A Comprehensive Mapping of the Druggable Cavities within the SARS-CoV-2 Therapeutically Relevant Proteins by Combining Pocket and Docking Searches as Implemented in Pockets 2.0

 , , , , and
, , , , and 
Abstract
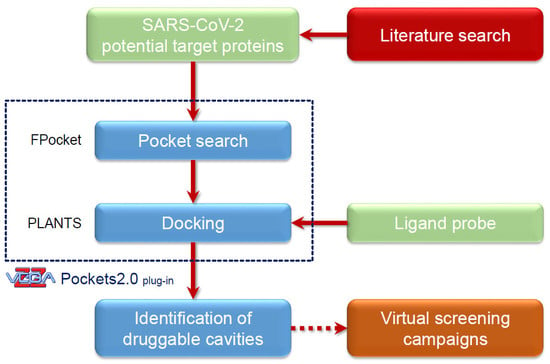
:
1. Introduction
2. Results
2.1. Overview of the Available Data on SARS-CoV-2 Binding Pockets
2.2. Pockets 2.0 Performances
3. Materials and Methods
3.1. Protein Structures
3.2. Preliminary Simulations
3.3. The Pockets 2.0 Approach
3.4. Pocket Search and Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3WL | 5,6,7-trihydroxy-2-phenyl-4H-chromen-4-one |
| 4VA | (2S)-4-amino-N-[(1R)-1-(4-chloro-2-fluoro-3-phenoxyphenyl)propyl]-4-oxobutan-2-aminium |
| 5GP | guanosine-5’-monophosphate |
| 77Z | 2-({(3R)-3-[(3S)-1-(3-methylbutyl)-2,4-dioxo-1,2,3,4-tetrahydroquinolin-3-yl]-1,1-dioxido-3,4-dihydro-2H-1,2,4-benzothiadiazin-7-yl}oxy)acetamide |
| ACE2 | angiotensin-converting enzyme 2 |
| AG7 | 4-{2-(4-fluoro-benzyl)-6-methyl-5-[(5-methyl-isoxazole-3-carbonyl)-amino]-4-oxo-heptanoylamino}-5-(2-oxo-pyrrolidin-3-yl)-pentanoic acid ethyl ester |
| C5P | cytidine-5’-monophosphate |
| G3A | guanosine-P3-adenosine-5’,5’-triphosphate |
| GTA | P1-7-methylguanosine-P3-adenosine-5’,5’-triphosphate |
| HCV | Hepatitis C virus |
| K22 | (Z)-N-(3-(4-(4-bromophenyl)-4-hydroxypiperidin-1-yl)-3-oxo-1-phenylprop-1-en-2-yl) benzamide |
| MERS-Cov | Middle-East Respiratory Syndrome Coronavirus |
| MGT | 7N-methyl-8-hydroguanosine-5’-triphosphate |
| N7-MTase | N-7 Methyl transferase |
| NSP | Non-structural protein |
| ORF | Opening Reading frame |
| P34 | 2-(dimethylamino)-N-(6-oxo-5H-phenanthridin-2-yl)acetamide (PJ34) |
| PFI | (6S)-6-cyclopentyl-6-[2-(3-fluoro-4-isopropoxyphenyl)ethyl]-4-hydroxy-5,6-dihydro-2h-pyran-2-one |
| PL-Pro | Papain-like protease |
| POO | 3-cyclohexyl-1-(2-{methyl[(1-methylpiperidin-3-yl)methyl]amino}-2-oxoethyl)-2-phenyl 1H-indole-6-carboxylic acid |
| RBD | receptor binding domain |
| RdRp | RNA-dependent RNA polymerase |
| SAH | S-Adenosyl homocysteine |
| SAM | S-Adenosyl methionine |
| SARS-CoV | Severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| TTT | 5-amino- 2-methyl-N-[(1R)-1-naphthalen-1-ylethyl]benzamide |
| U3P | uridine-3’-monophosphate |
| U5P | uridine-5’-monophosphate |
References
- Macari, G.; Toti, D.; Polticelli, F. Computational methods and tools for binding site recognition between proteins and small molecules: From classical geometrical approaches to modern machine learning strategies. J. Comput. Mol. Des. 2019, 33, 887–903. [Google Scholar] [CrossRef] [PubMed]
