
The Perspective of Vitamin D on suPAR-Related AKI in COVID-19
by
 , ,
, ,
Tzu-Hsien Liao
1,†,
Hsien-Chang Wu
1,2,
Min-Tser Liao
3,4,
Wan-Chung Hu
5,
Kuo-Wang Tsai
6,
Ching-Chieh Lin
7,† and
Kuo-Cheng Lu
8,9,* 1
Department of Chinese Medicine, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei City 231, Taiwan
2
School of Post-Baccalaureate Chinese Medicine, Tzu Chi University, Hualien 970, Taiwan
3
Department of Pediatrics, Taoyuan Armed Forces General Hospital Hsinchu Branch, Hsinchu City 300, Taiwan
4
Department of Pediatrics, Tri-Service General Hospital, National Defense Medical Center, Taipei 114, Taiwan
5
Department of Clinical Pathology and Medical Research, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei City 231, Taiwan
6
Department of Research, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei City 231, Taiwan
7
Department of Chest Medicine, Taoyuan Armed Forces General Hospital Hsinchu Branch, Hsinchu City 300, Taiwan
8
Division of Nephrology, Department of Medicine, Taipei Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, New Taipei City 231, Taiwan
9
Division of Nephrology, Department of Medicine, Fu-Jen Catholic University Hospital, School of Medicine, Fu-Jen Catholic University, New Taipei City 242, Taiwan
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2022, 23(18), 10725; https://doi.org/10.3390/ijms231810725
Submission received: 31 August 2022
/
Revised: 8 September 2022
/
Accepted: 13 September 2022
/
Published: 14 September 2022
(This article belongs to the Special Issue The Role of Vitamin D in Human Health and Diseases 2.0)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The coronavirus disease 2019 (COVID-19) pandemic has claimed the lives of millions of people around the world. Severe vitamin D deficiency can increase the risk of death in people with COVID-19. There is growing evidence that acute kidney injury (AKI) is common in COVID-19 patients and is associated with poorer clinical outcomes. The kidney effects of SARS-CoV-2 are directly mediated by angiotensin 2-converting enzyme (ACE2) receptors. AKI is also caused by indirect causes such as the hypercoagulable state and microvascular thrombosis. The increased release of soluble urokinase-type plasminogen activator receptor (suPAR) from immature myeloid cells reduces plasminogen activation by the competitive inhibition of urokinase-type plasminogen activator, which results in low plasmin levels and a fibrinolytic state in COVID-19. Frequent hypercoagulability in critically ill patients with COVID-19 may exacerbate the severity of thrombosis. Versican expression in proximal tubular cells leads to the proliferation of interstitial fibroblasts through the C3a and suPAR pathways. Vitamin D attenuates the local expression of podocyte uPAR and decreases elevated circulating suPAR levels caused by systemic inflammation. This decrease preserves the function and structure of the glomerular barrier, thereby maintaining renal function. The attenuated hyperinflammatory state reduces complement activation, resulting in lower serum C3a levels. Vitamin D can also protect against COVID-19 by modulating innate and adaptive immunity, increasing ACE2 expression, and inhibiting the renin–angiotensin–aldosterone system. We hypothesized that by reducing suPAR levels, appropriate vitamin D supplementation could prevent the progression and reduce the severity of AKI in COVID-19 patients, although the data available require further elucidation.
1. Introduction
Approximately 75% of all patients with coronavirus disease 2019 (COVID-19) have acute kidney injury (AKI) and/or abnormal urine dipstick findings [1]. The analysis of some, although not all, urine and postmortem kidney tissue revealed the presence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), confirming the kidney is one of the main targets of COVID-19 [2]. Epidemiological studies in Europe and China have reported AKI in approximately half of the patients with fulminant sepsis [3,4]. Another clinical study identifying resuscitation strategies for severe sepsis also reported that AKI occurs in approximately half of all emergency department patients [5]. Furthermore, almost one-third of patients with community-acquired pneumonia develop AKI even without evidence of severe sepsis [6]. Before the pandemic, a clue showed the incidence of COVID-19-associated AKI exceeds that of sepsis-associated AKI. However, the incidence of AKI was not significantly different from other forms of sepsis over time.
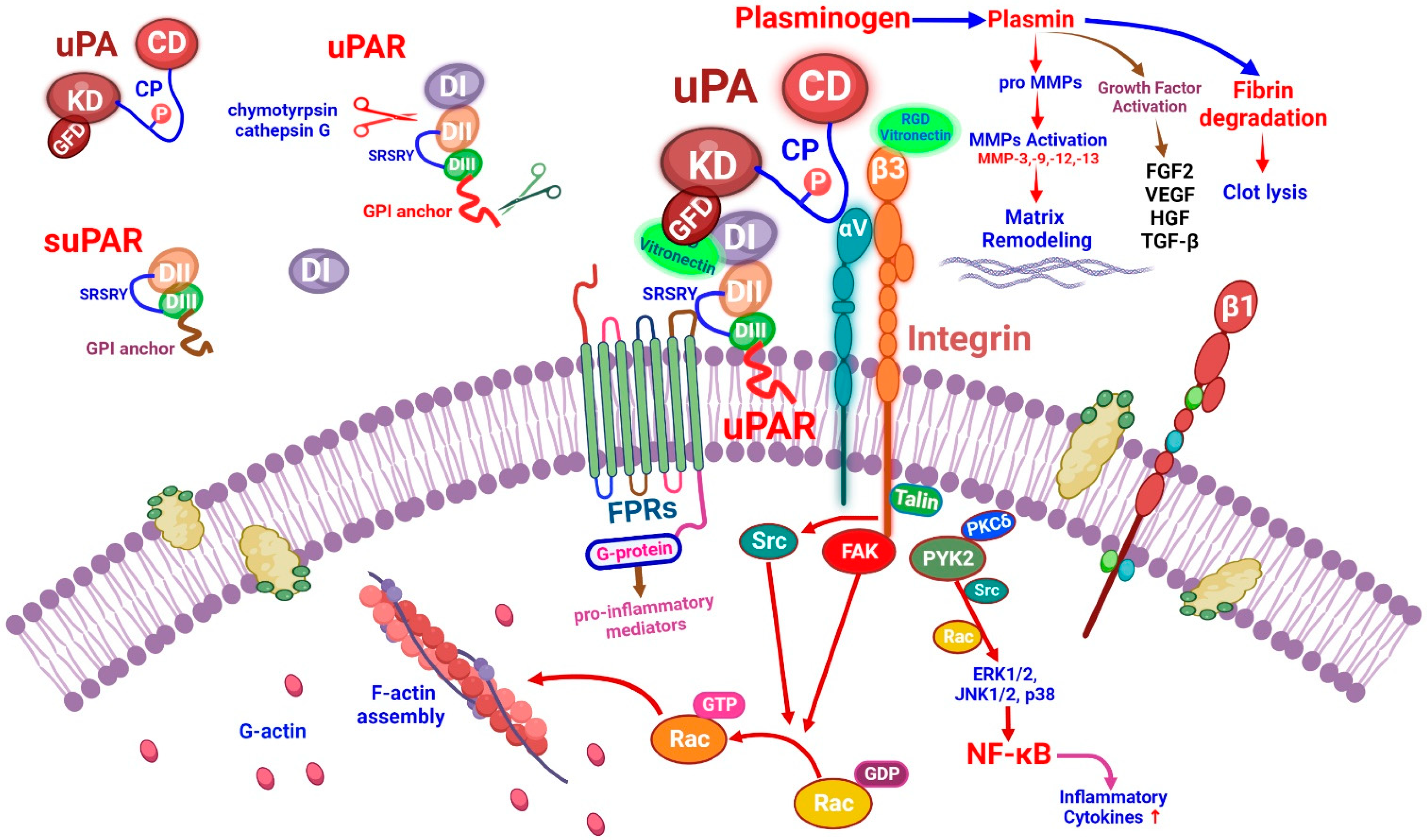
The uPA (urokinase plasminogen activator) and uPA receptor (uPAR or CD87) form a multimolecular complex at the cell surface which can cause fibrinolysis and perform immune functions [7]. The uPAR may be detached from the cellular membrane glycosylphosphatidylinositol (GPI) anchorage, resulting in the release of the soluble form of uPAR (suPAR) [8]. The uPA localized in podocytes/tubular cells can bind to the uPAR domain, then the uPAR can interact with other molecules such as integrins and vitronectin. Damaged endothelial cells produce and secrete plasminogen activator-inhibitor 1 (PAI-1). The binding of PAI-1 to the uPA-uPAR complex can inhibit plasminogen activation [9]. In COVID-19, excess suPAR may bind to uPA, thus preventing uPA from activating plasminogen [10]. By binding to uPAR, uPA catalyzes the conversion of plasminogen to plasmin, which contributes to the breakdown of the extracellular matrix (ECM) and promotes cell motility [11]. Once the uPA binds to the uPAR, a structure configuration transition occurs and it can play a role in linking with other co-receptors such as integrins and vitronectin. The uPAR acts on αvβ3 integrins and can promote cell motility in kidney podocytes [12]. The binding of the vitronectin with uPAR and β3 integrins is necessary for the activation of catabolic proteases such as MMP-3 and MMP-13 [13].
The suPAR has a potential prognostic value in critically ill patients [14]. In clinical laboratory medicine, it has the potential to predict critical illness and renal failure in intensive care unit (ICU) patients [15]. Low levels of suPAR have been reported as a positive predictor of critical care survival in seriously ill patients [16]. Afro-Caribbean patients with COVID-19 and increased suPAR levels exhibit a high risk of AKI [17]. Because elevated levels of suPAR can damage kidney cells directly, they can also increase the incidence of hypofibrinolysis and thrombogenesis, which can contribute to the severity of AKI. In COVID-19 inpatients, circulating suPAR levels on admission can predict the development of AKI and the need for renal replacement therapy [18].
Vitamin D deficiency is associated with severe COVID-19 in adult patients [19]. In children, a potential association between a profound vitamin D deficiency and the severity of COVID-19 multisystem inflammatory syndrome (C-MIS) has been reported [20]. Vitamin D levels can be used to predict the risk for severe C-MIS, and correcting abnormal levels in severe cases may help reduce the severity of C-MIS [21]. Vitamin D can attenuate the probability of breast tumor metastasis by inhibiting uPAR expression [22]. Vitamin D can also prevent podocyte injury and proteinuria development by inhibiting podocyte uPAR expression [23]. In mice with chronic kidney disease (CKD) and septicemia, treatment with vitamin D was found to inhibit podocyte uPAR expression and provide an obvious anti-proteinuric effect [24]. The renal tissue study of vitamin D receptor knockout diabetic mice showed that the glomerular basement membrane was significantly thickened and the podocytes were significantly reduced, resulting in more severe glomerulosclerosis and obvious proteinuria [25]. We hypothesized that the potential beneficial effects of vitamin D can be directly mitigated through uPA/uPAR-related integrin signaling and indirect anti-inflammatory response. However, the molecular mechanism by which vitamin D inhibits podocyte uPAR in COVID-19-related AKI remains to be fully elucidated. Therefore, we conducted this narrative review to explore the potential role of vitamin D in preventing AKI in COVID-19 patients. We specifically focused on the uPA/suAPR pathway and its possible relationship with vitamin D in patients with COVID-19-related AKI.
2. COVID-19-Associated AKI
2.1. Clinical Manifestations
Nearly 5% of patients with COVID-19 have serious associated conditions such as septicemia or organ failure. Proteinuria, hematuria, and AKI are the main features of kidney injury [26]. Kidney damage caused by COVID-19 manifests as increased serum levels of creatinine, proteinuria, hematuria, and AKI, and some patients require renal replacement therapy (RRT) [1,27]. Based on KDIGO’s (Kidney Disease: Improving Global Outcomes) AKI criteria, approximately half of such patients are in stage 3 [28]. In a cohort study, a high incidence of proteinuria and hematuria was reported and linked to mortality in patients with critical COVID-19 [29]. The qualitative and quantitative assessments of renal impairment are performed by determining decreased renal parenchymal attenuation on non-enhanced computed tomography, which shows a weak linear and inverse correlation with serum creatinine levels in COVID-19 patients [30].
Previous retrospective studies have shown that AKI is common in hospitalized patients due to COVID-19 and is associated with elevated mortality. The kidney function at discharge is restored in only 30% of patients with AKI [31]. An elevated incidence of AKI is found in patients with other comorbidities such as high blood pressure, diabetes, and pre-existing kidney disease [32]. Kidney function was not restored to baseline in one-third of patients with AKI and COVID-19. Considering the overall severity of AKI and the high prevalence of severe tubular lesions, underlying vascular micro-thrombus, and protein in COVID-19 patients, lower renal recovery rates are expected [33]. Another retrospective observational study evaluating clinical outcomes in adults with AKI with COVID-19 suggested that vital signs and laboratory data at admission may be useful for the risk stratification of severe AKI. The best-known risk factors associated with COVID-19-mediated AKI are obesity, diabetes, hypertension, aging, and pre-existing CKD [34].
2.2. Histopathological Characteristics and Pathophysiology
Initial histopathological analyses of 26 autopsy samples obtained from the kidneys of patients with COVID-19 provide direct evidence of SARS-CoV-2 invasion of the kidney tissue [33]. SARS-CoV-2 can cause kidney damage in a variety of ways, and it can directly infect renal tubular cells and podocytes through the ACE2 receptor, resulting in increased mitochondrial/endoplasmic reticulum (ER)-related oxidative stress, acute tubular necrosis, Bowman’s capsule leak, and collapsing glomerulopathy. Renal damage in patients with preexisting renal disease and increased expressions of ACE2 and CD147 in the renal tissue may be aggravated [35]. The histological analysis of renal biopsy samples from patients with COVID-19 showed tubular epithelium with reactive nuclei, significantly simplified cytoplasm, and obvious denudation of brush borders. Glomeruli showed a sagging of the tuft and hypertrophy and hyperplasia of mesangial cells (MCs) and epithelial cells. The ultrastructural study revealed extensive foot process effacement and tubuloreticular inclusions in a glomerular endothelial cell [36,37].
The detailed pathophysiology of COVID-19-related AKI is unclear [38]. Dysregulated immune reactions induced by SARS-CoV-2 may be a cause of AKI. In addition, endothelial dysfunction, prominent hypercoagulability, and microbial infection-related sepsis are other important potential causes of AKI. Furthermore, hypoxemia in the kidneys may lead to ischemic damage [39]. Potentially modifiable factors such as complement activation, hypercoagulability/hypofibrinolysis, and hyperinflammatory reaction should be explored in detail [34,40]. Whether targeting these pathways using vitamin D, anticoagulants, or cytokine inhibitors will reduce the severity of AKI is unclear. Moreover, the detailed mechanisms of antiviral therapy in the manipulation of COVID-19-related AKI remain unclear [41,42].
Hypercoagulability and microvascular thrombus formation are common mechanisms for acute respiratory distress syndrome and AKI. Immunofluorescence tests have shown the SARS-CoV-2 protein in different renal cells. However, D-dimer concentration has not been directly linked with AKI [38,43]. AKI in COVID-19-related sepsis has a multifactorial etiology, and suPAR is a crucial potential mediator [44,45]. As AKI is associated with local intrarenal and systemic inflammation and intravascular hypercoagulability; therefore, further exploration of the underlying molecular responses may help in the search for effective treatments to reduce the severity of AKI.
