
Manuscript accepted on :8-Dec-2020
Published online on: --
Plagiarism Check: Yes
Reviewed by: Dr. John George Hardy
Second Review by: Dr. Ahmed Salah
Final Approval by: Dr Ayush Dogra
Vishal Trivedi1*, Kush Biswas2, Santosh Fattepur3 and Nagaraja Sreeharsha4,5
1Department of Pharmacy, Madhav University, Abu Road, Pindwara, Rajasthan, India
2Department of Pharmaceutical technology (Biotechnology), National institute of Pharmaceutical Education and Research, Mohali, sector 67, SAS Nagar, Punjab, India
3School of Pharmacy, Management and Science University,Seksyen 13, 40100 Shah Alam, Selangor, Malaysia
4Department of Pharmaceutical Sciences, College of Clinical Pharmacy, King Faisal University, Al-Ahsa-31982, Saudi Arabia.
5Department of Pharmaceutics, Vidya Siri College of Pharmacy, Off Sarjapura Road, Bengaluru - 560 035, Karnataka, India
Corresponding Author Email: vishaltrivediqa@gmail.comDOI : https://dx.doi.org/10.13005/bpj/2080
Abstract
In Present time Novel coronavirus (SAR-CoV-2) was the biggest outbreak on human kind. SARS recognized febrile respiratory illness. It was first discovered in southern China in November 2002, and spread all other countries.Now SARS-CoV-2 was also originated from Wuhan, china. It was transmitted from human to human. For developing a molecular diagnostic for 2019-nCoV a PCR detection assay was developed. Sanger, Illumina, and Oxford nanopore techniques are used for sequencing analysis for 2019-nCoV.Based on different types of data base it was concluded that the SARS-CoV-2 was less mutated than other seasonal flu. This study shows the complete genome sequence and characteristic of SAR-CoV-2. SAR-CoV-2 has 29,903 nucleotides in length which is quite similar to others coronavirus. A complete genome sequence of different-different countries was studied.
Keywords
Coronavirus (SAR-Cov-2); Complete Genome Sequencing And Characterizations; PCR Detection
Download this article as:| Copy the following to cite this article: Trivedi V, Biswas K, Fattepur S, Sreeharsha N. Study on Genome Sequence of Novel Corona virus (Sars-Cov-2) Strains in Different Countries. Biomed Pharmacol J 2020;13(4). |
| Copy the following to cite this URL: Trivedi V, Biswas K, Fattepur S, Sreeharsha N. Study on Genome Sequence of Novel Corona virus (Sars-Cov-2) Strains in Different Countries. Biomed Pharmacol J 2020;13(4). Available from: https://bit.ly/3aFO9TC |
Introduction
2019-nCoV are large about 120 to 160nm long, spherical particles with a linear, non-segmented, capped, and polyadenylated positive-sense.1,2 It is a single-stranded RNA genome that isencapsulated in a helical nucleocapsid. The envelope is made up of intracellular membranes and have a characteristic crown of widely spaced club-shaped spikes that are 12 to 24 nmlong.3,4 Based ongenetic and serological relationships corona virus can be classified into three groups.5,6
Table 1: Classification of Corona virus.