- Stank, A.; Kokh, D.B.; Fuller, J.; Wade, R.C. Protein Binding Pocket Dynamics. Accounts Chem. Res. 2016, 49, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Pérot, S.; Sperandio, O.; Miteva, M.A.; Camproux, A.-C.; Villoutreix, B.O. Druggable pockets and binding site centric chemical space: A paradigm shift in drug discovery. Drug Discov. Today 2010, 15, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Le Guilloux, V.; Schmidtke, P.; Tufféry, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendlich, M.; Rippmann, F.; Barnickel, G. LIGSITE: Automatic and efficient detection of potential small molecule-binding sites in proteins. J. Mol. Graph. Model. 1997, 15, 359–363. [Google Scholar] [CrossRef]
- Dias, S.; Martins, A.M.; Nguyen, Q.T.; Gomes, A.J. GPU-based detection of protein cavities using Gaussian surfaces. BMC Bioinform. 2017, 18, 493. [Google Scholar] [CrossRef] [Green Version]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T. A geometric approach to macromolecule-ligand interactions. J. Mol. Boil. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Kawabata, T. Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins Struct. Funct. Bioinform. 2009, 78, 1195–1211. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, E.; Müller, P.; Schalon, C.; Bret, G.; Foata, N.; Rognan, D. sc-PDB: An Annotated Database of Druggable Binding Sites from the Protein Data Bank. J. Chem. Inf. Model. 2006, 46, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.; Simões, T.; Fernandes, F.; Martins, A.M.; Ferreira, A.; Jorge, J.; Gomes, A.J.P. CavBench: A benchmark for protein cavity detection methods. PLoS ONE 2019, 14, e0223596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussein, H.A.; Geneix, C.; Petitjean, M.; Borrel, A.; Flatters, D.; Camproux, A. Global vision of druggability issues: Applications and perspectives. Drug Discov. Today 2017, 22, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA: A versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J. Mol. Graph. Model. 2002, 21, 47–49. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from COVID-19 virus and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, eabb3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-K.; Hou, M.-H.; Chang, C.-F.; Hsiao, C.-D.; Huang, T.-H. The SARS coronavirus nucleocapsid protein—Forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary Analysis of SARS-2-CoV: How Mutation of Non-Structural Protein 6 (NSP6) Could Affect Viral Autophagy. SSRN Electron. J. 2020, 0163–4453. [Google Scholar] [CrossRef]
- Sutton, G.; Fry, E.; Carter, L.; Sainsbury, S.; Walter, T.; Nettleship, J.; Berrow, N.; Owens, R.; Gilbert, R.; Davidson, A.; et al. The nsp9 Replicase Protein of SARS-Coronavirus, Structure and Functional Insights. Structure 2004, 12, 341–353. [Google Scholar] [CrossRef]
- Egloff, M.-P.; Ferron, F.; Campanacci, V.; Longhi, S.; Rancurel, C.; Dutartre, H.; Snijder, E.J.; Gorbalenya, A.E.; Cambillau, C.; Canard, B. The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a single-stranded RNA-binding subunit unique in the RNA virus world. Proc. Natl. Acad. Sci. USA 2004, 101, 3792–3796. [Google Scholar] [CrossRef] [Green Version]