2.2.1. Direct Effects of SARS-CoV-2 on AKI
AKI observed in the context of COVID-19 is associated with a direct viral invasion of renal cells. The invasion of renal cells by SARS-CoV-2 is mediated by ACE2 receptors, resulting in acute tubular necrosis, filtration barrier disruption, and micro thrombosis with collapsing glomerulopathy [39,46]. The entry of SARS-CoV-2 into cells requires the interaction of ACE2, TMPRSS2 (transmembrane protease serine 2), ADAM17 (a disintegrin, metalloproteinase 17), and possibly cell membrane CD147 [47]. TMPRSS2 expression is lower in proximal tubules; therefore, its role in regulating the priming process is unclear [48]. Distal tubules express sufficient TMPRSS2 to facilitate the entry of SARS-CoV-2 into host cells [49]. With the help of TMPRSS2, the SARS-CoV-2 spike protein interacts with the ACE2 in the cell membrane, thereby promoting viral internalization. ADAM17 can eliminate circulating SARS-CoV-2 by splitting the ACE2 in the membrane into a soluble form which can capture SARS-CoV-2 particles. However, the expression of CD147 is primarily distributed on the basolateral side of the proximal tubes. Enhanced CD147 expression can further promote SARS-CoV-2 entry into renal tubular cells with pre-existing lesions [50].
Conventional renin–angiotensin–aldosterone system (RAAS) activation is common in CKD patients, and localized reduction in ACE2 expression increases hypercoagulability and induces the formation of microthrombi [51]. In the kidney, glomerular-filtered SARS-CoV-2 can invade proximal tubular epithelial cells via luminal ACE2 receptors. Entrance via luminal or basolateral routes can exacerbate tubule-interstitial inflammation and decrease cell survival. Damaged glomerular podocytes increase CD147 expression, which further enhances SARS-CoV-2 invasion. After SARS-CoV-2 enters the host cell, the virus downregulates the expression of ACE2 in the host cell, thereby increasing the expression of angiotensin II (Ang II) [52]. In addition to increased blood pressure, Ang II through nuclear factor κB signaling will enhance the expression of several inflammatory cytokine genes [53]. Over-activation of the RAAS can further enhance the expression of CD147. Therefore, increasing SARS-CoV-2 entry and inducing podocyte damage is one of the key mechanisms for inducing AKI [35]. Previous reports have also suggested that genetic variation in the ACE receptor is another key factor. In addition, the genotype of apolipoprotein L1 variant (APOL1) is another genetic risk factor for collapsing glomerulopathy in COVID-19 patients [54,55].
2.2.2. Indirect Effects of SARS-CoV-2 on AKI
Injured lung from COVID-19 releases damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) into circulation, which are then filtered in the glomerulus and can interact with pattern recognition receptors (PRRs) on tubular epithelial cells, resulting in renal tubular epithelial damage. DAMPs also elicit systemic inflammation that can damage the kidneys and other organs [56]. The release of chemokines can attract additional inflammatory cells to the site of inflammation, thereby exacerbating the cytokine storm, and can have indirect effects on several organs, especially on the kidneys [39]. Some COVID-19 patients experience the fulminant activation of macrophages following a cytokine storm. They often have high levels of ferritin in the plasma [57]. In addition, myocardial lesions secondary to pulmonary lesions can develop in critically ill patients [58]. Interactions between damaged lungs, heart, and kidneys can adversely affect clinical outcomes.
In addition, potential nephrotoxic drugs are commonly used in critical patients with COVID-19 [59]. An increase in right heart ventricle pressure can result from left heart ventricle failure or high positive expiratory pressure on mechanical ventilation in patients with respiratory failure. Mitochondrial dysfunction induced by SARS-CoV-2 can damage kidney cells directly and indirectly by enhancing systemic oxidative stress and inflammation [60]. Other epigenetic alterations, such as those in miRNAs, influence the pathogenesis of COVID-19 [61]. Consequently, the molecular mechanisms of AKI in COVID-19 patients would be similar to those caused by other forms of sepsis.
3. Role of suPAR in COVID-19-Related AKI
Infected critically ill patients usually have higher systemic suPAR concentrations than uninfected patients [16,62]. A meta-analysis of biological markers in adults disclosed that suPAR can be used as a diagnostic and prognostic marker for microbial infection [63]. However, the role of suPAR in critically ill patients with AKI is unclear [64]. An observational study has also shown that elevated suPAR levels predict AKI and the need for dialysis. As a result, suPAR may play an important role in the pathophysiology of AKI in COVID-19 [18].
3.1. Function of the Urokinase Receptor System
The uPAR is a glycosylphosphatidylinositol (GPI)-anchored urokinase receptor. The uPA can bind to uPAR and integrin through a growth-factor-like domain and connecting peptide (Figure 1). Three different suPARs exist in the circulation (suPAR DI-III, DI, and DII-III). Among them, suPAR DI-III is considered the active form of suPAR due to its strong affinity for uPA binding [65]. A recent report found that a dimer form of the mouse isoform 2 uPAR can induce kidney disease [66]. The overexpression of this dimer can cause kidney disease in mice by activating glomerular Src kinase through β3 integrin signaling [66]. The activation of uPA and plasminogen-dependent latent transforming growth factor β1 (TGF-β) yields active TGF-β, a powerful driver of endothelial–mesenchymal transition in the kidney [67].
The uPA localizes in podocytes/tubular cells by binding to uPAR domain 1 (DI). The uPA domains are represented as the growth-factor-like domain (GFD), the kringle domain (KD), the interdomain linker or “connecting peptide” (CP, residues 132–158), and the serine protease domain (CD). The uPA interacts with the GPI-anchored uPAR through GFD and with integrin through CP, bridging the two receptors together [67]. The amino-terminal region of uPA can bind to the DI region of uPAR. The DII:DIII region of uPAR can interact with co-receptors such as the vitronectin and membrane integrin (αvβ3, αvβ5) families. The binding of plasminogen activator-inhibitor 1 (PAI-1) to the uPA–uPAR complex can not only inhibit the uPA activation of plasminogen but also promote the internalization of uPA–uPAR–PAI-1 complex and β1-integrin, which then recycles uPAR back to the cell surface [9]. Under physiological conditions, after binding to uPAR, the uPA can catalyze the conversion of the inactive plasminogen to active plasmin [11]. Several proteins expressed during fibrogenesis can be cleaved by plasmin. In addition, plasmin can also activate matrix metalloproteinases (MMPs) and activate or release growth factors such as hepatocyte growth factor. Once uPA binds to uPAR, the structure configuration will be changed, and the uPAR SRSRY (88–92) sequence can bind with other co-receptors (Figure 1). Chymotrypsin and cathepsin G can hydrolyze uPAR at the DI:DII junction region, resulting in fragmented DII:DIII GPI-anchored uPAR. This modified uPAR would then detach from the GPI anchor, resulting in the release of the soluble form of uPAR (suPAR). Since uPAR lacks an intracellular domain, it will interact with other transmembrane receptors such as formyl peptide receptors (FPRs) and integrins. FPRs can also be activated by the DII:DIII SRSRY fragment. Briefly, the activation of these co-receptors will elicit intracellular signaling which is related to the synthesis of pro-angiogenesis and pro-inflammatory mediators [8].
The arginine–glycine–aspartic acid (RGD)-containing vitronectin fragments can induce podocyte damage through β3 integrin signaling (Figure 1). In addition, protein kinase C-delta (PKCδ) is the rate-limiting factor at the signaling transduction from vitronectin, which can activate PKCδ. PKCδ can activate NF-κB and mitogen-activated protein kinase (MAPK) signaling. The activation of MAPKs (ERK1/2, JNK1/2, and p38) will inhibit anabolic signaling such as IGF-1 and BMP7 signaling, increasing inflammatory cytokine levels, and upregulating catabolic proteases such as MMP-3 and MMP-13 [13]. The uPAR can act on β3 integrins to enhance cell motility in kidney podocytes. However, the binding of vitronectin to uPAR and β3 integrins is required for the activation of this pathway. This uPAR–β3 integrin interaction activates Src kinases, which leads to the phosphorylation of the focal adhesion kinase (FAK)-associated adapter protein. The activation of Rac by Src or FAK can stimulate actin polymerization, eliciting cell motility. In mouse podocytes, the uPAR–αvβ3 integrin signaling can activate the Rho family GTPase Cdc42 in a vitronectin-dependent manner. However, uPAR signaling through αvβ3 integrin could be suppressed by uPA-mediated uPAR cleavage [12] (Figure 1).
Integrin is composed of non-covalently linked α and β subunits which can be categorized into laminin, collagen, and RGD-binding receptors [68]. In addition to anchoring cells to the ECM (extracellular matrix), integrins also signal bidirectionally through the plasma membrane [69].
In the kidney, major laminin-binding receptors include integrins α3β1, α6β1, and α6β4. Effective adhesion of podocytes to the glomerular basement membrane (GBM) requires integrin α3β1 [69]. There are two major collagen receptors in the kidney, integrin α1β1, and α2β1, which play a crucial pathogenic role in inflammatory forms of kidney damage. In MCs, the binding of integrin α2β1 to collagen activates intracellular signaling, resulting in the increased production of collagen and reactive oxygen species (ROS) [70]. The collagen receptors in the kidney, integrin α1β1, and α2β1, play an important pathogenic role in inflammatory forms of kidney damage. However, uPA/uPAR mainly acts on the αvβ3 integrin. The RGD-containing ligands such as fibronectin and vitronectin can bind to β1, αv, and αIIbβ3 integrins. The αv subunit of integrin is ubiquitously expressed in adult kidneys, and it can modulate glomerular extracellular matrix homeostasis. The activation of integrin αvβ3 by uPAR and suPAR in podocytes has been reported to result in podocyte dysfunction [23]. Since COVID-19-related AKI may have high serum suPAR levels, we speculate that these high suPARs can activate integrin signaling that may cause kidney injury.
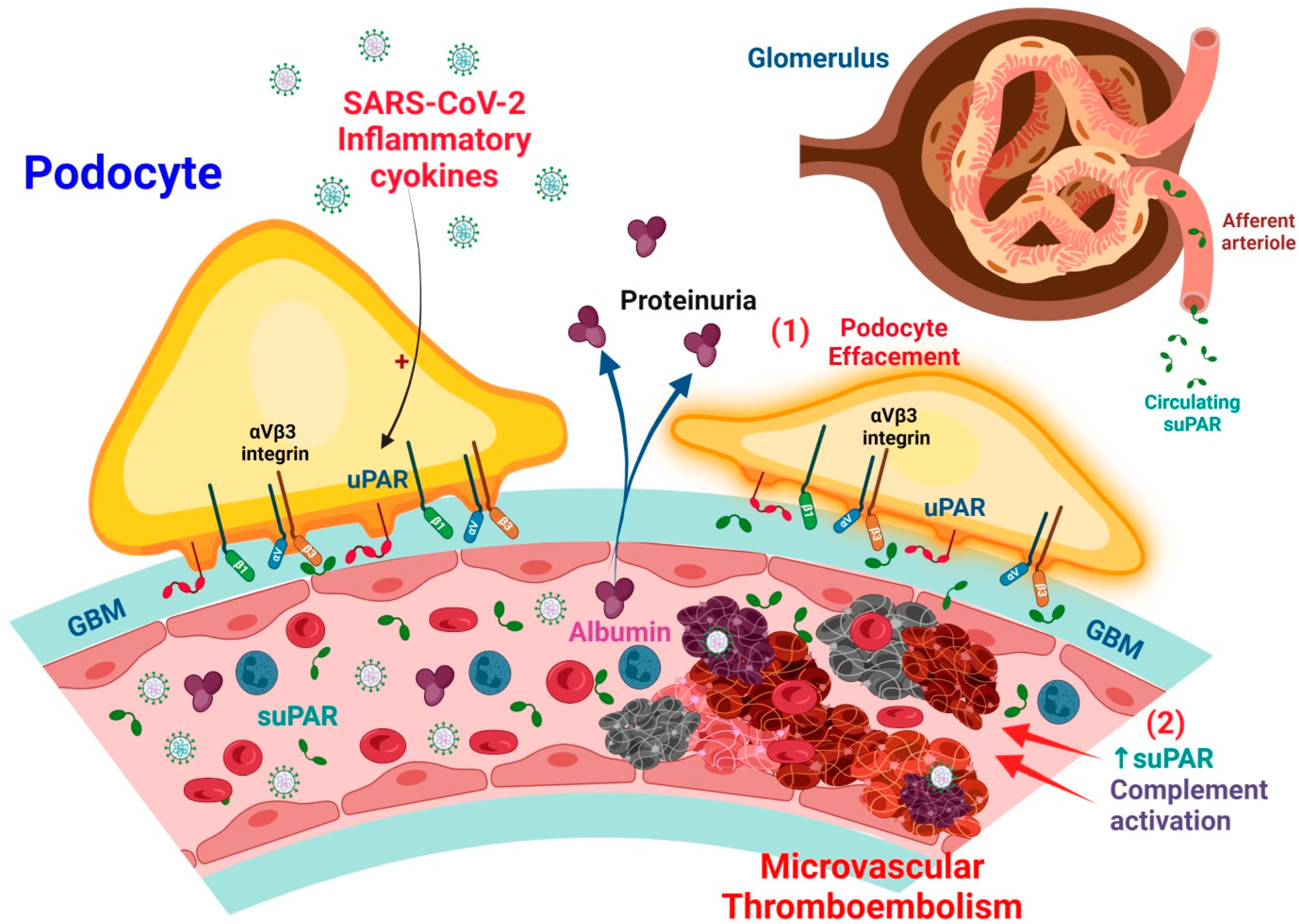
3.2. Putative Mechanism of suPAR-Related Kidney Injury in COVID-19-Related AKI
The suPAR is a circulating biomarker that can cause glomerular podocyte injury. The suPAR levels are increased in the plasma of COVID-19 patients, which provides mechanistic insights into how increased suPAR levels can lead to AKI. In this case, suPAR is produced mainly by immature Gr-1lo myeloid cells, neutrophils, monocytes, and perhaps T lymphocytes. Through blood circulation, suPAR enters the glomerulus of the kidneys and binds and activates β3 integrin. High plasma levels of suPAR enhanced β3 integrin signaling activation, leading to podocyte dysfunction, effacement, and proteinuria. The activation of integrin αvβ3 by uPAR and suPAR in podocytes impairs the podocyte function [69]. The damaged podocytes repress vascular endothelial growth factor expression which leads to endothelial dysfunction. The damaged endothelial cells produce and secrete PAI-1, which binds to circulating uPA either in the capillaries or on the podocyte surface, which makes a complex with uPAR. The complex can bind to β1-integrin on the surface of the podocyte, then β1-integrin translocates into the cytoplasm through endocytosis. Then, the β1-integrin-lost podocytes will detach from the glomerular basement membrane [71].
In terms of microvascular thrombosis, elevated suPAR levels in COVID-19 and AKI are associated with increased extra mitochondrial enzyme oxidation and oxidative stress in renal cells. Increased suPAR release in the bloodstream reduces plasmin production by competitively inhibiting uPA function on the renal cell surface. This may promote hypofibrinolytic status in critically ill patients with COVID-19, which will synergistically exacerbate thrombosis severity [10]. Furthermore, increased C3a production in patients with severe COVID-19 induces the release of activated CD16+ cytotoxic T cells from bone marrow. The proportion of activated CD16+ T cells and plasma C3a levels are associated with lethal thromboembolic outcomes in COVID-19 (Figure 2).
The uPAR primary ligand is the uPA, which converts plasminogen into plasmin. The uPA activates plasminogen in solution or when associated with its cellular receptor uPAR in a fibrin-independent manner [72]. The uPAR is a multifunctional receptor that coordinates pericellular proteolysis related to cell membrane integrin signal transduction, affecting normal physiology and leading to abnormal pathological mechanisms [67] (Figure 1).