| Group 1 | Group 2 | Group 3 |
| Porcine epidemic diarrhea virus (PEDV)
Porcine transmissible gastroenteritis virus (TGEV) Canine coronavirus (CCoV) Feline infectious peritonitis virus (FIPV) Human coronavirus 229E (HCoV-229E) Human coronavirus NL63 (HCoV-NL63) |
Murine hepatitis virus (MHV)
Bovine coronavirus (BCoV) Human coronavirus OC43 (HCoV-OC43) Rat sialodacryoadenitis virus (SDAV) Porcine hemagglutinating encephalomyelitis virus (PHEV) Canine respiratory coronavirus Equine coronavirus (ECoV) |
Avian infectious bronchitis virus (IBV)
Turkey coronavirus(TCoV) |
The corona virus genomes are the largest of the known RNA viruses (27 to 31.5 kb) and are polycistronic, generating a nested set of sub- genomic RNAs with common 5 and 3 sequences.7,8 The 5 two-thirds of the genome consists of two large replicase open reading frames (ORFs), ORF1a and ORF1b. The ORF1a polyprotein (pp1a) can be extended with ORF1b-encoded sequences via a 1 ribosomal frame shift at a conserved slippery site, generating the 7,000-amino-acid polyprotein pp1ab, which includes the putative RNA-dependent RNA polymerase (RdRp) and RNA helicase (HEL) activity.9,10,11
Virus isolation
A Pathogen free human airway epithelial (HAE)cells were used for virus isolation.12 Briefly, bronchoalveolar lavage fluids or throat swabs from the patients were inoculated into the HAE cells through the apical surfaces.HAE cells were maintained in an air–liquid interface incubated at 37°C. The cells were monitored daily for cytopathic effects by light microscopy and the cell super natants were collected for use in quantitative RT-PCR assays.13,14,15 After three passages, apical samples were collected for sequencing.16
Genome Sequences of SAR-CoV-2
The genome of SARS-CoV-2 is a 29,903 nucleotide, polyadenylated RNA, and 41% ofthe residues are G or C.17 The genomic organization of corona viruses, having the characteristic gene order [5 replicase (rep), spike (S), envelope (E),membrane (M), and nucleocapsid (N)-3] and short untranslated regions at both termini.18,19 The SARS-CoV-2 rep gene,which comprises approximately two-thirds ofthe genome, is predicted to encode two polyproteins ORF1a and ORF1b that undergo cotranslational proteolytic processing.20 Downstream of four open reading frames (ORFs)are used to encodethe structural proteins S, E, M, and N, which are common to all known corona viruses.21,22 Hemagglutinin – esterase, are present between ORF1b and S of corona viruses. These corona viruses encoded non-structural proteins which are located between S and E, between M and N, or downstream of N.23,24The genome of SARS-CoV-2 contains ORFs for five potential non-structural proteins that are more than 50 amino acids long inthese intergenic regions.25 overlapping of ORFs encoding show the proteins of 274 and 154 amino acids are located between S and E. Three non-structural genes, X3, X4, and X5 are located between M and N.26,27 GenBank database (BLAST and FastA) show that there is no significant sequence similarity between these potential non-structural proteins ofSARS-CoV-2 and any other proteins.28,29 ORFs encoding proteins are more than 50 amino acids long in the structural genes of SARS-CoV-2 (such as N, S, and rep).30,31SARS-CoV-2 repgene products have genomic RNA, and the remaining viral proteins are translated from sub genomicm RNAs that form a 3-coterminal nested set,each with a 5-end derived from the genomic 5 leader sequence.32,33Sub genomicm RNAs are synthesized from a discontinuous transcription process. The SARS-CoV-2 leader sequence was mapped by comparing the sequence of rapid amplification of cDNA ends.34,35The products which are synthesized from the N gene mRNA with those synthesized from genomic RNA.36 A sequence, AAACGAAC (genomic nucleotides 65to 72), was identified where the N gene mRNA and genomic sequences diverged.37 This sequence was also present upstream of ORF1a and immediately up stream of five other ORFs suggesting that it functions as the conserved core of the transcription-regulating sequences (TRSs).38,39 The nucleotides required for TRS function must be identified experimentally.40 The production of sub genomic mRNAs of SAR-CoV-2 proposes that discontinuous transcription has occurs during the synthesis of negative strand. Sub-genomic negative strands containing a complimentary copy of the leader sequence at their 3 termini serve as templates for synthesis of sub-genomic mRNAs.41,42 At the site of 5 terminus of the