- Davis, B.C.; Brown, J.A.; Thorpe, I.F. Allosteric Inhibitors Have Distinct Effects, but Also Common Modes of Action, in the HCV Polymerase. Biophys. J. 2015, 108, 1785–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marco, S.; Volpari, C.; Tomei, L.; Altamura, S.; Harper, S.; Narjes, F.; Koch, U.; Rowley, M.; De Francesco, R.; Migliaccio, A.R.; et al. Interdomain Communication in Hepatitis C Virus Polymerase Abolished by Small Molecule Inhibitors Bound to a Novel Allosteric Site. J. Boil. Chem. 2005, 280, 29765–29770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Tatlock, J.; Linton, A.; González, J.; Borchardt, A.; Dragovich, P.; Jewell, T.; Prins, T.; Zhou, R.; Blazel, J.; et al. Identification and structure-based optimization of novel dihydropyrones as potent HCV RNA polymerase inhibitors. Bioorganic Med. Chem. Lett. 2006, 16, 4834–4838. [Google Scholar] [CrossRef]
- Shadrick, W.R.; Ndjomou, J.; Kolli, R.; Mukherjee, S.; Hanson, A.M.; Frick, D.N. Discovering new medicines targeting helicases: Challenges and recent progress. J. Biomol. Screen. 2013, 18, 761–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, W.; Wojdyla, J.A.; Zhao, R.; Han, R.; Das, R.; Zlatev, I.; Manoharan, M.; Wang, M.; Cui, S. Crystal structure of Middle East respiratory syndrome coronavirus helicase. PLoS Pathog 2017, 13, e1006474. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Jayachandran, U.; Bonneau, F.; Fiorini, F.; Basquin, C.; Domcke, S.; Le Hir, H.; Conti, E. Molecular Mechanisms for the RNA-Dependent ATPase Activity of Upf1 and Its Regulation by Upf2. Mol. Cell 2011, 41, 693–703. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14–nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef] [Green Version]
- Baez-Santos, Y.M.; John, S.S.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2014, 115, 21–38. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-Y.; Liu, C.-L.; Chang, Y.-M.; Zhao, J.; Perlman, S.; Hou, M.-H. Structural Basis for the Identification of the N-Terminal Domain of Coronavirus Nucleocapsid Protein as an Antiviral Target. J. Med. Chem. 2014, 57, 2247–2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef] [Green Version]
- Lundin, A.; Dijkman, R.; Bergström, T.; Kann, N.; Adamiak, B.; Hannoun, C.; Kindler, E.; Jonsdottir, H.R.; Muth, D.; Kint, J.; et al. Targeting Membrane-Bound Viral RNA Synthesis Reveals Potent Inhibition of Diverse Coronaviruses Including the Middle East Respiratory Syndrome Virus. PLoS Pathog 2014, 10, e1004166. [Google Scholar] [CrossRef] [Green Version]