The uPA and uPAR are expressed by different cells in the hematopoietic lineage. Multiple observations linked the fibrinolytic system to innate and adaptive immunity [73]. In general, systemic inflammation resulting from various infections leads to the activation of the coagulation system through the over-production of thrombin, attenuation of anticoagulant mechanisms, and inhibition of fibrinolysis. The proinflammatory cytokines (IL-1β, TNFα) play a crucial role in both the coagulation and fibrinolysis pathways [74]. Severe or critical COVID-19 is associated with an increased secondary infection rate and could lead to a significantly worsened prognosis. Secondary infection risks increased after receiving invasive respiratory ventilation and intravascular devices. The most common infections were respiratory, and the most detected pathogens were gram-negative bacteria, followed by gram-positive bacteria, viruses, and fungi [75]. During bacterial infection, circulating levels of uPA and its inhibitor PAI-1 increase significantly. These increases are also triggered by releases of bacterial endotoxins and cytokines due to the activation of innate immunity [76]. The conversion of uPA-related plasminogen into plasmin promotes the release of proinflammatory cytokines and activates phagocytic monocyte/macrophage (PMM) zymogenes, thus amplifying acute inflammatory responses [77] (Figure 1).
3.3. suPAR as a Biomarker of Infection in AKI
It is well known that circulating suPAR levels can represent the overall activity of the uPA/uPAR system that is related to innate immunity, proteolytic enzyme activity, and ECM remodeling capability [12,78]. The uPAR can be induced by leukocyte activation and differentiation in response to smoking and certain RNA viruses [79,80]. The suPAR has been identified as a functional link between the bone marrow (BM) and kidneys, and an increase in immature Gr-1lo myeloid cells in the BM is a key factor in the subsequent development of glomerular dysfunction caused by increased suPAR in proteinuric mice [81]. Therefore, BM-derived immature myeloid cells serve as the primary source of suPAR and may contribute to renal pathologies [82].
3.3.1. Effects of suPAR on Podocyte Injury
The suPAR is a risk factor in several human clinical conditions [14,83,84], such as AKI [85,86], and CKD [87]. In native and grafted kidneys, suPAR circulating has been shown to activate podocyte β integrin3, thus causing foot process erasure, proteinuria, and focal segmental glomerular glomerulopathy [88]. Hence, a high incidence of kidney disease is observed in patients with a high proinflammatory state with high levels of suPAR [89,90,91,92]. In podocytes, the induction of uPAR leads to foot process disappearance and the loss of urinary protein through the activation of αvβ3 integrin signaling [23] (Figure 2). High levels of circulating suPAR strongly predict renal function progression; further, chronic exposure directly activates podocyte αvβ3 integrin, resulting in proteinuria and ultimately, chronic renal dysfunction [66,81]. A previous study on transgenic mice with an over-expression of suPAR demonstrated the development of proteinuria and severe AKI after contrast injection [85].
Circulating suPAR affects podocyte configuration and viability, resulting in foot process effacement and cell apoptosis [88,93]. The structure–function association of suPAR with αvβ3 integrin can be regulated by sphingomyelinase-like phosphodiesterase 3b (SMPDL3b). SMPDL3b may modify the glomerular filtration barrier by affecting podocyte function due to the movement of a migratory phenotype to an apoptotic phenotype [94,95] (Figure 2). The lipid-modulating enzyme SMPDL3b is also a known off-target therapeutic of the anti-CD20 antibody rituximab [96].
A study of lipopolysaccharide (LPS)-mediated proteinuria in mice showed that uPAR is required for the activation of αvβ3 integrin signaling, enhancing the motility and activation of the small GTPases Cdc42 and Rac1 in podocytes (Figure 2). The blockade of αvβ3 integrin signaling will reduce podocyte motility and proteinuria in mice [23]. In addition, APOL1 gene variants G1 and G2 exhibit high affinity for activated αvβ3 integrin, which would synergize with suPAR to further activate αvβ3 integrin signaling on podocytes, thereby promoting the development of CKD [97].
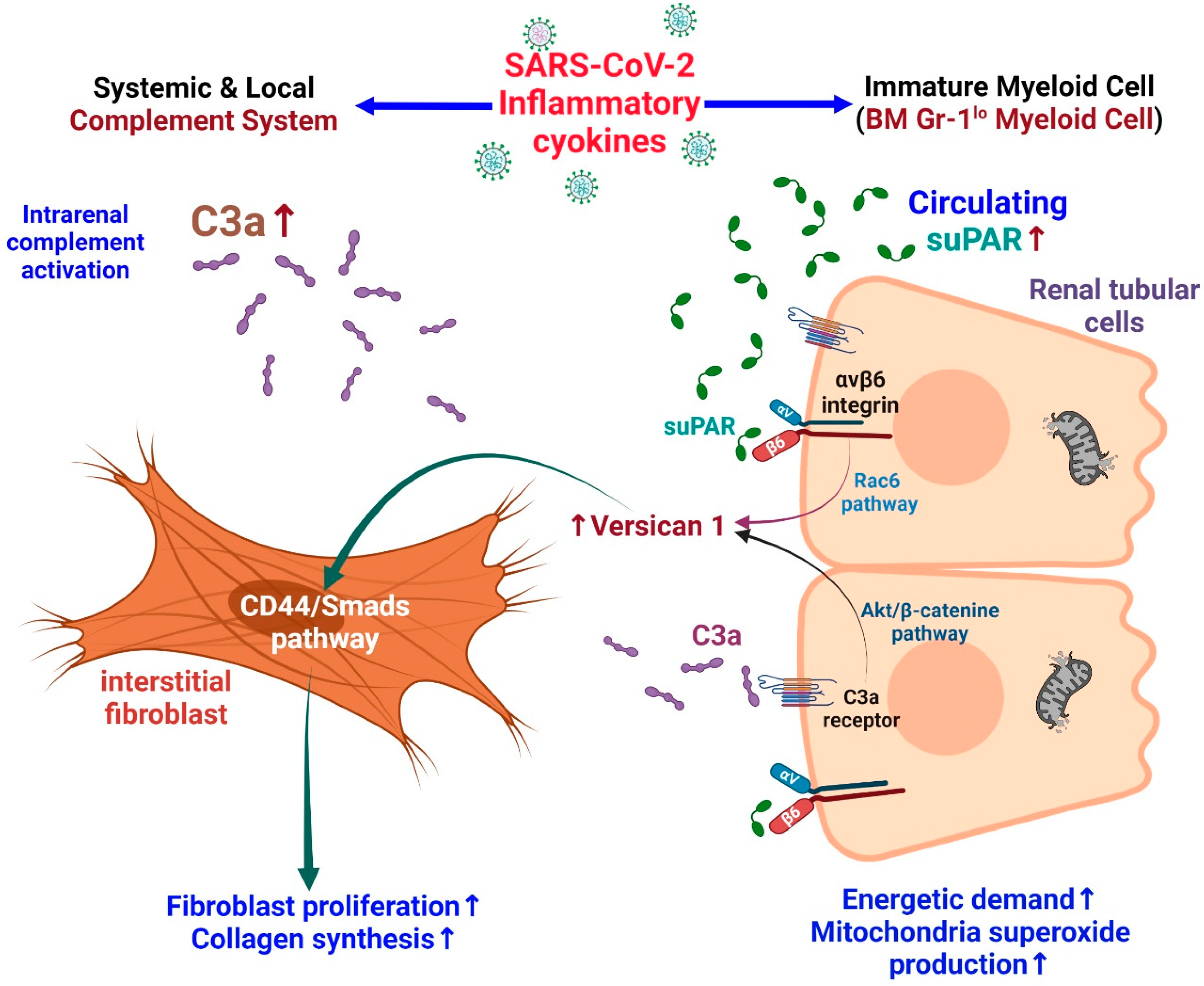
3.3.2. Effects of suPAR on Tubular Cell Injury
A study in mice with high expression of suPAR showed that they are vulnerable to contrast-mediated kidney injury. Further, proximal tubular cell cultures showed altered mitochondrial respiration with increased energy demand and the production of oxygen free radicals [85]. Elevated suPAR expression may induce tubulointerstitial fibrosis in an integrin-dependent manner. Versican is a large ECM proteoglycan found in different human tissues. In patients with focal segmental glomerular sclerosis (FSGS), renal tissue showed high levels of versican. Rienstra et al. showed that renal tubular-cell-derived versican can induce fibroblast proliferation and collagen production by activating the CD44/Smad3 pathway in renal interstitial fibroblasts [98]. In patients with COVID-19, an increased tubular bond between urinary C3a and suPAR enhances tubular versican expression and subsequently promotes renal interstitial fibrosis [99] (Figure 3).
3.3.3. The αvβ3-Integrin-Dependent Activation and Mesangial Fibrosis
MCs are stromal cells and are important for glomerular homeostasis and reaction to injury [100]. The plasma-derived fibrinogen is deposited in the mesangium of certain renal lesions. The adhesion of MCs to fibrinogen is mediated by αvβ3 integrin [101]. The histological analysis of samples obtained from a kidney biopsy performed in a patient with COVID-19 showed mesangial hyperplasia with fibrocystic crescents in some glomeruli. Occasionally, various degrees of secondary sclerotic ischemic changes are seen in the mesangial stroma associated with GBM thickening and podocyte detachment [102]. This sclerotic change is associated with TGF-β1 signaling. In cultured renal MCs, TGF-β1 stimulates collagen expression through the αvβ3-integrin-dependent activation of FAK and subsequent ERK/MAPK activity. This causes the phosphorylation of the Smad3 link region, which leads to collagen production and mesangial fibrosis [103]. High PAI-1 expression in MCs activates the MAPK signaling pathway and stimulates TGF-β1 expression, leading to accumulation in the mesangial matrix [104]. In rats with diabetes mellitus, uPA can improve the diabetic mesangial condition by binding to uPAR for capturing PAI-1 and accelerating its degradation, thereby attenuating the expression of MCs and their matrix [105].
3.3.4. Synergistic Effects of suPAR and C3a on Kidney Injury
In the kidney, human C3a and its receptor C3aR are mainly expressed in the proximal renal tubular cells [106,107]. In renal diseases, the expression of C3aRa and levels of C3a in plasma and urine are increased, and both are associated with the progression of renal disease. This reveals that the C3a/C3aR route facilitates the progression of glomerular and tubule-interstitial pathologies [108]. Patients infected with SARS-CoV-2 are at increased risk of dying due to the hyperactivity and consumption of C3 [109]. Dysregulated pro-inflammatory response may activate the complement system, which is associated with increased mortality and thromboembolic complications [110]. The increased production of C3a in severe COVID-19 can activate cytotoxic CD16+ T lymphocytes. Both CD16+ activated T-cell proportions and C3a plasma concentrations are associated with a fatal COVID-19 outcome [111].
C3a activates the AKT/β-catenin signaling pathway and can promote the transcription of versican in renal tubular cells. Furthermore, suPAR can bind to tubular cell integrin β6 and activate Rac1, thereby forming versican. In short, C3a and suPAR can result in versican expression in tubular cells, which may lead to renal fibrosis [99]. In experimental models, anti-uPAR monoclonal antibodies have been shown to mitigate these adverse effects in podocytes and tubular cells [85].
3.4. suPAR Levels and CKD
An observational study in patients with mild to moderate CKD showed a gradual association of suPAR levels with an increased risk of incident cardiovascular disease [112]. Furthermore, circulating suPAR levels were independently associated with an increased risk of progression to end-stage renal disease (ESRD) in Chinese patients with CKD, especially in patients with pre-existing glomerulonephritis [113]. A clinical study also showed the suPAR level can predict CKD incidence and renal dysfunction progression [114]. The suPAR levels are positively correlated with IgA severity, reflected by pathology findings and subsequent proteinuria [115]. Elevated plasma suPAR levels were also significantly associated with enhanced crescent formation in IgA nephritis [116]. In a large cohort of lupus nephritis patients, elevated suPAR levels were reported to have a higher rate of renal involvement [117].
Elevated plasma suPAR level is strongly associated with incident kidney disease. From a Cardiovascular Biobank analysis, higher suPAR concentrations at baseline are associated with a more prominent deterioration in renal function during the follow-up visits which is also accompanied by a potential for progression to CKD [87]. A study explored the suPAR levels in various kidney pathologies and found minimal change disease has the highest levels and chronic interstitial nephritis has the lowest levels; however, it was elevated in all studied patients with kidney diseases [118]. A meta-analysis revealed that suPAR levels may serve as a prognostic biomarker in CKD patients. The higher suPAR level has been shown to increase the risk of mortality, cardiovascular accidents, and ESRD occurrence [119].
Clinically, COVID-19 may pose an increased risk for severe sequelae (e.g., long COVID-19). Patients who have survived COVID-19 are at increased risk for renal outcomes. Therefore, post-acute care for COVID-19 should address kidney disease [120]. In a cohort study of inpatients with AKI, COVID-19-related acute respiratory infection (ARI) was associated with higher rates of post-discharge estimated glomerular filtration rate (eGFR) decline than in patients with AKI without COVID-19 [121]. Thus, we propose that higher levels of suPAR in COVID-19-related AKI may have a higher incidence of long COVID kidney outcomes. However, this possible point of view needs to be further elucidated.
4. High Prevalence of Vitamin D Deficiency in COVID-19 Patients
4.1. Vitamin D and COVID-19
COVID-19 remains a persistent global health crisis. Type II alveolar epithelial cells develop local innate immunity after the inhalation of SARS-CoV-2. These viral particles may infect circulating macrophages, and T lymphocytes may harbor SARS-CoV-2 antigens. SARS-CoV-2 inhibits diverse immune processes such as pathogen recognition and signaling, the production of IFN, and various interferon-stimulated genes (ISGs) [122]. As various types of T cells are activated and differentiated, the uncontrolled release of cytokines (called a cytokine storm) leads to an exaggerated immune response and tissue destruction. Vitamin D strengthens innate immunity against COVID-19 through activating toll-like receptor 2 and accentuating antimicrobial properties, such as enhancing the synthesis of lysosomes in macrophages, promoting the synthesis and secretion of β-defensins and cathelicidins, and augmenting autophagy through the autophagosomes pathway. Vitamin D also induces adaptive immunity by increasing the population of CD4+ T cells, activating T cell-dependent B cells to promote virus-specific antibody production, and suppressing T helper 17 cells. Furthermore, vitamin D decreases the release of proinflammatory cytokines by CD4+ T cells and inhibits the probable exacerbation of a cytokine storm. After SARS-CoV-2 enters cells by binding to the ACE2 receptor with its spike protein, vitamin D improves the bioavailability of soluble ACE2 to capture and inactivate the virus. SARS-CoV-2 activates the RAAS, which is responsible for not only inflammation but also tissue destruction and probable organ failure. Vitamin D inhibits renin expression and acts as a negative regulator of the RAAS [123]. Therefore, vitamin D protects from SARS-CoV-2 by activating intricate mechanisms including both innate and adaptive immunity, enhancing ACE2 expression, and efficiently inhibiting the RAAS. Serum 25-hydroxyvitamin D levels are reported to be negatively associated with the incidence or severity of COVID-19. Although experimental validation is insufficient [53], the clues collected to date probably meet Hill’s criteria for causality in biological systems [124].