genome, the TRS conserved core sequence appears six times in the remainder of the genome.43 The positions of TRS in the genome of SARS-CoV-2 predict that sub-genomic mRNAs of 8.3, 4.5, 3.4, 2.5, 2.0, and1.7 kb, not including the poly(A) tail. At least five sub-genomic mRNAs were detected by hybridization of RNA from SARS-CoV–2 infected cells.44,45 Sizes of the five predominant bands correspond to the sizes of five of the predictedsub genomic mRNAs of SARS-CoV-2. Full-length of genomic RNA was not detected, probably because it is the least prevalent viral RNA in infected cells.46,47 The predicted 2.0-kb transcript was also not detected, which suggests that the consensus TRS at 27,771 to 27,778 is not used or that it is a low-abundance mRNA.48 Byanalogy with other corona viruses, the 8.3-kband 1.7-kb sub-genomic mRNAs are predicted to be monocistronic, directing translation of S andN, respectively, whereas multiple proteins could be translated from the 4.5-kb,3.4-kb and 2.5-kb mRNAs. It is possible that the 3.4-kb band contained more than one mRNA species that was not resolved in the gel or that the monocistronicm RNA.49,50,51
CDC sequencing strategy
The whole-genome sequences of SAR-CoV-2 were generated by a combination. Sanger, Illumina, and Oxford nanopore sequencing were used for this detection.52 In this CDC sequencing strategy at First viral RNAs were extracted directly from clinical samples with the QIAamp Viral RNA Mini Kit, and then used to synthesise cDNA with the SuperScript III Reverse Transcriptase and N6 random primers, followed by second-strand synthesis with DNA Polymerase I, Large Fragment.53,54 Viral cDNA libraries were prepared with use of the Nextera XT Library Prep Kit (Illumina), then purified with Agencourt AMPure XP beads followed by quantification with an Invitrogen Qubit 2. 0F luorometer.55,56 The resulting DNA libraries were sequenced on either the MiSeq or iSeq platforms (Illumina) using a300-cycle reagent kit. About 1·2–5 GB of data were obtained.57 The raw fast Q files were filtered then subjected tode novo assembly with the CLCBio software. Mapped assemblies were also done using the bat derived SARS-like corona virus isolate bat-SL-CoVZC45 as a reference. Variant calling, genome alignments, and sequence illustrations were generated with CLCBio software, and the assembled genome sequences were confirmed by Sanger sequencing.58,59Rapid amplification of cDNA ends (RACE) was doneto obtain the sequences of the 5ʹ and 3ʹ termini, using the Invitrogen 5ʹ RACE System and 3ʹ RACE System, according to the manufacturer’s instructions.60 Gene-specific primers for 5ʹ and 3ʹ RACE PCR amplification were designed to obtain a fragment of approximately 400–500 bp for the two regions. Purified PCR products were cloned into the pMD18-T Simple Vector and chemically competent Escherichia coli. PCR products were sequenced with use of M13 forward and reverse primers.61,62,63
Development of molecular diagnostics for 2019-nCoV
COVID-19 disease is spreads all over the world. IAEA and FAO (Food and Agriculture Organization)are supporting the most important laboratory technique Real time reverse transcription–Polymerase Chain Reaction (real time RT-PCR)for detecting, tracking, and studying of COVID-19. 64,65,66
On the basis of the genome sequences obtained, a real-time PCR detection assay was developed. PCR primers and probes were designed using Applied Bio systems Primer Express Software on the basis of our sequenced virus genomes. Primers and probes were synthesised by BGI. RT-PCR was done with an Applied Bio systems 7300 Real-Time PCR.67,68 It is a nuclear-derived method for detection of genetic material from any pathogen. Radioactive isotope markers are used to detect targeted genetic materials. Fluorescent dyes were used to replace the isotopic labelling. While real time RT-PCR is now the most widely used method for detecting corona viruses in many countries.69,70,71(SARS-CoV-2) only contains RNA. In SARS-CoV-2 RNA take control and reprogrammed the cells so that they multiple and make more viruses. SAR-CoV-2 detected in the body using real time RT-PCR. In which a sample is taken from infected person body like nose or throat.72,73,74 The taken sample is treated with several chemical solutions that remove substances,only the RNA is been extracted from the taken sample.The RNA is reverse transcription process. By using specific enzymes this RNA was converted into DNA. short fragments of DNA