- Ponnusamy, R.; Moll, R.; Weimar, T.; Mesters, J.R.; Hilgenfeld, R. Variable Oligomerization Modes in Coronavirus Non-structural Protein 9. J. Mol. Boil. 2008, 383, 1081–1096. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.A.; Dragovich, P.S.; Webber, S.E.; Fuhrman, S.A.; Patick, A.K.; Zalman, L.S.; Hendrickson, T.F.; Love, R.A.; Prins, T.J.; Marakovits, J.T.; et al. Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. USA 1999, 96, 11000–11007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deore, R.R.; Chern, J.W. NS5B RNA dependent RNA polymerase inhibitors: The promising approach to treat hepatitis C virus infections. Curr. Med. Chem. 2010, 17, 3806–3826. [Google Scholar] [CrossRef] [PubMed]
- Ismail, N.S.M.; Elzahabi, H.S.A.; Sabry, P.; Baselious, F.N.; Abdelmalak, A.S.; Hanna, F. A study of the allosteric inhibition of HCV RNA-dependent RNA polymerase and implementing virtual screening for the selection of promising dual-site inhibitors with low resistance potential. J. Recept. Signal Transduct. 2016, 37, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Tomei, L.; Altamura, S.; Bartholomew, L.; Biroccio, A.; Ceccacci, A.; Pacini, L.; Narjes, F.; Gennari, N.; Bisbocci, M.; Incitti, I.; et al. Mechanism of action and antiviral activity of benzimidazole-based allosteric inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Virol. 2003, 77, 13225–13231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jácome, R.; Becerra, A.; De León, S.P.; Lazcano, A. Structural Analysis of Monomeric RNA-Dependent Polymerases: Evolutionary and Therapeutic Implications. PLoS ONE 2015, 10, e0139001. [Google Scholar] [CrossRef] [Green Version]
- Shaw, A.N.; Tedesco, R.; Bambal, R.; Chai, D.; Concha, N.O.; Darcy, M.G.; Dhanak, D.; Duffy, K.; Fitch, D.M.; Gates, A.; et al. Substituted benzothiadizine inhibitors of Hepatitis C virus polymerase. Bioorganic Med. Chem. Lett. 2009, 19, 4350–4353. [Google Scholar] [CrossRef] [PubMed]
- Abdurakhmanov, E.; Solbak, S.M.Ø.; Danielson, U.H. Biophysical Mode-of-Action and Selectivity Analysis of Allosteric Inhibitors of Hepatitis C Virus (HCV) Polymerase. Viruses 2017, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Saalau-Bethell, S.M.; Woodhead, A.J.; Chessari, G.; Carr, M.G.; Coyle, J.; Graham, B.; Hiscock, S.D.; Murray, C.W.; Pathuri, P.; Rich, S.J.; et al. Discovery of an allosteric mechanism for the regulation of HCV NS3 protein function. Nat. Methods 2012, 8, 920–925. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Jedrzejczak, R.; Maltseva, N.I.; Wilamowski, M.; Endres, M.; Godzik, A.; Michalska, K.; Joachimiak, A. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV -2. Protein Sci. 2020, 29, 1596–1605. [Google Scholar] [CrossRef]