The risk factors of COVID-19 include male sex, obesity, hypertension, diabetes, and vitamin D deficiency [125,126]. It is common knowledge that vitamin D deficiency is associated with an increased incidence of microbial infections. Generally, higher risk occurred at 25-hydroxyvitamin D concentrations below 20 ng/mL, but in a retrospective study from the National Health and Nutrition Examination Survey, levels < 30 ng/mL were associated with 58% higher odds of ARI [127,128]. Vitamin D treatment can prevent ARI, especially in patients with severe vitamin D deficiency [129]. In addition, countries with patients having lower average 25-hydroxyvitamin D levels have higher mortality rates from COVID-19 [130]. Patients admitted due to COVID-19 have low levels of 25-hydroxy vitamin D [131]. A meta-analysis study demonstrated that vitamin D deficiency may exacerbate COVID-19 [132]. The administration of high-dose calcidiol or 25-hydroxyvitamin D significantly reduces the need for ICU care in patients hospitalized with COVID-19 [133]. We believe that adequate vitamin D administration is important in attenuating the progression and severity of COVID-19, especially in a population with vitamin D deficiency.
4.2. Vitamin D and suPAR
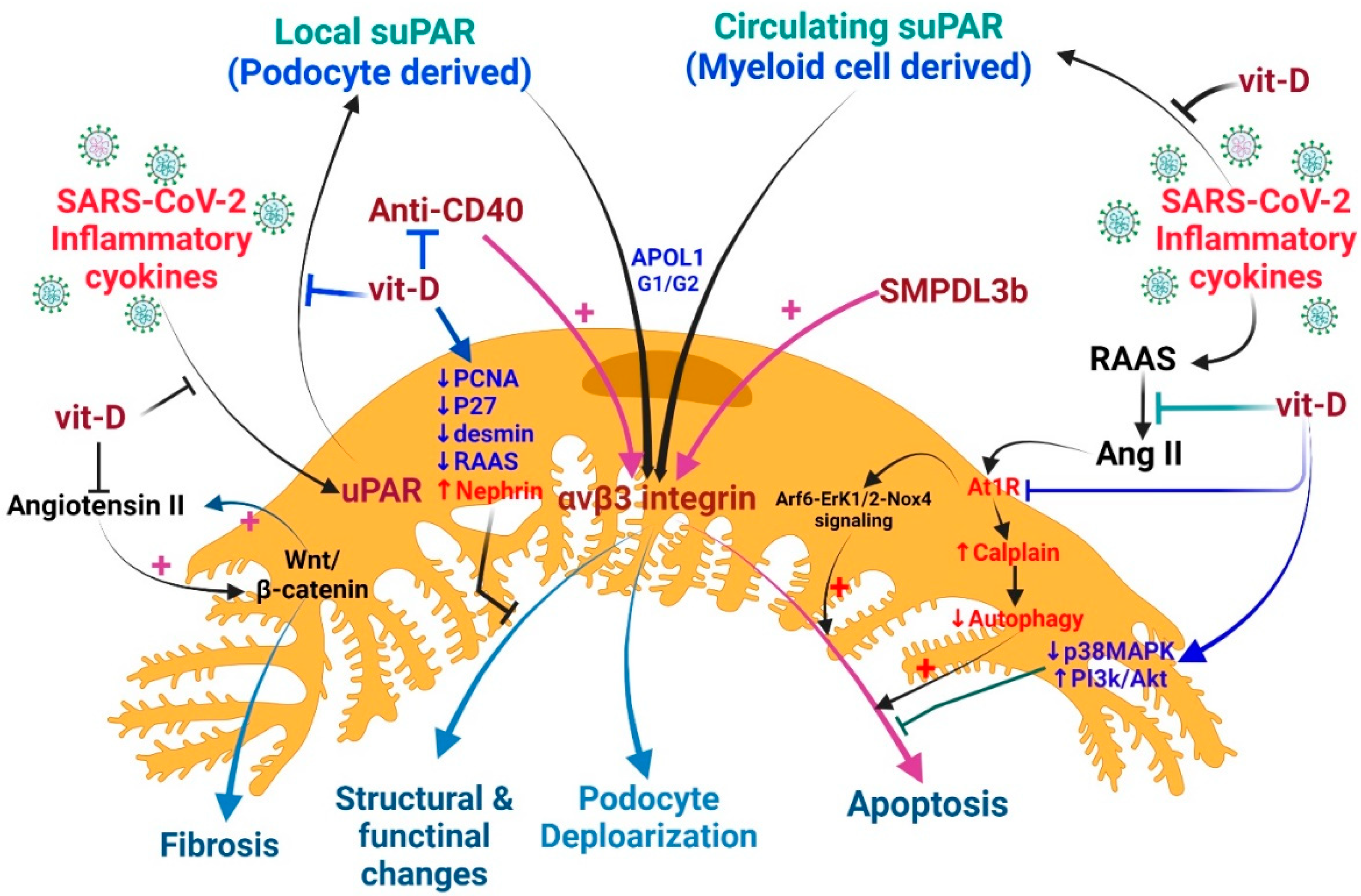
Vitamin D can inhibit the expression of uPAR in podocytes to prevent damage to podocytes and the subsequent development of proteinuria [23]. In mice with CKD and septicemia, vitamin D inhibits the production of podocyte uPAR, resulting in an anti-proteinuria effect [24]. Vitamin D also attenuated podocyte foot process loss in endotoxemic mice and reduced glomerulosclerosis in nephrectomy rats [24,134]. Amiloride, which can inhibit uPAR synthesis, can be used as a drug to reduce uPAR damage to podocytes in patients with COVID-19 [135]. Due to this inhibitory effect, αVβ3 integrin activation is inhibited and podocyte detachment is reduced [136,137]. Concerning the potential risk of hyperkalemia in AKI, it is not appropriate to administer amiloride instead of vitamin D. Amiloride and vitamin D can inhibit uPAR expression by an off-target mechanism. Whether or not vitamin D can drive uPAR suppression through the vitamin D receptor deserves further evaluation.
4.3. Proposed Cross-Talk among Vitamin D, suPAR, and COVID-19-Related AKI
As vitamin D has pleiotropic effects on human pathology [53,138], it can effectively prevent the loss of podocytes, enhance the expression of slit membrane proteins, and maintain the integrity of the renal filtration barrier by attenuating the activity of the RAAS, down-regulating the Wnt/β-catenin signaling, and attenuating the pro-apoptotic pathway [139]. Autophagy is an important protective mechanism of podocytes that is related to angiotensin II (Ang II) levels. Mouse podocytes treated with Ang II exhibited increased calpain activity, inhibited podocyte autophagy, and promoted apoptosis [140]. In cultured human podocytes, Ang II promotes ROS production and podocyte apoptosis by activating ADP-ribosylation factor 6 (Arf6)-Erk1/2-Nox4 signaling [141]. Since COVID-19 patients always have an activation RAAS and a high level of Ag II, this signaling pathway may also occur in COVID-19 patients. Both in vitro and in vivo studies confirmed that Wnt/β-catenin is an important upstream regulator that controls the expression of all RAAS components in the kidneys. The inhibition of Wnt/β-catenin suppresses RAAS activation and ameliorates proteinuria and renal injury [142]. Ang II increasing β-catenin signaling in mouse collection cells leads to increased expression of collagen I, fibronectin, cyclin D1, and c-myc. A reciprocal stimulation relationship between Wnt/β-catenin and Ang II signaling during renal fibrosis seems probable [143] (Figure 4).
In various animal models, vitamin D reduced desmine (a marker of early podocyte lesion), proliferative nuclear cell antigen (PCNA), p27 kinase inhibitor levels, and the local intrarenal activation of the RAAS [139,144,145]. In diabetic nephropathy, vitamin D and its receptor have multiple roles in podocyte, tubulopathy, and interstitial inflammation/fibrosis damage [146]. Though VDR knock-out diabetic mice have developed severe proteinuria and glomerulosclerosis, this study failed to suggest that the action of vitamin D through its receptor and the incidental renoprotective effects may be off-target effects [25]. In doxorubicin-induced podocyte injury, active vitamin D inhibits p38 MAPK signaling and activates the PI3K/AKT survival signaling to prevent podocyte injury [147].
Vitamin D can improve the podocyte damage and proteinuria caused by puromycin aminonucleoside (PAN); this beneficial effect may be due to the direct regulation of TGF-β1/BMP-7 signaling [148]. The PAN-induced podocyte damage can also be ameliorated by treating it with the Smad3 inhibitor SIS3. Since the expression of the actin-binding protein transgelin is induced by the Smad3 signaling pathway during the PAN-induced podocyte lesion, it can be reversed by blocking this pathway [149]. The effects of vitamin D through the nephrin signaling pathway were demonstrated by assessing the expression of podocalyxin and nephrin in isolated rat glomeruli. Previous studies also showed that active vitamin D treatment can enhance the vitamin D receptor (VDR) expression, which will facilitate the co-localization of VDR and RXR (retinoid X receptor) in the nucleus, resulting in the increased expression of nephrin, which promotes glomerular survival [150] (Figure 4).
Vitamin D is an inhibitor of renin biosynthesis and a modulator of RAAS activity; therefore, VDR dysregulation will increase renin and angiotensin II levels, which in turn leads to hypertension and cardiac hypertrophy [151,152,153]. In addition, VDR is involved in controlling the degree of inflammation, epithelial-mesenchymal transition, and maintaining podocyte integrity [154,155]. Vitamin D can effectively suppress kidney inflammation thanks to VDR sequestration of NF-κB signaling [156]. Vitamin D also inhibits NF-κB transaction by modulating advanced glycation end products (AGE) and receptors for AGE (AGE-RAGE system) [157]. In vitamin D deficiency, vitamin D supplementation can lower AGE levels and increase soluble RAGE levels [158].
Vitamin D helped in protecting mitochondrial morphology in kidney tissue by reversing high AT1 receptor expression and low Hsp70 expression in spontaneously hypertensive rats [159]. Mitochondrial preservation underlies the cellular functions of Ca2+ signaling regulation, ROS status regulation, adenosine triphosphate production, and redox reactions [151,160]. VDR activation also promotes mitochondrial integrity through the ligand-independent control of the permeability transition pore [161].
5. AKI Management in COVID-19
5.1. Targeting AKI in COVID-19
Several experimental drugs are currently being developed as anti-viral treatments for COVID-19, but their effects on AKI are unknown [162]. There are currently no specific treatment options for COVID-19-related AKI, so intensive care is largely supportive. At present, approaches to managing AKI and identifying potential indications for RRT are largely based on non-COVID-19 clinical experience. Furthermore, AKI strategies are empirically tailored to patients with COVID-19 [163].
5.2. Adjunctive Therapy with Vitamin D
It is unclear whether recovery from COVID-19-related AKI differs from that of other forms of AKI. Further studies are needed to better understand the direct effects of SARS-CoV-2 on long-term renal injury and recovery [34]. Some patients with COVID-19 develop pulmonary fibrosis after recovery [164]. While we do not know whether progressive renal fibrosis develops in patients recovering from COVID-19-related AKI, the development of fibrosis and progression to CKD in patients recovering from other forms of AKI suggests that this should likely occur. Patients with COVID-19-related AKI must be monitored for a minimum of two to three months or more after discharge to assess renal recovery [165]. Vitamin D attenuates systemic inflammation and thus decreases circulating myeloid-cell-derived suPAR, resulting in the amelioration of kidney tubule, glomerulus, and podocyte injury. Furthermore, vitamin D attenuates the local renal production of uPAR (at least in podocytes) and preserves the glomerular barrier function and structure, responsible for maintaining adequate renal function. Furthermore, vitamin D may exhibit a powerful influence on podocytes by inhibiting apoptosis and structural fibrosis [34].
Recent association studies explore the possible role of vitamin D deficiency in COVID-19. Low levels of 25(OH)D may be a result of COVID-19 [166]. Therapeutic doses of vitamin D supplements may not be dangerous for people with COVID-19, and may even reduce disease progression or severity. Given the urgency of the current situation, sufficient evidence to support this claim remains to be proven. The recommended intake of 10,000 IU/day for 7–14 days, followed by 5000 IU per day for individuals at high risk of COVID-19 to achieve target blood 25(OH)D concentrations of over 40 to 60 ng/mL has been suggested [167].
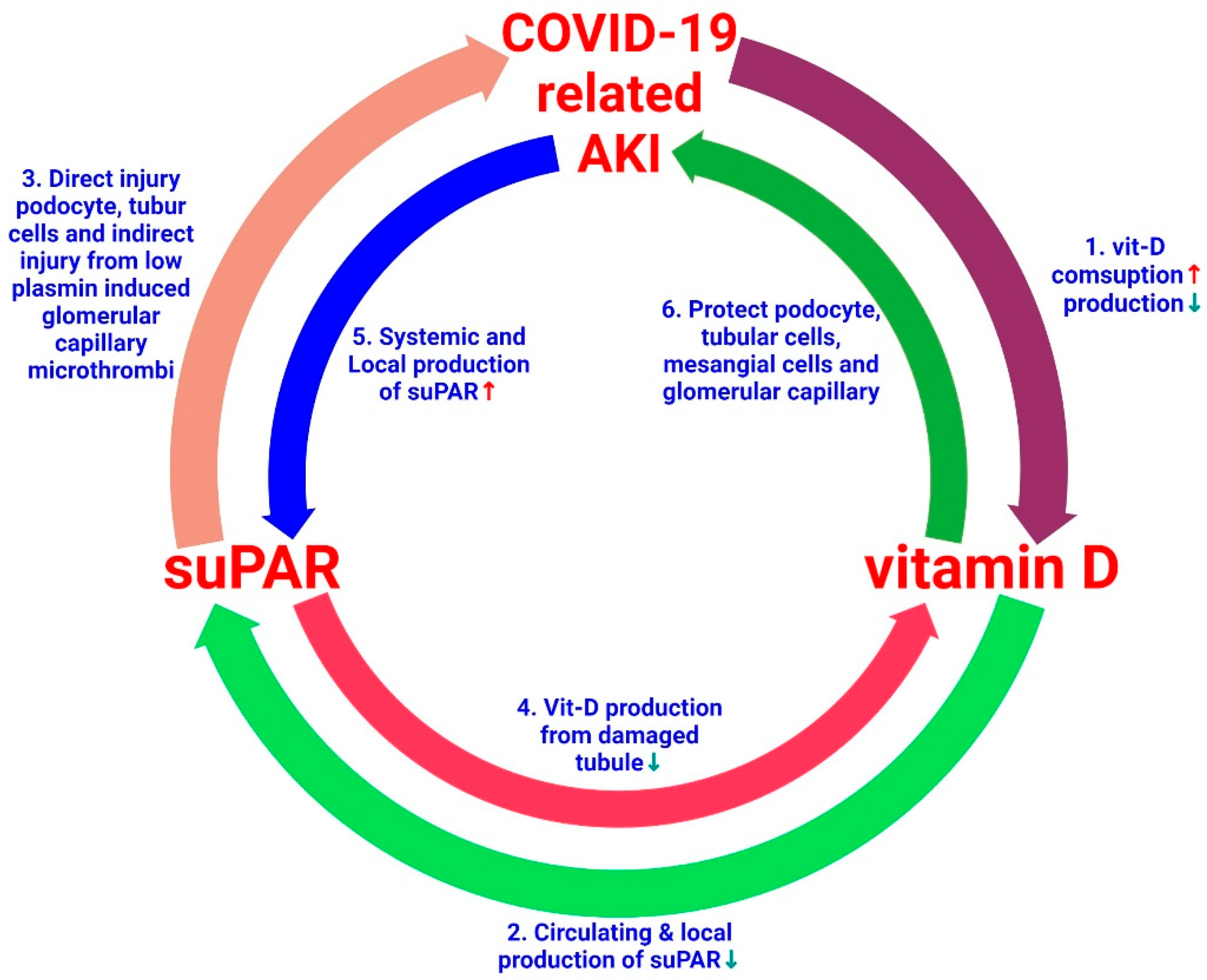
6. Conclusions
Current evidence suggests that high levels of suPAR are probably a risk factor for AKI in patients with COVID-19 [18] and this AKI may be related to vitamin D status [168]. Clinically, higher levels of suPAR are linked to a rapid deterioration of renal function in children with CKD [169]. Measuring suPAR levels in patients with COVID-19 may be a crucial tool for improving disease severity accuracy and predicting complications of SARS-CoV-2 infection, especially in comorbid AKI and micro- and macro-thrombotic events. Increased suPAR concentrations increase oxidative stress from mitochondrial and extra-mitochondrial enzymes in renal cells, [170,171], which synergizes with direct SARS-CoV-2 renal injury and indirect micro-thrombosis in the renal vasculature, therefore increasing the risk and severity of AKI [172]. Elevated circulating suPAR levels may decrease active plasmin production via the competitive inhibition of cell membrane uPA [173]. Ultimately, the development of hypercoagulability in gravely ill COVID-19 patients leads to severe thrombosis [10,174,175].