are added that are complementary to specific parts of the transcribed viral DNA. These fragments of DNA to target sections of the viral DNA.75,76,77 The genetic fragments are for building DNA strands during amplification, while the others are for building the DNA and adding marker labels to the strands, which are then used to detect the virus. These mixers then placed in a RT-PCR machine.78,79 The machine cycles heat and cool the mixture chemical reactions this create new, identical copies of the target sections of viral DNA. This cycle can repeat over and over to continue copying the target sections of viral DNA. Each cycle doubles the amount: two copies become four, four copies become eight, and so on.80,81 A standard real time RT-PCR setup goes through 35 cycles, which means that by the end of the process, around 35 billion new copies of the sections of viral DNA are created. the marker labels and a fluorescent dye were used to measured.82,83 Computer’s has been used for tracking the amount of fluorescence in the taken sample after each cycle. If the amount of fluorescence goes over a certain level, it gives the conformation of virus present.84 This technique is highly sensitive and specific. It can deliver a reliable diagnosis as fast as three hours. by Comparing with other available virus isolation methods, real time RT-PCR is much faster and it has a lower potential for contamination or errors. the entire process can be done within a closed tube. It is the most accurate method available for detection of the SAR-CoV-2.85,86,87
Effects of Genome sequence COVID-19 on different countries
Table 2: Effect of different genome sequences on different countries
| Countries | Total cases | Active cases | Total recovered | Total death |
| Europe | 1,322,784 | 7,36,539 | 4,59,814 | 1,26,431 |
| North America | 1,120,597 | 8,79,941 | 1,76,244 | 64,412 |
| Asia | 4,95,108 | 2,30,909 | 2,46,272 | 17,927 |
| South America | 1,55,592 | 93,708 | 54,294 | 7,590 |
| Africa | 35,779 | 22,383 | 11,868 | 1,528 |
| Oceania | 8,314 | 1,274 | 6,933 | 107 |
This study shows the effects on changing in genomic sequences of COVID 19 on different countries. It was stated from Wuhan city, china and spread all over the world. In December 2019, an outbreak of apparently viral pneumonia of unknown ethology emerged in the city of Wuhan, in the Chinese province of Hubei. Chinese scientist done 7,700 genome sequences of COVID-19. These genome sequences help to vital tracking, how the virus mutates time by time as it spread and also used for development of diagnostic tests and vaccines.88
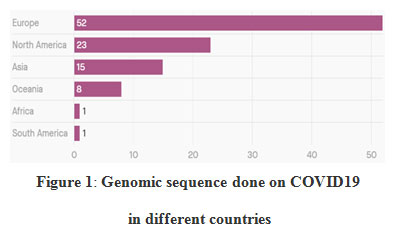 |
Figure 1: Genomic sequence done on COVID19 in different countries |
Data shows that Africa and south America has only 1% of genome sequencing done on COVID-19. Whereas Europe and north America has the highest genome sequencing done. There should be more genomic data from Africa and south America for vaccine development.89
9 January, 2020, health authorities of Chinese and the World Health Organization (WHO) found and announces the novel corona virus, known as SAR-CoV-2, these agents responsible for the pneumonia cases. On 11 and 12 January, the health authorities of Chinese shared the full genome sequence of SAR-CoV-2. The genome sequences of SAR-CoV-2 is crucial for the development of specific diagnostic tests and the identification of potential treatment options. Chinese scientists claim there are two main strain of SAR-CoV-2 in humans, indicating that the virus is mutating.Researchers of china say the COVID-19 virus, which has since been renamed SARS-CoV2, has evolved into two major lineages, known as “L” and “S” types.The newer and more aggressive L type strain accounted for about 70 per cent of the analysed cases, the researchers said, while the rest were linked to the older S type version.90,91
On Friday 24 January, 2020, detection of the corona virus was confirmed in France.
India researchers [National Institute of Virology (NIV), Pune and Gujarat Biotechnology Research Centre (GBRC)] decoding the entire genome sequence of the novel corona virus, and confirmed that there are three new mutations has occurred.These viruses mutate in order to adapt to and survive in different situations. It was mutated when it will try to control by medicines. This virus is mutating fast. After establishing the genome sequence then analyse the genome in them and after that formulate a strategy to fight the virus.