- Ricagno, S.; Egloff, M.-P.; Ulferts, R.; Coutard, B.; Nurizzo, D.; Campanacci, V.; Cambillau, C.; Ziebuhr, J.; Canard, B. Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc. Natl. Acad. Sci. USA 2006, 103, 11892–11897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decroly, E.; Debarnot, C.; Ferron, F.; Bouvet, M.; Coutard, B.; Imbert, I.; Gluais, L.; Papageorgiou, N.; Sharff, A.; Bricogne, G.; et al. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2′-o-methyltransferase nsp10/nsp16 complex. PLoS Pathog 2011, 7, e1002059. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, L.; Yan, L.; Ming, Z.; Jia, Z.; Lou, Z.; Rao, Z. Structural and Biochemical Characterization of Endoribonuclease Nsp15 Encoded by Middle East Respiratory Syndrome Coronavirus. J. Virol. 2018, 92, e00893-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodel, A.E.; Gershon, P.D.; Quiocho, F.A. Structural basis for sequence-nonspecific recognition of 5′-Capped mRNA by a cap-modifying enzyme. Mol. Cell. 1998, 1, 443–447. [Google Scholar] [CrossRef]
- Aminjafari, A.; Ghasemi, S. The possible of immunotherapy for COVID-19: A systematic review. Int. Immunopharmacol. 2020, 83, 106455. [Google Scholar] [CrossRef]
- Fan, H.-H.; Wang, L.-Q.; Liu, W.-L.; An, X.-P.; Liu, Z.-D.; He, X.-Q.; Song, L.-H.; Tong, Y.-G. Repurposing of clinically approved drugs for treatment of coronavirus disease 2019 in a 2019-novel coronavirus-related coronavirus model. Chin. Med. J. 2020, 133, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Remmert, M.; Biegert, A.; Hauser, A.; Soeding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef]
- Xu, J.; Jiao, F.; Berger, B. A tree-decomposition approach to protein structure prediction. Proceedings of 2005 IEEE Computational Systems Bioinformatics Conference (CSB’05), Stanford, CA, USA, 8–12 August 2005; IEEE Computer Society Press: Los Alamitos, CA, USA; pp. 247–256. [Google Scholar] [CrossRef]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L.; Dunbrack, R.L. Improved prediction of protein side-chain conformations with SCWRL4. Proteins Struct. Funct. Bioinform. 2009, 77, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Boil. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Jr, A.D.M.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2010, 27, 343–350. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo-distance constraints applied on model quality estimation. Bioinformatis 2020, 36, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. Modeling of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Proteins by Machine Learning and Physics-Based Refinement. bioRxiv. Preprint. 2020. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2012, 19, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Pedretti, A.; Villa, L.; Vistoli, G. VEGA—An open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J. Comput. Mol. Des. 2004, 18, 167–173. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
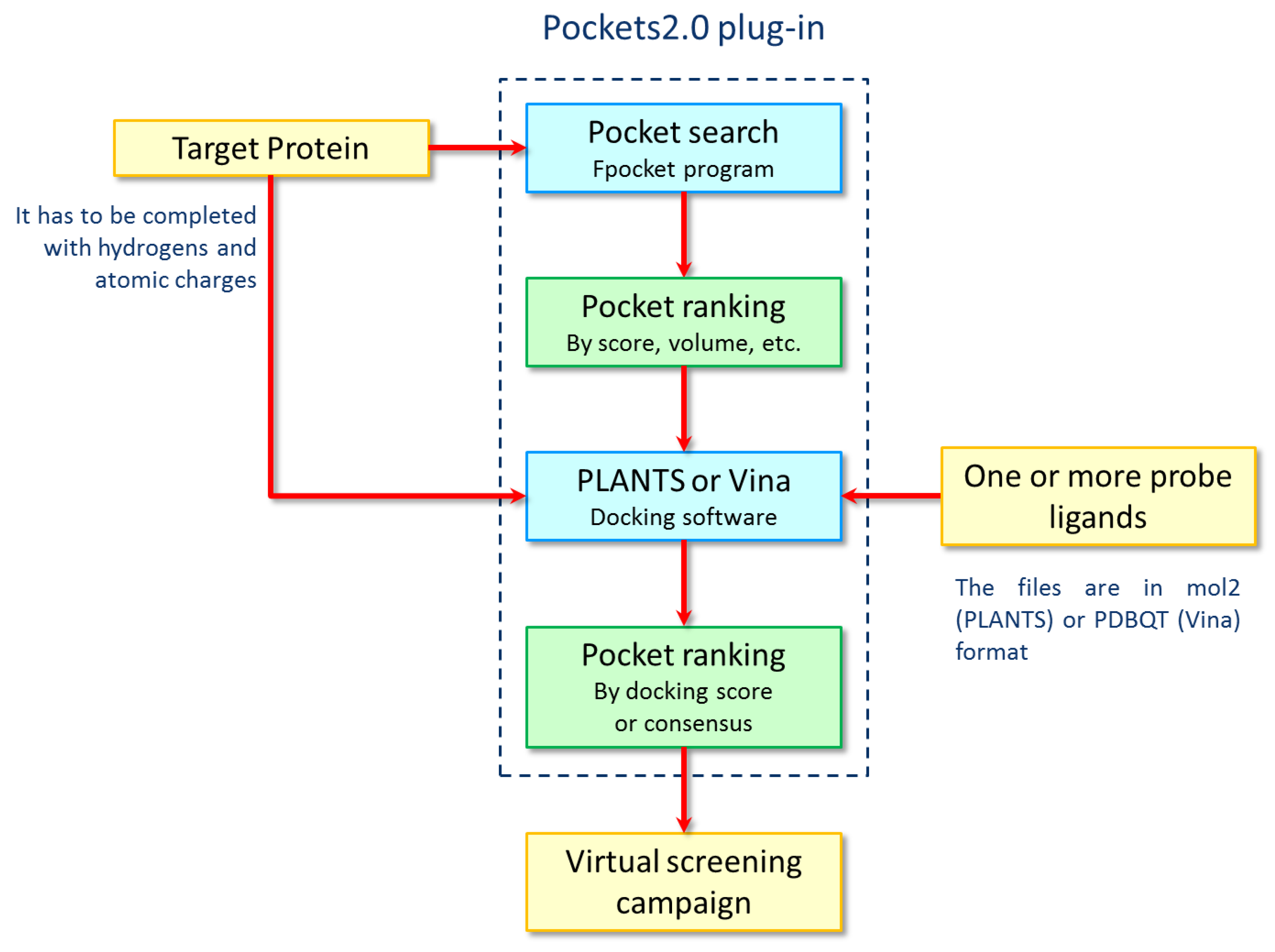{kind=link}
| Protein | Source a | Function | Reference Protein | PDB Id | Site | Ligand c | Ref. |
|---|---|---|---|---|---|---|---|
| 3CL-PRO | See Table 2 | protease | ---b | ---b | orthosteric | 3WL | [15,16] |