Vitamin D deficiency occurs frequently in patients with COVID-19 [176]. Patients with COVID-19-related AKI have increased consumption and reduced kidney capacity to produce vitamin D, which may aggravate the vitamin D insufficiency. In these patients, all have increased inflammatory cytokine levels, which can accentuate local and systemic suPAR levels. Vitamin-D-deficiency-induced inflammation further promotes suPAR production from myeloid cells and podocytes. A high level of suPAR causes damage to kidney cells such as tubular cells, podocytes, endothelial cells, and MCs. High suPAR levels damage renal tubular cells, which further decreases vitamin D production. The administration of vitamin D inhibits local and systemic suPAR production by reducing the severity of inflammation [53,176], protects podocytes, tubular cells, and MCs, and prevents microthrombus formation in the glomerular capillaries and other vascular structures [35]. Based on this basic and clinical evidence, here we propose a possible link between vitamin D, suPAR, and COVID-19-related AKI. Indeed, the mechanisms underlying these hypotheses require further study (Figure 5). Given the well-established role of vitamin D in COVID-19, among high-risk SARS-CoV-2-positive patients, individuals and older adults with comorbidities are expected to receive vitamin D supplementation during the COVID-19 pandemic [177]. Moreover, vitamin D is comparatively inexpensive, safe, and widely available. Therefore, supplementation could be considered in these high-risk groups, especially in patients with AKI [53,168]. However, well-designed research is needed to better explore the possible linkage between vitamin D deficiency and COVID-19 morbidity and mortality, and to assess whether the correction of this deficiency can avoid or lessen the severity of COVID-19-related AKI.
Author Contributions
T.-H.L., H.-C.W., M.-T.L., W.-C.H. and K.-C.L. drew up the manuscript and obtained revision documents. K.-W.T., C.-C.L. and K.-C.L. Conduct data analysis. H.-C.W., M.-T.L., W.-C.H. and K.-C.L. carried out the research. H.-C.W., M.-T.L., W.-C.H. and K.-C.L. were responsible to collect examination data. T.-H.L., H.-C.W., M.-T.L., W.-C.H. and K.-C.L. designed this research. All authors have read and agreed to the published version of the manuscript.
Funding
The study was funded by the Taipei Tzu Chi Hospital, Tzu Chi Buddhist Health Foundation (TCRD-TPE-111-01(1/3) & TCRD-TPE-111-RT-4(1/3)) & The Wu Xiulan Education Foundation (WXL-TPE-111-01).
Data Availability Statement
This is a narrative review article. The primary collection of documents for analysis and review comes from PubMed.
Acknowledgments
This research was fully supported administratively by the Department of Medical Research and Department of Chinese Medicine at Taipei Tzu Chi Hospital. They not only provided the necessary assistance in terms of workforce, but also in terms of finances, and provided considerable assistance in the publication of this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pei, G.; Zhang, Z.; Peng, J.; Liu, L.; Zhang, C.; Yu, C.; Ma, Z.; Huang, Y.; Liu, W.; Yao, Y.; et al. Renal Involvement and Early Prognosis in Patients with COVID-19 Pneumonia. J. Am. Soc. Nephrol. 2020, 31, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhu, A.; Li, H.; Zheng, K.; Zhuang, Z.; Chen, Z.; Shi, Y.; Zhang, Z.; Chen, S.-B.; Liu, X.; et al. Isolation of infectious SARS-CoV-2 from urine of a COVID-19 patient. Emerg. Microbes Infect. 2020, 9, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.-L.; Sakr, Y.; Sprung, C.L.; Ranieri, V.M.; Reinhart, K.; Gerlach, H.; Moreno, R.; Carlet, J.; Le Gall, J.-R.; Payen, D.; et al. Sepsis in European intensive care units: Results of the SOAP study. Crit. Care Med. 2006, 34, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Nie, S.; Liu, Z.; Chen, C.; Xu, G.; Zha, Y.; Qian, J.; Liu, B.; Han, S.; Xu, A.; et al. Epidemiology and Clinical Correlates of AKI in Chinese Hospitalized Adults. Clin. J. Am. Soc. Nephrol. 2015, 10, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Chawla, L.S.; Keener, C.; Singbartl, K.; Palevsky, P.M.; Pike, F.L.; Yealy, D.M.; Huang, D.T.; Angus, D.C. The Effects of Alternative Resuscitation Strategies on Acute Kidney Injury in Patients with Septic Shock. Am. J. Respir. Crit. Care Med. 2016, 193, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Murugan, R.; Karajala-Subramanyam, V.; Lee, M.; Yende, S.; Kong, L.; Carter, M.; Angus, D.C.; Kellum, J.A. Acute kidney injury in non-severe pneumonia is associated with an increased immune response and lower survival. Kidney Int. 2010, 77, 527–535. [Google Scholar] [CrossRef]
- Del Rosso, M.; Margheri, F.; Serrati, S.; Chilla, A.; Laurenzana, A.; Fibbi, G. The Urokinase Receptor System, A Key Regulator at the Intersection between Inflammation, Immunity, and Coagulation. Curr. Pharm. Des. 2011, 17, 1924–1943. [Google Scholar] [CrossRef]
- D’Alonzo, D.; De Fenza, M.; Pavone, V. COVID-19 and pneumonia: A role for the uPA/uPAR system. Drug Discov. Today 2020, 25, 1528–1534. [Google Scholar] [CrossRef]
- Pappot, H.; Gårdsvoll, H.; Rømer, J.; Pedersen, A.N.; Grøndahl-Hansen, J.; Pyke, C.; Brünner, N. Plasminogen activator inhibitor type 1 in cancer: Therapeutic and prognostic implications. Biol. Chem. Hoppe-Seyler 1995, 376, 259–267. [Google Scholar] [CrossRef]
- Lippi, G.; Henry, B.M.; Favaloro, E.J. Elevated soluble urokinase plasminogen activator receptor (suPAR) in COVID-19 patients. Clin. Chem. Lab. Med. (CCLM) 2021, 59, e413–e415. [Google Scholar] [CrossRef]
- Schuliga, M.; Grainge, C.; Westall, G.; Knight, D. The fibrogenic actions of the coagulant and plasminogen activation systems in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2018, 97, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Jiang, S.; Wang, R.; Zhang, Y.; Dong, J.; Li, Y. Mechanistic Insight Into the Roles of Integrins in Osteoarthritis. Front. Cell Dev. Biol. 2021, 9, 693484. [Google Scholar] [CrossRef] [PubMed]
- Hamie, L.; Daoud, G.; Nemer, G.; Nammour, T.; El Chediak, A.; Uthman, I.W.; Kibbi, A.G.; Eid, A.; Kurban, M. SuPAR, an emerging biomarker in kidney and inflammatory diseases. Postgrad. Med. J. 2018, 94, 517–524. [Google Scholar] [CrossRef]
- Reisinger, A.C.; Niedrist, T.; Posch, F.; Hatzl, S.; Hackl, G.; Prattes, J.; Schilcher, G.; Meißl, A.-M.; Raggam, R.B.; Herrmann, M.; et al. Soluble urokinase plasminogen activator receptor (suPAR) predicts critical illness and kidney failure in patients admitted to the intensive care unit. Sci. Rep. 2021, 11, 17476. [Google Scholar] [CrossRef]
- Koch, A.; Voigt, S.; Kruschinski, C.; Sanson, E.; Dückers, H.; Horn, A.; Yagmur, E.; Zimmermann, H.; Trautwein, C.; Tacke, F. Circulating soluble urokinase plasminogen activator receptor is stably elevated during the first week of treatment in the intensive care unit and predicts mortality in critically ill patients. Crit. Care 2011, 15, R63. [Google Scholar] [CrossRef]
- Padelli, M.; Gueye, P.; Guilloux, D.; Banydeen, R.; Campana, V.; Cabie, A.; Neviere, R. Soluble urokinase plasminogen activator receptor levels are predictive of COVID-19 severity in Afro-Caribbean patients. Biomarkers Med. 2022, 16, 169–177. [Google Scholar] [CrossRef]
- Azam, T.U.; Shadid, H.R.; Blakely, P.; O’Hayer, P.; Berlin, H.; Pan, M.; Zhao, P.; Zhao, L.; Pennathur, S.; Pop-Busui, R.; et al. Soluble Urokinase Receptor (SuPAR) in COVID-19–Related AKI. J. Am. Soc. Nephrol. 2020, 31, 2725–2735. [Google Scholar] [CrossRef]
- Mercola, J.; Grant, W.B.; Wagner, C.L. Evidence Regarding Vitamin D and Risk of COVID-19 and Its Severity. Nutrients 2020, 12, 3361. [Google Scholar] [CrossRef]
- Rivera, D.T.; Misra, A.; Sanil, Y.; Sabzghabaei, N.; Safa, R.; Garcia, R.U. Vitamin D and morbidity in children with Multisystem inflammatory syndrome related to COVID-19. Prog. Pediatr. Cardiol. 2022, 66, 101507. [Google Scholar] [CrossRef]
- Feketea, G.; Vlacha, V.; Bocsan, I.C.; Vassilopoulou, E.; Stanciu, L.A.; Zdrenghea, M. Vitamin D in Corona Virus Disease 2019 (COVID-19) Related Multisystem Inflammatory Syndrome in Children (MIS-C). Front. Immunol. 2021, 12, 648546. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, E.M.; Nonogaki, S.; Katayama, M.L.H.; Folgueira, M.A.A.K.; Brentani, M.M. Vitamin D3 modulation of plasminogen activator inhibitor type-1 in human breast carcinomas under organ culture. Virchows Arch. 2004, 444, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Möller, C.C.; Altintas, M.; Li, J.; Schwarz, K.; Zacchigna, S.; Xie, L.; Henger, A.; Schmid, H.; Rastaldi, M.P.; et al. Modification of kidney barrier function by the urokinase receptor. Nat. Med. 2008, 14, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, B.; Liu, S.; Xie, S.; Yang, Y.; Ma, J.; Deng, Y.; Wang, W.; Xu, L.; Li, R.; et al. 1,25-Dihydroxyvitamin D(3) Inhibits Podocyte uPAR Expression and Reduces Proteinuria. PLoS ONE 2013, 8, e64912. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, L.; Wang, Y.; Ning, G.; Minto, A.; Kong, J.; Quigg, R.; Li, Y. Renoprotective role of the vitamin D receptor in diabetic nephropathy. Kidney Int. 2008, 73, 163–171. [Google Scholar] [CrossRef]
- Kant, S.; Menez, S.P.; Hanouneh, M.; Fine, D.M.; Crews, D.C.; Brennan, D.C.; Sperati, C.J.; Jaar, B.G. The COVID-19 nephrology compendium: AKI, CKD, ESKD and transplantation. BMC Nephrol. 2020, 21, 449. [Google Scholar] [CrossRef]
- Ertuğlu, L.A.; Kanbay, A.; Afşar, B.; Elsürer Afşar, R.; Kanbay, M. COVID-19 and acute kidney injury. Tuberk. Toraks 2020, 68, 407–418. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef]
- Han, S.S.; Ahn, S.Y.; Ryu, J.; Baek, S.H.; Chin, H.J.; Na, K.Y.; Chae, D.-W.; Kim, S. Proteinuria and hematuria are associated with acute kidney injury and mortality in critically ill patients: A retrospective observational study. BMC Nephrol. 2014, 15, 93. [Google Scholar] [CrossRef]
- Middaugh, R.E.; Middaugh, D.J.; Menk, E.J. Current considerations in respiratory and acid-base management during cardiopulmonary resuscitation. Crit. Care Nurs. Q. 1988, 10, 25–34. [Google Scholar] [CrossRef]
- Chan, L.; Chaudhary, K.; Saha, A.; Chauhan, K.; Vaid, A.; Zhao, S.; Paranjpe, I.; Somani, S.; Richter, F.; Miotto, R.; et al. AKI in Hospitalized Patients with COVID-19. J. Am. Soc. Nephrol. 2021, 32, 151–160. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Qu, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yang, M.; Wan, C.; Yi, L.-X.; Tang, F.; Zhu, H.-Y.; Yi, F.; Yang, H.-C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Nadim, M.K.; Forni, L.G.; Mehta, R.L.; Connor, M.J., Jr.; Liu, K.D.; Ostermann, M.; Rimmelé, T.; Zarbock, A.; Bell, S.; Bihorac, A.; et al. COVID-19-associated acute kidney injury: Consensus report of the 25th Acute Disease Quality Initiative (ADQI) Workgroup. Nat. Rev. Nephrol. 2020, 16, 747–764. [Google Scholar] [CrossRef] [PubMed]
- Chueh, T.-I.; Zheng, C.-M.; Hou, Y.-C.; Lu, K.-C. Novel Evidence of Acute Kidney Injury in COVID-19. J. Clin. Med. 2020, 9, 3547. [Google Scholar] [CrossRef]
- Kissling, S.; Rotman, S.; Gerber, C.; Halfon, M.; Lamoth, F.; Comte, D.; Lhopitallier, L.; Sadallah, S.; Fakhouri, F. Collapsing glomerulopathy in a COVID-19 patient. Kidney Int. 2020, 98, 228–231. [Google Scholar] [CrossRef]
- Deshmukh, S.; Zhou, X.J.; Hiser, W. Collapsing glomerulopathy in a patient of Indian descent in the setting of COVID-19. Ren. Fail. 2020, 42, 877–880. [Google Scholar] [CrossRef]
- Batlle, D.; Soler, M.J.; Sparks, M.A.; Hiremath, S.; South, A.M.; Welling, P.A.; Swaminathan, S.; COVID-19 and ACE2 in Cardiovascular, Lung, and Kidney Working Group. Acute Kidney Injury in COVID-19: Emerging Evidence of a Distinct Pathophysiology. J. Am. Soc. Nephrol. 2020, 31, 1380–1383. [Google Scholar] [CrossRef]
- Ahmadian, E.; Khatibi, S.M.H.; Soofiyani, S.R.; Abediazar, S.; Shoja, M.M.; Ardalan, M.; Vahed, S.Z. COVID-19 and kidney injury: Pathophysiology and molecular mechanisms. Rev. Med Virol. 2020, 31, e2176. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Leaf, R.S.K.; Dzik, W.H.; Carlson, J.C.T.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- Adamsick, M.L.; Gandhi, R.G.; Bidell, M.R.; Elshaboury, R.H.; Bhattacharyya, R.P.; Kim, A.Y.; Nigwekar, S.; Rhee, E.P.; Sise, M.E. Remdesivir in Patients with Acute or Chronic Kidney Disease and COVID-19. J. Am. Soc. Nephrol. 2020, 31, 1384–1386. [Google Scholar] [CrossRef] [PubMed]
- Major, R.; Selvaskandan, H.; Makkeyah, Y.M.; Hull, K.; Kuverji, A.; Graham-Brown, M. The Exclusion of Patients with CKD in Prospectively Registered Interventional Trials for COVID-19—a Rapid Review of International Registry Data. J. Am. Soc. Nephrol. 2020, 31, 2250–2252. [Google Scholar] [CrossRef] [PubMed]
- Puelles, V.G.; Lütgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964. [Google Scholar] [CrossRef]
