin II regulates ACE and ACE2 in neurons through p38 mitogen-activated protein kinase and extracellular signal-regulated kinase 1/2 signaling. Am J Physiol Cell Physiol 304(11):C1073–C1079. https://doi.org/10.1152/ajpcell.00364.2012
Xu J, Zhong S, Liu J, Li L, Li Y, Wu X, Li Z, Deng P, Zhang J, Zhong N, Ding Y, Jiang Y (2005) Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the Chemokine Mig in pathogenesis. Clin Infect Dis 41(8):1089–1096. https://doi.org/10.1086/444461
Xu P, Sriramula S, Lazartigues E (2011) ACE2/ANG-(1–7)/Mas pathway in the brain: The axis of good. Am J Physiol Regul Integr Comp Physiol 300(4):R804–R817. https://doi.org/10.1152/ajpregu.00222.2010
Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367(6485):1444–1448. https://doi.org/10.1126/science.abb2762
Yesilkaya UH, Balcioglu YH (2020) Neuroimmune correlates of the nervous system involvement of COVID-19: A commentary. J Clin Neurosci 78:449–450. https://doi.org/10.1016/j.jocn.2020.05.056
Zhao H, Shen D, Zhou H, Liu J, Chen S (2020) Guillain-Barré syndrome associated with SARS-CoV-2 infection: Causality or coincidence? Lancet Neurol 19(5):383–384. https://doi.org/10.1016/S1474-4422(20)30109-5
Zhou Z, Kang H, Li S, Zhao X (2020) Understanding the neurotropic characteristics of SARS-CoV-2: From neurological manifestations of COVID-19 to potential neurotropic mechanisms. J Neurol 1–6. https://doi.org/10.1007/s00415-020-09929-7
Zubair AS, McAlpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S (2020) Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: a review. JAMA Neurology. https://doi.org/10.1001/jamaneurol.2020.2065
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest/Competing Interests
The authors declare that there is no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Simões, J.L.B., Bagatini, M.D. Purinergic Signaling of ATP in COVID-19 Associated Guillain-Barré Syndrome. J Neuroimmune Pharmacol 16, 48–58 (2021). https://doi.org/10.1007/s11481-020-09980-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-020-09980-1