| N-Protein | 6M3M 6VYO | Nucleocapsid protein | HCoV-OC43 N-NTD | 4LMC | orthosteric | C5P | [17] |
| 4LM9 | 5GP | ||||||
| 4LM7 | U5P | ||||||
| 4LI4 | AMP | ||||||
| 4KXJ | P34 | ||||||
| Nsp3 | 6W02 | ADP ribose phosphatase | --- | --- | orthosteric | ADP | *** d |
| Nsp6 | DN | Membrane-spanning protein | No experimental information apart from mutants analysis | --- | allosteric | K22 | [18] |
| Nsp9 | 6W4B | Replicase | Coronavirus NSP9 | 1QZ8 | orthosteric | SO4 | [19] |
| Type 2 rhinovirus 3C protease | 1CQQ | orthosteric | AG7 | [20] | |||
| Nsp12 | 7BV2 | RNA-dependent RNA polymerase (RdRp) | --- | --- | orthosteric | F86 | *** |
| Hepatitis C RdRp | 2BRL | allosteric1 (thumb) | POO | [21] | |||
| Hepatitis C NS5B polymerase | 2HAI | alllosteric2 (thumb) | PFI | [22] | |||
| Hepatitis C NS5 polymerase | 3HHK | allosteric3 (palm) | 77Z | [23] | |||
| Nsp13 | HM | Helicase | RNA-Dependent ATPase Upf1 | 2XZL | orthosteric | ADP-ALF | [24] |
| Hepatitis C virus NS3 protein | 4B75 | Allosteric | 4VA | [25] | |||
| Nsp14 | HM | Methyltransferase | SARS-CoV | 5C8S | orthosteric | SAH, G3A | [26] |
| Nsp15 | 6W01 | Endoribonuclease | SARS-CoV | 2H85 | orthosteric | U3M | [27] |
| Nsp16 | 6WKS 6W4H | Methyltransferase | --- | --- | orthosteric | SAM, GTA | *** |
| PL-pro | 6W9C | Papain-like protease | SARS-CoV | 3E9S | orthosteric | TTT | *** |
| Spike | Xray with ACE2 | Viral entry glycoprotein | --- | --- | Protein–protein interaction | YMZ | [28,29,30,31,32,33,34,35,36] |
| Protein | Protein Data | Data for the Search of the Correct Pocket | ||||||
|---|---|---|---|---|---|---|---|---|
| Source/ID | Ligand | N Pockets | Rank by Fpocket | Rank by PLANTS | Rank by Consensus | Volume (Å3) | ChemPLP (kcal/mol) | |
| 3CL-PRO | 5R7Y | 3WL | 16 | 1 | 1 | 1 | 3604.93 | −76.15 |
| 5R7z | 3WL | 16 | 1 | 1 | 1 | 4246.06 | −73.85 | |
| 5R80 | 3WL | 20 | 2 | 2 | 1 | 1833.23 | −76.48 | |
| 5R81 | 3WL | 17 | 1 | 1 | 1 | 3141.06 | −80.27 | |
| 5R82 | 3WL | 19 | 1 | 4 | 1 | 3989.84 | −68.23 | |
| 5R83 | 3WL | 21 | 1 | 1 | 1 | 4079.17 | −77.59 | |
| 5R84 | 3WL | 21 | 1 | 1 | 1 | 2562.02 | −80.88 | |
| 6LU7 | 3WL | 14 | 1 | 1 | 1 | 4239.62 | −77.83 | |
| 6MN2 dimer | 3WL | 80 | 1 | 1 | 1 | 3344.72 | −86.30 | |
| 6M03 | 3WL | 24 | 3 | 3 | 3 | 2175.81 | −69.70 | |
| 6Y2E | 3WL | 18 | 1 | 4 | 1 | 2629.29 | −75.02 | |
| 6Y2F | 3WL | 20 | 1 | 1 | 1 | 2581.33 | −77.61 | |
| 6Y2G | 3WL | 16 | 2 | 3 | 2 | 5074.55 | −75.78 | |
| 6Y84 | 3WL | 20 | 1 | 2 | 1 | 3068.30 | −73.94 | |
| Nsp3 | 6W02 | APR | 6 | 2 | 1 | 1 | 2247.84 | −119.05 |
| Nsp6 | DN | K22 | 25 | 3 | 1 | 1 | 2364.82 | −75.26 |
| DN | 27 | 4 | 1 | 1 | 3692.30 | −76.86 | ||
| Nsp9 | 6W4B | AG7 | 13 | 2 | 3 | 1 | 2740.53 | −97.41 |
| Nsp12-Nsp7-Nsp8 | 7BV2 trimer | ATP (ortho) | 79 | 1 | 2 | 1 | 4397.28 | −94.18 |