- Rabb, H.; Griffin, M.D.; McKay, D.B.; Swaminathan, S.; Pickkers, P.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J. Am. Soc. Nephrol. 2016, 27, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S. Coronavirus (COVID-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 69, 1077–1085. [Google Scholar] [CrossRef]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26, and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy 2020, 75, 2829–2845. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Wilson, P.C.; Wu, H.; Kirita, Y.; Uchimura, K.; Ledru, N.; Rennke, H.G.; Welling, P.A.; Waikar, S.S.; Humphreys, B.D. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2019, 116, 19619–19625. [Google Scholar] [CrossRef]
- Sehirli, A.O.; Sayiner, S.; Serakinci, N. Role of melatonin in the treatment of COVID-19; as an adjuvant through cluster differentiation 147 (CD147). Mol. Biol. Rep. 2020, 47, 8229–8233. [Google Scholar] [CrossRef]
- Shi, W.; Lv, J.; Lin, L. Coagulopathy in COVID-19: Focus on vascular thrombotic events. J. Mol. Cell. Cardiol. 2020, 146, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Banu, N.; Panikar, S.S.; Leal, L.R.; Leal, A.R. Protective role of ACE2 and its downregulation in SARS-CoV-2 infection leading to Macrophage Activation Syndrome: Therapeutic implications. Life Sci. 2020, 256, 117905. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.-Y.; Liu, W.-C.; Zheng, J.-Q.; Lu, C.-L.; Hou, Y.-C.; Zheng, C.-M.; Song, J.-Y.; Lu, K.-C.; Chao, Y.-C. Immunological Aspects of SARS-CoV-2 Infection and the Putative Beneficial Role of Vitamin-D. Int. J. Mol. Sci. 2021, 22, 5251. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Larsen, C.P.; Hernandez-Arroyo, C.F.; Mohamed, M.M.; Caza, T.; Sharshir, M.; Chughtai, A.; Xie, L.; Gimenez, J.M.; Sandow, T.A.; et al. AKI and Collapsing Glomerulopathy Associated with COVID-19 and APOL1 High-Risk Genotype. J. Am. Soc. Nephrol. 2020, 31, 1688–1695. [Google Scholar] [CrossRef]
- Shetty, A.A.; Tawhari, I.; Safar-Boueri, L.; Seif, N.; Alahmadi, A.; Gargiulo, R.; Aggarwal, V.; Usman, I.; Kisselev, S.; Gharavi, A.G.; et al. COVID-19–Associated Glomerular Disease. J. Am. Soc. Nephrol. 2021, 32, 33–40. [Google Scholar] [CrossRef]
- Gomez, H.; Ince, C.; De Backer, D.; Pickkers, P.; Payen, D.; Hotchkiss, J.; Kellum, J.A. A Unified Theory of Sepsis-Induced Acute Kidney Injury: Inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 2014, 41, 3–11. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Puri, V.; Singh, V.P.; Naura, A.S. COVID-19 Severity: Lung-Heart Interplay. Curr. Cardiol. Rev. 2021, 17, e230421189016. [Google Scholar] [CrossRef]
- Pecly, I.M.D.; Azevedo, R.B.; Muxfeldt, E.S.; Botelho, B.G.; Albuquerque, G.G.; Diniz, P.H.P.; Silva, R.; Rodrigues, C.I.S. A review of COVID-19 and acute kidney injury: From pathophysiology to clinical results. J. Bras. Nefrol. 2021, 43, 551–571. [Google Scholar] [CrossRef]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef]
- Zhang, S.; Amahong, K.; Sun, X.; Lian, X.; Liu, J.; Sun, H.; Lou, Y.; Zhu, F.; Qiu, Y. The miRNA: A small but powerful RNA for COVID-19. Briefings Bioinform. 2021, 22, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Galliera, E.; Drago, L.; Marazzi, M.G.; Romanò, C.; Vassena, C.; Romanelli, M.M.C. Soluble urokinase-type plasminogen activator receptor (suPAR) as new biomarker of the prosthetic joint infection: Correlation with inflammatory cytokines. Clin. Chim. Acta 2015, 441, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Han, Y.; Zhao, J.; Cui, J.; Wang, K.; Wang, R.; Liu, Y. Serum soluble urokinase-type plasminogen activator receptor as a biological marker of bacterial infection in adults: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 39481. [Google Scholar] [CrossRef]
- Hall, A.; Crichton, S.; Varrier, M.; Bear, D.E.; Ostermann, M. suPAR as a marker of infection in acute kidney injury—A prospective observational study. BMC Nephrol. 2018, 19, 191. [Google Scholar] [CrossRef] [PubMed]
- Huai, Q.; Mazar, A.P.; Kuo, A.; Parry, G.C.; Shaw, D.E.; Callahan, J.; Li, Y.; Yuan, C.; Bian, C.; Chen, L.; et al. Structure of Human Urokinase Plasminogen Activator in Complex with Its Receptor. Science 2006, 311, 656–659. [Google Scholar] [CrossRef]
- Wei, C.; Li, J.; Adair, B.D.; Zhu, K.; Cai, J.; Merchant, M.; Samelko, B.; Liao, Z.; Koh, K.H.; Tardi, N.J.; et al. uPAR isoform 2 forms a dimer and induces severe kidney disease in mice. J. Clin. Investig. 2019, 129, 1946–1959. [Google Scholar] [CrossRef]
- Alfano, D.; Franco, P.; Stoppelli, M.P. Modulation of Cellular Function by the Urokinase Receptor Signalling: A Mechanistic View. Front. Cell Dev. Biol. 2022, 10, 818616. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Pozzi, A.; Zent, R. Integrins in Kidney Disease. J. Am. Soc. Nephrol. 2013, 24, 1034–1039. [Google Scholar] [CrossRef]
- Kaneko, Y.; Otsuka, T.; Tsuchida, Y.; Gejyo, F.; Narita, I. Integrin α1/β1 and α2/β1 as a receptor for IgA1 in human glomerular mesangial cells in IgA nephropathy. Int. Immunol. 2012, 24, 219–232. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Ueno, T.; Ohashi, K.; Yamashita, H.; Takahashi, Y.; Sakamoto, K.; Manabe, S.; Hara, S.; Takashima, Y.; Dan, T.; et al. Podocyte injury-driven intracapillary plasminogen activator inhibitor type 1 accelerates podocyte loss via uPAR-mediated β1-integrin endocytosis. Am. J. Physiol. Renal. Physiol. 2015, 308, F614–F626. [Google Scholar] [CrossRef] [PubMed]
- Whyte, C.S.; Mutch, N.J. uPA-mediated plasminogen activation is enhanced by polyphosphate. Haematologica 2021, 106, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Plesner, T.; Behrendt, N.; Ploug, M. Structure, Function and Expression on Blood and Bone Marrow Cells of the Urokinase-Type Plasminogen Activator Receptor, uPAR. Stem Cells 1997, 15, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Schultz, M.; van der Poll, T. Disseminated Intravascular Coagulation in Infectious Disease. Semin. Thromb. Hemost. 2010, 36, 367–377. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Y.; Wu, J.; Li, Y.; Zhou, X.; Li, X.; Chen, H.; Guo, M.; Chen, S.; Sun, F.; et al. Risks and features of secondary infections in severe and critical ill COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 1958–1964. [Google Scholar] [CrossRef]
- Blasi, F. Urokinase and urokinase receptor: A paracrine/autocrine system regulating cell migration and invasiveness. BioEssays 1993, 15, 105–111. [Google Scholar] [CrossRef]
- Vaday, G.G.; Lider, O. Extracellular matrix moieties, cytokines, and enzymes: Dynamic effects on immune cell behavior and inflammation. J. Leukoc. Biol. 2000, 67, 149–159. [Google Scholar] [CrossRef]
- Isselbacher, E.M. Thoracic and Abdominal Aortic Aneurysms. Circulation 2005, 111, 816–828. [Google Scholar] [CrossRef]
- Solberg, H.; Ploug, M.; Høyer–Hansen, G.; Nielsen, B.S.; Lund, L.R. The Murine Receptor for Urokinase-Type Plasminogen Activator Is Primarily Expressed in Tissues Actively Undergoing Remodeling. J. Histochem. Cytochem. 2001, 49, 237–246. [Google Scholar] [CrossRef]
- Nykjaer, A.; Moller, B.; Todd, R.F., III; Christensen, T.; Andreasen, P.A.; Gliemann, J.; Petersen, C.M. Urokinase receptor. An activation antigen in human T lymphocytes. J. Immunol. 1994, 152, 505–516. [Google Scholar]
- Hahm, E.; Wei, C.; Fernandez, I.; Li, J.; Tardi, N.J.; Tracy, M.; Wadhwani, S.; Cao, Y.; Peev, V.; Zloza, A.; et al. Bone marrow-derived immature myeloid cells are a main source of circulating suPAR contributing to proteinuric kidney disease. Nat. Med. 2017, 23, 100–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankland, S.J.; Jefferson, J.A. A bone marrow factor contributes to kidney disease. Nat. Med. 2017, 23, 13–14. [Google Scholar] [CrossRef]
- Reiser, J.; Wei, C.; Tumlin, J. Soluble urokinase receptor and focal segmental glomerulosclerosis. Curr. Opin. Nephrol. Hypertens. 2012, 21, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.H.D.M.; Da Silva, C.A.; Monteiro, M.L.G.D.R.; Araújo, L.S.; Rocha, L.P.; Reis, M.B.D.R.; Ramalho, F.S.; Corrêa, R.R.M.; Silva, M.V.; Reis, M.A.; et al. Podocin and uPAR are good biomarkers in cases of Focal and segmental glomerulosclerosis in pediatric renal biopsies. PLoS ONE 2019, 14, e0217569. [Google Scholar] [CrossRef] [PubMed]
- Hayek, S.S.; Leaf, D.E.; Tahhan, A.S.; Raad, M.; Sharma, S.; Waikar, S.S.; Sever, S.; Camacho, A.; Wang, X.; Dande, R.R.; et al. Soluble Urokinase Receptor and Acute Kidney Injury. N. Engl. J. Med. 2020, 382, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Mossanen, J.C.; Pracht, J.; Jansen, T.U.; Buendgens, L.; Stoppe, C.; Goetzenich, A.; Struck, J.; Autschbach, R.; Marx, G.; Tacke, F. Elevated Soluble Urokinase Plasminogen Activator Receptor and Proenkephalin Serum Levels Predict the Development of Acute Kidney Injury after Cardiac Surgery. Int. J. Mol. Sci. 2017, 18, 1662. [Google Scholar] [CrossRef] [PubMed]
- Hayek, S.S.; Sever, S.; Ko, Y.-A.; Trachtman, H.; Awad, M.; Wadhwani, S.; Altintas, M.M.; Wei, C.; Hotton, A.L.; French, A.L.; et al. Soluble Urokinase Receptor and Chronic Kidney Disease. N. Engl. J. Med. 2015, 373, 1916–1925. [Google Scholar] [CrossRef]
- Wei, C.; El Hindi, S.; Li, J.; Fornoni, A.; Goes, N.; Sageshima, J.; Maiguel, D.; Karumanchi, S.A.; Yap, H.-K.; Saleem, M.; et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nat. Med. 2011, 17, 952–960. [Google Scholar] [CrossRef]
- Gussen, H.; Hohlstein, P.; Bartneck, M.; Warzecha, K.T.; Buendgens, L.; Luedde, T.; Trautwein, C.; Koch, A.; Tacke, F. Neutrophils are a main source of circulating suPAR predicting outcome in critical illness. J. Intensiv. Care 2019, 7, 26. [Google Scholar] [CrossRef]
- Diederichsen, M.Z.; Diederichsen, S.Z.; Mickley, H.; Steffensen, F.H.; Lambrechtsen, J.; Sand, N.P.R.; Christensen, K.L.; Olsen, M.H.; Diederichsen, A.; Grønhøj, M.H. Prognostic value of suPAR and hs-CRP on cardiovascular disease. Atherosclerosis 2018, 271, 245–251. [Google Scholar] [CrossRef]
- Andersen, O.; Eugen-Olsen, J.; Kofoed, K.; Iversen, J.; Haugaard, S.B. Soluble urokinase plasminogen activator receptor is a marker of dysmetabolism in HIV-infected patients receiving highly active antiretroviral therapy. J. Med. Virol. 2008, 80, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Langkilde, A.; Petersen, J.; Klausen, H.H.; Henriksen, J.H.; Eugen-Olsen, J.; Andersen, O. Inflammation in HIV-Infected Patients: Impact of HIV, Lifestyle, Body Composition, and Demography—A Cross Sectional Cohort Study. PLoS ONE 2012, 7, e51698. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Trachtman, H.; Li, J.; Dong, C.; Friedman, A.L.; Gassman, J.J.; McMahan, J.L.; Radeva, M.; Heil, K.M.; Trautmann, A.; et al. Circulating suPAR in Two Cohorts of Primary FSGS. J. Am. Soc. Nephrol. 2012, 23, 2051–2059. [Google Scholar] [CrossRef]
- Mallela, S.K.; Merscher, S.; Fornoni, A. Implications of Sphingolipid Metabolites in Kidney Diseases. Int. J. Mol. Sci. 2022, 23, 4244. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.-H.; Pedigo, C.E.; Guzman, J.; Correa-Medina, M.; Wei, C.; Villarreal, R.; Mitrofanova, A.; Leclercq, F.; Faul, C.; Li, J.; et al. Sphingomyelinase-Like Phosphodiesterase 3b Expression Levels Determine Podocyte Injury Phenotypes in Glomerular Disease. J. Am. Soc. Nephrol. 2015, 26, 133–147. [Google Scholar] [CrossRef]
- Ahmad, A.; Mitrofanova, A.; Bielawski, J.; Yang, Y.; Marples, B.; Fornoni, A.; Zeidan, Y.H. Sphingomyelinase-like phosphodiesterase 3b mediates radiation-induced damage of renal podocytes. FASEB J. 2017, 31, 771–780. [Google Scholar] [CrossRef]
- Hayek, S.S.; Koh, K.H.; E Grams, M.; Wei, C.; Ko, Y.-A.; Li, J.; Samelko, B.; Lee, H.; Dande, R.R.; Lee, H.W.; et al. A tripartite complex of suPAR, APOL1 risk variants and αvβ3 integrin on podocytes mediates chronic kidney disease. Nat. Med. 2017, 23, 945–953. [Google Scholar] [CrossRef]
- Rienstra, H.; Katta, K.; Celie, J.W.A.M.; Van Goor, H.; Navis, G.; Born, J.V.D.; Hillebrands, J.-L. Differential Expression of Proteoglycans in Tissue Remodeling and Lymphangiogenesis after Experimental Renal Transplantation in Rats. PLoS ONE 2010, 5, e9095. [Google Scholar] [CrossRef]
- Han, R.; Hu, S.; Qin, W.; Shi, J.; Hou, Q.; Wang, X.; Xu, X.; Zhang, M.; Zeng, C.; Liu, Z.; et al. C3a and suPAR drive versican V1 expression in tubular cells of focal segmental glomerulosclerosis. JCI Insight 2019, 4, e130986. [Google Scholar] [CrossRef]
- Avraham, S.; Korin, B.; Chung, J.-J.; Oxburgh, L.; Shaw, A.S. The Mesangial cell—The glomerular stromal cell. Nat. Rev. Nephrol. 2021, 17, 855–864. [Google Scholar] [CrossRef]
- Fischer, E.G. Glomerular mesangial cell adhesion to fibrinogen is mediated by αvβ3 integrin. Biochem. Cell Biol. 2004, 82, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Serafinelli, J.; Mastrangelo, A.; Morello, W.; Cerioni, V.F.; Salim, A.; Nebuloni, M.; Montini, G. Kidney involvement and histological findings in two pediatric COVID-19 patients. Pediatr. Nephrol. 2021, 36, 3789–3793. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Wu, M.-H.; Pierce, A.; Poncelet, A.-C.; Varga, J.; Schnaper, H.W. MAP-kinase activity necessary for TGFβ1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J. Cell Sci. 2007, 120, 4230–4240. [Google Scholar] [CrossRef]