| POO/ PFI (allo) | 2 | 1 | 1 | 3545.24 | −97.80 | |||
| 77Z (allo) | 1 | 4 | 2 | 4397.28 | −85.22 | |||
| Nsp13 | HM | ADP (ortho) | 40 | 3 | 2 | 2 | 4359.36 | −79.98 |
| 4VA (allo) | 5 | 10 | 5 | 2526.96 | −74.28 | |||
| Nsp14-Nsp10 | HM dimer | SAH | 49 | 2 | 9 | 4 | 5307.98 | −80.25 |
| SAM | 2 | 2 | 1 | 5307.98 | −88.09 | |||
| G3A | 2 | 1 | 1 | 5307.98 | −119.09 | |||
| Nsp15 | 6W01 hexamer | U3P | 170 | 5 | 3 | 1 | 2792.58 | −80.39 |
| 6VWW dimer | U3P | 52 | 6 | 1 | 3 | 2275.76 | −81.75 | |
| Nsp16-Nsp10 | 6WKS dimer | SAM | 63 | 1 | 8 | 1 | 4772.88 | −88.43 |
| GTA | 1 | 1 | 1 | 4772.88 | −118.80 | |||
| 6W4H dimer | SAM | 31 | 1 | 1 | 1 | 3243.06 | −87.81 | |
| GTA | 1 | 2 | 1 | 3243.06 | −112.33 | |||
| N-protein | 6M3M | C5P, 5GP, U5P, AMP, P34 | 7 | 3 | 2 | 2 | 1252.56 | −66.69 |
| 6VYO | 7 | 3 | 3 | 3 | 1961.99 | −70.57 | ||
| PL-Pro | 6W95 | TTT | 23 | 3 | 2 | 1 | 2134.23 | −94.17 |
| SPIKE–ACE2 | 6LZG dimer | YMZ | 56 | 2 | 2 | 1 | 4792.66 | −80.66 |
| 6M0J dimer | 58 | 1 | 5 | 1 | 4407.92 | −74.29 | ||
| 6M17 hexamer | 275 | 1 11 | 1 1 | 1 2 | 3767.99 | −81.79 | ||
| 6VW1 dimer | 61 | 1 | 2 | 1 | 4920.74 | −77.78 | ||
| Correctly Identified Pockets | 20 | 18 | 30 | |||||
| Correct Pockets Ranked as #2 | 9 | 10 | 5 | |||||
| Correct Pockets Ranked as #3 | 6 | 5 | 3 | |||||
| Correct Rockets Out of the Podium | 5 | 7 | 2 | |||||
| Average Rank | 2.18 | 2.43 | 1.45 | |||||
| Precision | 0.5 | 0.45 | 0.75 | |||||
| Accuracy | 0.97 | 0.97 | 0.99 | |||||
| MCC | 0.48 | 0.43 | 0.74 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gervasoni, S.; Vistoli, G.; Talarico, C.; Manelfi, C.; Beccari, A.R.; Studer, G.; Tauriello, G.; Waterhouse, A.M.; Schwede, T.; Pedretti, A. A Comprehensive Mapping of the Druggable Cavities within the SARS-CoV-2 Therapeutically Relevant Proteins by Combining Pocket and Docking Searches as Implemented in Pockets 2.0. Int. J. Mol. Sci. 2020, 21, 5152. https://doi.org/10.3390/ijms21145152
Gervasoni S, Vistoli G, Talarico C, Manelfi C, Beccari AR, Studer G, Tauriello G, Waterhouse AM, Schwede T, Pedretti A. A Comprehensive Mapping of the Druggable Cavities within the SARS-CoV-2 Therapeutically Relevant Proteins by Combining Pocket and Docking Searches as Implemented in Pockets 2.0. International Journal of Molecular Sciences. 2020; 21(14):5152. https://doi.org/10.3390/ijms21145152
Chicago/Turabian StyleGervasoni, Silvia, Giulio Vistoli, Carmine Talarico, Candida Manelfi, Andrea R. Beccari, Gabriel Studer, Gerardo Tauriello, Andrew Mark Waterhouse, Torsten Schwede, and Alessandro Pedretti. 2020. "A Comprehensive Mapping of the Druggable Cavities within the SARS-CoV-2 Therapeutically Relevant Proteins by Combining Pocket and Docking Searches as Implemented in Pockets 2.0" International Journal of Molecular Sciences 21, no. 14: 5152. https://doi.org/10.3390/ijms21145152