- Nicholas, S.B.; Aguiniga, E.; Ren, Y.; Kim, J.; Wong, J.; Govindarajan, N.; Noda, M.; Wang, W.; Kawano, Y.; Collins, A.; et al. Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int. 2005, 67, 1297–1307. [Google Scholar] [CrossRef]
- Cheng, H.; Chen, C.; Wang, S. Effects of uPA on mesangial matrix changes in the kidney of diabetic rats. Ren. Fail. 2014, 36, 1322–1327. [Google Scholar] [CrossRef]
- Klos, A.; Wende, E.; Wareham, K.J.; Monk, P. International Union of Basic and Clinical Pharmacology. LXXXVII. Complement Peptide C5a, C4a, and C3a Receptors. Pharmacol. Rev. 2013, 65, 500–543. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.C.; Reins, R.Y.; Li, T.-B.; Hollmann, T.J.; Dutta, R.; Rick, W.A.; Teng, B.-B.; Ke, B. Renal Expression of the C3a Receptor and Functional Responses of Primary Human Proximal Tubular Epithelial Cells. J. Immunol. 2004, 173, 4190–4196. [Google Scholar] [CrossRef]
- Gao, S.; Cui, Z.; Zhao, M.-H. The Complement C3a and C3a Receptor Pathway in Kidney Diseases. Front. Immunol. 2020, 11, 1875. [Google Scholar] [CrossRef]
- Sinkovits, G.; Mező, B.; Réti, M.; Müller, V.; Iványi, Z.; Gál, J.; Gopcsa, L.; Reményi, P.; Szathmáry, B.; Lakatos, B.; et al. Complement Overactivation and Consumption Predicts In-Hospital Mortality in SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 663187. [Google Scholar] [CrossRef]
- De Nooijer, A.H.; Grondman, I.; Janssen, N.A.F.; Netea, M.G.; Willems, L.; van de Veerdonk, F.L.; Giamarellos-Bourboulis, E.J.; Toonen, E.J.M.; Joosten, L.A.B.; Jaeger, M.; et al. Complement Activation in the Disease Course of Coronavirus Disease 2019 and Its Effects on Clinical Outcomes. J. Infect. Dis. 2021, 223, 214–224. [Google Scholar] [CrossRef]
- Georg, P.; Astaburuaga-García, R.; Bonaguro, L.; Brumhard, S.; Michalick, L.; Lippert, L.J.; Kostevc, T.; Gäbel, C.; Schneider, M.; Streitz, M.; et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell 2022, 185, 493–512. [Google Scholar] [CrossRef]
- Meijers, B.; Poesen, R.; Claes, K.; Dietrich, R.; Bammens, B.; Sprangers, B.; Naesens, M.; Storr, M.; Kuypers, D.; Evenepoel, P. Soluble urokinase receptor is a biomarker of cardiovascular disease in chronic kidney disease. Kidney Int. 2015, 87, 210–216. [Google Scholar] [CrossRef]
- Lv, L.; Wang, F.; Wu, L.; Wang, J.-W.; Cui, Z.; Hayek, S.S.; Wei, C.; Reiser, J.; He, K.; Zhang, L.; et al. Soluble urokinase-type plasminogen activator receptor and incident end-stage renal disease in Chinese patients with chronic kidney disease. Nephrol. Dial. Transplant. 2020, 35, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Zeier, M.; Reiser, J. suPAR and chronic kidney disease—A podocyte story. Pflugers Arch. 2017, 469, 1017–1020. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, L.; Huang, J.; Shi, S.; Lv, J.; Liu, G.; Zhao, M.; Zhang, H. Plasma Soluble Urokinase Receptor Level Is Correlated with Podocytes Damage in Patients with IgA Nephropathy. PLoS ONE 2015, 10, e0132869. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-M.; Han, M.; Chen, M.-X.; Ning, Y.; Pei, G.-C.; Li, Y.-Q.; Dai, W.; Ge, S.-W.; Deng, Y.-J.; Guo, Y.-Y.; et al. Soluble Urokinase Receptor Levels Are Correlated with Focal Segmental Glomerulosclerosis Lesions in IgA Nephropathy: A Cohort Study from China. PLoS ONE 2015, 10, e0138718. [Google Scholar] [CrossRef]
- Qin, D.D.; Song, D.; Huang, J.; Yu, F.; Zhao, M.H. Plasma-soluble urokinase-type plasminogen activator receptor levels are associated with clinical and pathological activities in lupus nephritis: A large cohort study from China. Lupus 2015, 24, 546–557. [Google Scholar] [CrossRef]
- Wu, C.-Z.; Chang, L.-C.; Lin, Y.-F.; Hung, Y.-J.; Pei, D.; Chu, N.-F.; Chen, J.-S. Urokinase plasminogen activator receptor and its soluble form in common biopsy-proven kidney diseases and in staging of diabetic nephropathy. Clin. Biochem. 2015, 48, 1324–1329. [Google Scholar] [CrossRef]
- Shuai, T.; Yan, P.; Xiong, H.; Huang, Q.; Zhu, L.; Yang, K.; Liu, J. Association between Soluble Urokinase-Type Plasminogen Activator Receptor Levels and Chronic Kidney Disease: A Systematic Review and Meta-Analysis. BioMed Res. Int. 2019, 2019, 6927456. [Google Scholar] [CrossRef]
- Bowe, B.; Xie, Y.; Xu, E.; Al-Aly, Z. Kidney Outcomes in Long COVID. J. Am. Soc. Nephrol. 2021, 32, 2851–2862. [Google Scholar] [CrossRef] [PubMed]
- Nugent, J.; Aklilu, A.; Yamamoto, Y.; Simonov, M.; Li, F.; Biswas, A.; Ghazi, L.; Greenberg, J.H.; Mansour, S.G.; Moledina, D.G.; et al. Assessment of Acute Kidney Injury and Longitudinal Kidney Function After Hospital Discharge Among Patients With and Without COVID-19. JAMA Netw. Open 2021, 4, e211095. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Del-Barco, D.; Risco-Acevedo, D.; Berlanga-Acosta, J.; Martos-Benítez, F.D.; Guillén-Nieto, G. Revisiting Pleiotropic Effects of Type I Interferons: Rationale for Its Prophylactic and Therapeutic Use Against SARS-CoV-2. Front. Immunol. 2021, 12, 655528. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Kong, J.; Wei, M.; Chen, Z.-F.; Liu, S.Q.; Cao, L.-P. 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin-angiotensin system. J. Clin. Investig. 2002, 110, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Walton, J. Chronic Aluminum Intake Causes Alzheimer’s Disease: Applying Sir Austin Bradford Hill’s Causality Criteria. J. Alzheimer’s Dis. 2014, 40, 765–838. [Google Scholar] [CrossRef]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; the Northwell COVID-19 Research Consortium. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized with COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Bilezikian, J.P.; Bikle, D.; Hewison, M.; Lazaretti-Castro, M.; Formenti, A.M.; Gupta, A.; Madhavan, M.V.; Nair, N.; Babalyan, V.; Hutchings, N.; et al. Mechanisms in Endocrinology: Vitamin D and COVID-19. Eur. J. Endocrinol. 2020, 183, R133–R147. [Google Scholar] [CrossRef] [PubMed]
- Science, M.; Maguire, J.L.; Russell, M.L.; Smieja, M.; Walter, S.D.; Loeb, M. Low Serum 25-Hydroxyvitamin D Level and Risk of Upper Respiratory Tract Infection in Children and Adolescents. Clin. Infect. Dis. 2013, 57, 392–397. [Google Scholar] [CrossRef]
- Dancer, R.C.A.; Parekh, D.; Lax, S.; D’Souza, V.; Zheng, S.; Bassford, C.R.; Park, D.; Bartis, D.G.; Mahida, R.; Turner, A.M.; et al. Vitamin D deficiency contributes directly to the acute respiratory distress syndrome (ARDS). Thorax 2015, 70, 617–624. [Google Scholar] [CrossRef]
- Martineau, A.R.; Jolliffe, D.A.; Hooper, R.L.; Greenberg, L.; Aloia, J.F.; Bergman, P.; Dubnov-Raz, G.; Esposito, S.; Ganmaa, D.; Ginde, A.A.; et al. Vitamin D supplementation to prevent acute respiratory tract infections: Systematic review and meta-analysis of individual participant data. BMJ 2017, 356, i6583. [Google Scholar] [CrossRef]
- Ilie, P.C.; Stefanescu, S.; Smith, L. The role of vitamin D in the prevention of coronavirus disease 2019 infection and mortality. Aging Clin. Exp. Res. 2020, 32, 1195–1198. [Google Scholar] [CrossRef]
- Hutchings, N.; Babalyan, V.; Baghdasaryan, S.; Qefoyan, M.; Sargsyants, N.; Aghajanova, E.; Martirosyan, A.; Harutyunyan, R.; Lesnyak, O.; Formenti, A.M.; et al. Patients hospitalized with COVID-19 have low levels of 25-hydroxyvitamin D. Endocrine 2021, 71, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.; Dantas Damascena, A.D.; Galvão Azevedo, L.M.G.; de Almeida Oliveira, T.D.A.; da Mota Santana, J.D.M. Vitamin D deficiency aggravates COVID-19: Systematic review and meta-analysis. Crit. Rev. Food Sci. Nutr. 2022, 62, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Entrenas Castillo, M.E.; Entrenas Costa, L.M.E.; Vaquero Barrios, J.M.V.; Alcalá Díaz, J.F.A.; López Miranda, J.L.; Bouillon, R.; Quesada Gomez, J.M.Q. Effect of calcifediol treatment and best available therapy versus best available therapy on intensive care unit admission and mortality among patients hospitalized for COVID-19: A pilot randomized clinical study. J. Steroid Biochem. Mol. Biol. 2020, 203, 105751. [Google Scholar] [CrossRef]
- Sonneveld, R.; Ferrè, S.; Hoenderop, J.G.; Dijkman, H.B.; Berden, J.H.; Bindels, R.J.; Wetzels, J.F.; van der Vlag, J.; Nijenhuis, T. Vitamin D Down-Regulates TRPC6 Expression in Podocyte Injury and Proteinuric Glomerular Disease. Am. J. Pathol. 2013, 182, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Xie, S.; Shi, W.; Yang, Y. Amiloride off-target effect inhibits podocyte urokinase receptor expression and reduces proteinuria. Nephrol. Dial. Transplant. 2011, 27, 1746–1755. [Google Scholar] [CrossRef]
- Trimarchi, H.; Forrester, M.; Lombi, F.; Pomeranz, V.; Rana, M.S.; Karl, A.; Andrews, J. Amiloride as an Alternate Adjuvant Antiproteinuric Agent in Fabry Disease: The Potential Roles of Plasmin and uPAR. Case Rep. Nephrol. 2014, 2014, 854521. [Google Scholar] [CrossRef]
- Trimarchi, H.; Paulero, M.; Canzonieri, R.; Schiel, A.; Iotti, A.; Costales-Collaguazo, C.; Stern, A.; Forrester, M.; Lombi, F.; Pomeranz, V.; et al. In Acute IgA Nephropathy, Proteinuria and Creatinine Are in the Spot, but Podocyturia Operates in Silence: Any Place for Amiloride? Case Rep. Nephrol. 2017, 2017, 1292531. [Google Scholar] [CrossRef]
- Liu, W.-C.; Wu, C.-C.; Hung, Y.-M.; Liao, M.-T.; Shyu, J.-F.; Lin, Y.-F.; Lu, K.-C.; Yeh, K.-C. Pleiotropic effects of vitamin D in chronic kidney disease. Clin. Chim. Acta 2016, 453, 1–12. [Google Scholar] [CrossRef]
- Li, Y.C. Podocytes as target of vitamin D. Curr. Diabetes Rev. 2011, 7, 35–40. [Google Scholar] [CrossRef]
- Bensaada, I.; Robin, B.; Perez, J.; Salemkour, Y.; Chipont, A.; Camus, M.; Lemoine, M.; Guyonnet, L.; Lazareth, H.; Letavernier, E.; et al. Calpastatin prevents Angiotensin II–mediated podocyte injury through maintenance of autophagy. Kidney Int. 2021, 100, 90–106. [Google Scholar] [CrossRef]
- Che, G.; Gao, H.; Hu, Q.; Xie, H.; Zhang, Y. Angiotensin II promotes podocyte injury by activating Arf6-Erk1/2-Nox4 signaling pathway. PLoS ONE 2020, 15, e0229747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Liu, Y. Wnt/β-catenin signaling and renin–angiotensin system in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.A.; Gonzalez, A.A.; Inestrosa, N.C.; Vio, C.P.; Prieto, M.C. Angiotensin II increases fibronectin and collagen I through the β-catenin-dependent signaling in mouse collecting duct cells. Am. J. Physiol. Renal. Physiol. 2015, 308, F358–F365. [Google Scholar] [CrossRef]
- Kuhlmann, A.; Haas, C.S.; Gross, M.-L.; Reulbach, U.; Holzinger, M.; Schwarz, U.; Ritz, E.; Amann, K. 1,25-Dihydroxyvitamin D3decreases podocyte loss and podocyte hypertrophy in the subtotally nephrectomized rat. Am. J. Physiol. Renal. Physiol. 2004, 286, F526–F533. [Google Scholar] [CrossRef] [PubMed]
- Deb, D.K.; Sun, T.; Wong, K.E.; Zhang, Z.; Ning, G.; Zhang, Y.; Kong, J.; Shi, H.; Chang, A.; Li, Y.C. Combined vitamin D analog and AT1 receptor antagonist synergistically block the development of kidney disease in a model of type 2 diabetes. Kidney Int. 2010, 77, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Liu, Z.; Guo, J. The Emerging Role of Vitamin D and Vitamin D Receptor in Diabetic Nephropathy. BioMed Res. Int. 2020, 2020, 4137268. [Google Scholar] [CrossRef]
- Zou, M.-S.; Yu, J.; Nie, G.-M.; He, W.-S.; Luo, L.-M.; Xu, H.-T. 1, 25-dihydroxyvitamin D3 decreases adriamycin-induced podocyte apoptosis and loss. Int. J. Med Sci. 2010, 7, 290–299. [Google Scholar] [CrossRef]
- Xiao, H.-Q.; Shi, W.; Liu, S.-X.; Zhang, B.; Xu, L.-X.; Liang, X.-L.; Liang, Y.-Z. Podocyte injury is suppressed by 1,25-dihydroxyvitamin d3via modulation of transforming growth factor-β1/bone morphogenetic protein-7 signalling in puromycin aminonucleoside nephropathy rats. Clin. Exp. Pharmacol. Physiol. 2009, 36, 682–689. [Google Scholar] [CrossRef]
- Jiang, L.; Cui, H.; Ding, J.; Yang, A.; Zhang, Y. Puromycin aminonucleoside-induced podocyte injury is ameliorated by the Smad3 inhibitor SIS3. FEBS Open Bio 2020, 10, 1601–1611. [Google Scholar] [CrossRef]
- Trohatou, O.; Tsilibary, E.-F.; Charonis, A.; Iatrou, C.; Drossopoulou, G. Vitamin D3 ameliorates podocyte injury through the nephrin signalling pathway. J. Cell. Mol. Med. 2017, 21, 2599–2609. [Google Scholar] [CrossRef]
- Gembillo, G.; Siligato, R.; Amatruda, M.; Conti, G.; Santoro, D. Vitamin D and Glomerulonephritis. Medicina 2021, 57, 186. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Qiao, G.; Uskokovic, M.; Xiang, W.; Zheng, W.; Kong, J. Vitamin D: A negative endocrine regulator of the renin–angiotensin system and blood pressure. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 387–392. [Google Scholar] [CrossRef] [PubMed]
- McMullan, C.J.; Borgi, L.; Curhan, G.C.; Fisher, N.; Forman, J.P. The effect of vitamin D on renin–angiotensin system activation and blood pressure: A randomized control trial. J. Hypertens. 2017, 35, 822–829. [Google Scholar] [CrossRef]
- Agarwal, R. Vitamin D, Proteinuria, Diabetic Nephropathy, and Progression of CKD. Clin. J. Am. Soc. Nephrol. 2009, 4, 1523–1528. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Hernández, R.M.; García-Cantón, C.; Lorenzo, D.; Quevedo, V.; Bosch, E.; López-Ríos, L.; Riaño, M.; Boronat, M. The specific relationship between vitamin D deficiency and diabetic nephropathy among patients with advanced chronic kidney disease: A cross-sectional study in Gran Canaria, Spain. Clin. Nephrol. 2015, 83, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Wen, X.; Liu, Y. Paricalcitol Inhibits Renal Inflammation by Promoting Vitamin D Receptor–Mediated Sequestration of NF-κB Signaling. J. Am. Soc. Nephrol. 2008, 19, 1741–1752. [Google Scholar] [CrossRef] [PubMed]
- Fukami, K.; Taguchi, K.; Yamagishi, S.-I.; Okuda, S. Receptor for advanced glycation endproducts and progressive kidney disease. Curr. Opin. Nephrol. Hypertens. 2015, 24, 54–60. [Google Scholar] [CrossRef]
- Kheirouri, S.; Alizadeh, M. Vitamin D and advanced glycation end products and their receptors. Pharmacol. Res. 2020, 158, 104879. [Google Scholar] [CrossRef]
- García, I.M.; Altamirano, L.; Mazzei, L.; Fornés, M.; Cuello-Carrión, F.D.; Ferder, L.; Manucha, W. Vitamin D receptor-modulated Hsp70/AT1 expression may protect the kidneys of SHRs at the structural and functional levels. Cell Stress Chaperon 2014, 19, 479–491. [Google Scholar] [CrossRef]
- McCarron, J.G.; Wilson, C.; Sandison, M.E.; Olson, M.L.; Girkin, J.M.; Saunter, C.; Chalmers, S. From Structure to Function: Mitochondrial Morphology, Motion and Shaping in Vascular Smooth Muscle. J. Vasc. Res. 2013, 50, 357–371. [Google Scholar] [CrossRef]
- Silvagno, F.; Consiglio, M.; Foglizzo, V.; Destefanis, M.; Pescarmona, G. Mitochondrial Translocation of Vitamin D Receptor Is Mediated by the Permeability Transition Pore in Human Keratinocyte Cell Line. PLoS ONE 2013, 8, e54716. [Google Scholar] [CrossRef] [Green Version]
- Gabarre, P.; Dumas, G.; Dupont, T.; Darmon, M.; Azoulay, E.; Zafrani, L. Acute kidney injury in critically ill patients with COVID-19. Intensiv. Care Med. 2020, 46, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C.; Reis, T.; Husain-Syed, F. Management of acute kidney injury in patients with COVID-19. Lancet Respir. Med. 2020, 8, 738–742. [Google Scholar] [CrossRef]
- Spagnolo, P.; Balestro, E.; Aliberti, S.; Cocconcelli, E.; Biondini, D.; Della Casa, G.; Sverzellati, N.; Maher, T.M. Pulmonary fibrosis secondary to COVID-19: A call to arms? Lancet 2020, 8, 750–752. [Google Scholar] [CrossRef]
- Kashani, K.; Rosner, M.H.; Haase, M.; Lewington, A.J.; O’Donoghue, D.J.; Wilson, F.P.; Nadim, M.K.; Silver, S.; Zarbock, A.; Ostermann, M.; et al. Quality Improvement Goals for Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2019, 14, 941–953. [Google Scholar] [CrossRef] [PubMed]
- Isaia, G.; Medico, E. Associations between hypovitaminosis D and COVID-19: A narrative review. Aging Clin. Exp. Res. 2020, 32, 1879–1881. [Google Scholar] [CrossRef]
- Grant, W.B.; Lahore, H.; McDonnell, S.L.; Baggerly, C.A.; French, C.B.; Aliano, J.L.; Bhattoa, H.P. Evidence that Vitamin D Supplementation Could Reduce Risk of Influenza and COVID-19 Infections and Deaths. Nutrients 2020, 12, 988. [Google Scholar] [CrossRef]
- Hsieh, M.-C.; Hsiao, P.-J.; Liao, M.-T.; Hou, Y.-C.; Chang, Y.-C.; Chiang, W.-F.; Wu, K.-L.; Chan, J.-S.; Lu, K.-C. The Role of Vitamin D in SARS-CoV-2 Infection and Acute Kidney Injury. Int. J. Mol. Sci. 2022, 23, 7368. [Google Scholar] [CrossRef]
- Weidemann, D.K.; Abraham, A.G.; Roem, J.L.; Furth, S.L.; Warady, B.A. Plasma Soluble Urokinase Plasminogen Activator Receptor (suPAR) and CKD Progression in Children. Am. J. Kidney Dis. 2020, 76, 194–202. [Google Scholar] [CrossRef]
- Faubel, S. SuPAR: A potential predictive biomarker for acute kidney injury. Nat. Rev. Nephrol. 2020, 16, 375–376. [Google Scholar] [CrossRef]
- Saleem, M.A. What is the Role of Soluble Urokinase-Type Plasminogen Activator in Renal Disease? Nephron Exp. Nephrol. 2018, 139, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, F.; Lütgehetmann, M.; Pfefferle, S.; Wong, M.N.; Carsten, A.; Lindenmeyer, M.T.; Nörz, D.; Heinrich, F.; Meißner, K.; Wichmann, D.; et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet 2020, 396, 597–598. [Google Scholar] [CrossRef]
- Lin, H.; Xu, L.; Yu, S.; Hong, W.; Huang, M.; Xu, P. Therapeutics targeting the fibrinolytic system. Exp. Mol. Med. 2020, 52, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Sanchis-Gomar, F.; Favaloro, E.J.; Lavie, C.J.; Henry, B.M. Coronavirus Disease 2019–Associated Coagulopathy. Mayo Clin. Proc. 2021, 96, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Vadasz, Z.; Brenner, B.; Toubi, E. Immune-Mediated Coagulopathy in COVID-19 Infection. Semin. Thromb. Hemost. 2020, 46, 838–840. [Google Scholar] [CrossRef]
- Chiu, S.-K.; Tsai, K.-W.; Wu, C.-C.; Zheng, C.-M.; Yang, C.-H.; Hu, W.-C.; Hou, Y.-C.; Lu, K.-C.; Chao, Y.-C. Putative Role of Vitamin D for COVID-19 Vaccination. Int. J. Mol. Sci. 2021, 22, 8988. [Google Scholar] [CrossRef]
- Mohan, M.; Cherian, J.J.; Sharma, A. Exploring links between vitamin D deficiency and COVID-19. PLOS Pathog. 2020, 16, e1008874. [Google Scholar] [CrossRef]
Figure 1.
The urokinase plasminogen activator (uPA)/uPA receptor (uPAR) system function. The uPAR lacks an intracellular domain, however, it can interact with other transmembrane receptors such as formyl peptide receptors (FPRs) and integrins (mainly, αvβ3 integrin). The activation of these co-receptors then triggers intracellular signaling.
Figure 1.
The urokinase plasminogen activator (uPA)/uPA receptor (uPAR) system function. The uPAR lacks an intracellular domain, however, it can interact with other transmembrane receptors such as formyl peptide receptors (FPRs) and integrins (mainly, αvβ3 integrin). The activation of these co-receptors then triggers intracellular signaling.

Figure 2.
Putative molecular mechanism of suPAR-related podocyte injury and glomerular capillary microthrombus formation in COVID-19-related AKI. (1) High-level suPAR activation of integrin αvβ3 signaling impairs podocytes. (2) High levels of suPAR in combination with complement activation exacerbate the hypercoagulable state that leads to the development of AKI.
Figure 2.
Putative molecular mechanism of suPAR-related podocyte injury and glomerular capillary microthrombus formation in COVID-19-related AKI. (1) High-level suPAR activation of integrin αvβ3 signaling impairs podocytes. (2) High levels of suPAR in combination with complement activation exacerbate the hypercoagulable state that leads to the development of AKI.

Figure 3.
SARS-CoV-2-infection-induced injury to the renal tubular cells. Patients with severe SARS-CoV-2 infection are more likely to have high suPAR levels. In renal tubular cells, suPAR can bind to integrin β6 and activate Rac1 leading to the formation of versican V1. suPAR also affects proximal tubular cells, altering mitochondrial respiration and inducing oxidative stress. Patients with severe SARS-CoV-2 infection are also more likely to use C3. The increased generation of C3a in severe COVID-19 is accompanied by the release of activated CD16+ cytotoxic T cells. C3a promotes the transcription of versican by activating the AKT/β-catenin pathway. Tubular cell-derived versican V1 induces proliferation and collagen synthesis by activating the CD44/Smad3 pathway in fibroblasts. Briefly, C3a and suPAR can drive versican V1 expression in tubular cells, which contributes to fibrosis of renal interstitial tissues.
Figure 3.
SARS-CoV-2-infection-induced injury to the renal tubular cells. Patients with severe SARS-CoV-2 infection are more likely to have high suPAR levels. In renal tubular cells, suPAR can bind to integrin β6 and activate Rac1 leading to the formation of versican V1. suPAR also affects proximal tubular cells, altering mitochondrial respiration and inducing oxidative stress. Patients with severe SARS-CoV-2 infection are also more likely to use C3. The increased generation of C3a in severe COVID-19 is accompanied by the release of activated CD16+ cytotoxic T cells. C3a promotes the transcription of versican by activating the AKT/β-catenin pathway. Tubular cell-derived versican V1 induces proliferation and collagen synthesis by activating the CD44/Smad3 pathway in fibroblasts. Briefly, C3a and suPAR can drive versican V1 expression in tubular cells, which contributes to fibrosis of renal interstitial tissues.

Figure 4.
Potential mechanisms by which vitamin D protects podocytes and reduces proteinuria in COVID-19-related AKI. The SARS-CoV-2 infection causes renal podocytes to overproduce suPAR locally and systemically; suPAR activates αvβ3 integrins in podocytes and leads to apoptosis, depolarization, and structural and functional changes in podocytes. At this time, anti-CD40, SMPDL3, and G1/G2 with APOL1 significantly increase the activation of αvβ3 integrins and damage podocytes. Treatment with vitamin D may not only reduce systemic inflammation but also reduce BM-derived leukocyte suPAR. Vitamin D may also reduce the production of uPAR in podocyte cells and protect podocyte function by reducing the deleterious effect of angiotensin II and AT1R signaling.
Figure 4.
Potential mechanisms by which vitamin D protects podocytes and reduces proteinuria in COVID-19-related AKI. The SARS-CoV-2 infection causes renal podocytes to overproduce suPAR locally and systemically; suPAR activates αvβ3 integrins in podocytes and leads to apoptosis, depolarization, and structural and functional changes in podocytes. At this time, anti-CD40, SMPDL3, and G1/G2 with APOL1 significantly increase the activation of αvβ3 integrins and damage podocytes. Treatment with vitamin D may not only reduce systemic inflammation but also reduce BM-derived leukocyte suPAR. Vitamin D may also reduce the production of uPAR in podocyte cells and protect podocyte function by reducing the deleterious effect of angiotensin II and AT1R signaling.

Figure 5.
A putative cross-talk between vitamin D, suPAR, and COVID-19-related AKI. (1) Patients with COVID-19-related AKI have increased consumption and reduced kidney capacity to produce vitamin D, which may lead to vitamin D deficiency. (2) Vitamin D insufficiency or deficiency increases suPAR production from myeloid cells and podocytes. The addition of vitamin D suppresses local and systemic suPAR levels. (3) A high level of suPAR causes various lesions in the kidney cells such as tubular cells, podocytes, endothelial cells, and mesangial cells. (4) High levels of suPAR damage the renal tubular cells, resulting in a decrease in vitamin D production. (5) Patients with COVID-19-related AKI may have increased systemic and local suPAR levels. (6) Vitamin D supplementation can protect podocytes, tubular cells, and mesangial cells and prevent microthrombus formation in the glomerular capillaries and other vascular structures.
Figure 5.
A putative cross-talk between vitamin D, suPAR, and COVID-19-related AKI. (1) Patients with COVID-19-related AKI have increased consumption and reduced kidney capacity to produce vitamin D, which may lead to vitamin D deficiency. (2) Vitamin D insufficiency or deficiency increases suPAR production from myeloid cells and podocytes. The addition of vitamin D suppresses local and systemic suPAR levels. (3) A high level of suPAR causes various lesions in the kidney cells such as tubular cells, podocytes, endothelial cells, and mesangial cells. (4) High levels of suPAR damage the renal tubular cells, resulting in a decrease in vitamin D production. (5) Patients with COVID-19-related AKI may have increased systemic and local suPAR levels. (6) Vitamin D supplementation can protect podocytes, tubular cells, and mesangial cells and prevent microthrombus formation in the glomerular capillaries and other vascular structures.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Liao, T.-H.; Wu, H.-C.; Liao, M.-T.; Hu, W.-C.; Tsai, K.-W.; Lin, C.-C.; Lu, K.-C. The Perspective of Vitamin D on suPAR-Related AKI in COVID-19. Int. J. Mol. Sci. 2022, 23, 10725. https://doi.org/10.3390/ijms231810725
AMA Style
Liao T-H, Wu H-C, Liao M-T, Hu W-C, Tsai K-W, Lin C-C, Lu K-C. The Perspective of Vitamin D on suPAR-Related AKI in COVID-19. International Journal of Molecular Sciences. 2022; 23(18):10725. https://doi.org/10.3390/ijms231810725
Chicago/Turabian StyleLiao, Tzu-Hsien, Hsien-Chang Wu, Min-Tser Liao, Wan-Chung Hu, Kuo-Wang Tsai, Ching-Chieh Lin, and Kuo-Cheng Lu. 2022. "The Perspective of Vitamin D on suPAR-Related AKI in COVID-19" International Journal of Molecular Sciences 23, no. 18: 10725. https://doi.org/10.3390/ijms231810725
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.