World Health Organization, confirmed that the diagnosis of COVID-19 should be done by RT-PCR or gene sequencing.92 The RT- PCR test is a highly sensitive. It is a gene based diagnostic test. It is much more reliable and much better than the conventional test.In united states of America, Researchers have examined the genomes sequences of corona virus say the similarities between the cases suggest that the virus may have been spreading in the state for weeks. On 20 January, by the analysis of genome sequences, centre of disease control announced that Washington had the United States’ first confirmed case of corona virus. Peoples who live in the same county, but did not known to have any contact with one another. By the study of genomic sequences, it has been concluded that the virus has been spreading through other people in the community.U.S.A. scientist also concluded that these viruses are independently would arrive in the same geographical area and be genetically related unless they were connected.93
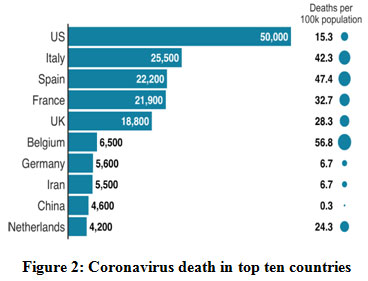 |
Figure 2: Coronavirus death in top ten countries. |
In united states death rate is very much high as compared to another countries. In USA, Human-to-human transmission has been reported too much.On 27 February, the first case of community spread in the US was reported, a person who did not have any travel history to the corona virus-affected regions.New York has reported the highest number of corona virus cases and deaths in the US and also accounts for 40% of deaths in the country. The government announced a disaster declaration for the state and the National Guard has been activated.The US passed a bill on 13 March making corona virus tests free for its residents.The CDC developed the RT-PCR diagnostic panel for testing patient specimens for COVID-19. It was approved for emergency use by the U.S. Food and Drug Administration (FDA) under Emergency Use Authorization (EUA).The CDC later announced that it would use its existing seasonal influenza surveillance system for testing corona virus cases in five states including Los Angeles, San Francisco, Seattle, Chicago, and New York.94,95
India has recorded 78,194 confirmed cases, 2,551 deaths, 26,400 cured cases, according to the latest update released by the Union Ministry for Health and Family Welfare.
Covid-19 is not easy to diagnose. Real Time-PCR can be used for virus testing followed by gene fingerprinting.There are 15 labs in India were the test has been done so far and Nineteen will be made operational as soon as possible. So far, specimens of suspected novel corona virus patients are tested at the National Centre for Disease Control (NCDC) in Delhi, Indian Council of Medical Research (ICMR)’s laboratories at Alappuzha, Bengaluru, Hyderabad and Mumbai, besides the National Institute of Virology (NIV), Pune. Under the guidance of NIV these labs are worked.In this test, a nucleic acid amplification-based assay called polymerase chain reaction (PCR) are used and a more sensitive form called reverse-transcription polymerase chain reaction(RTPCR).According to recommendations by the World Health Organization. COVID-19 diagnosis must be confirmed by RT-PCR or gene sequencing for respiratory or blood specimens. It is much more reliable and much better than the conventional test.96
China take immediate precautions to stop the spread of the virus and provide diagnosis and treatment in-time for reduction the impacts.On 23 January, the Chinese Civil Aviation Administration guided foreign airlines to reduce their scheduled flights to Wuhan, when necessary, to prevent and control the spread of pneumonia caused by corona virus. Chinese’s government take a number of steps to stop the outbreak, which are: –
Pooling trained medical personnel from across the country
Ensuring supplies to the most vulnerable and affected areas
Construction of new temporary coronavirus hospitals on a war-footing
Extending holidays for schools and businesses as well as the Chinese Lunar New Year holiday
Researchers has allowed for gene testing, epidemic control and research institutions to use Linear Fold, its RNA structure algorithm that will help to understand the virus and screening compounds in less than half-a-minute compared to approximately an hour earlier. Bangkok (Thailand) is currently at risk from a spread of the virus. Hong Kong (China) is second on the list, followed by Taipei (Taiwan, the Republic of China).
The most ‘at-risk’ countries or regions worldwide are Sydney, New York, London, Thailand, Japan and Hong Kong. USA, Australia and the UK. In mainland of China, the cities of Beijing, Guangzhou, Shanghai, Chongqing, Guangdong, Zhejiang, Sichuan and Henan are all identified as high-risk by the researchers. A common factor behind the deaths recorded in china was smoking, those who with already damaged lungs are more likely to succumb to pneumonia. According to WHO, Italy has been hit so badly, because of the age of its population. The country has the oldest average age in Europe, second oldest in the world and corona virus disproportionately affects the elderly, whose immune system may not strong enough to stave off the pneumonia the virus causes.97,98
First confirmed cases in Brazil came from Italy. On 28 January, the first Brazilian sequence closely resembles to the sequenced in Germany and it was different from the genome sequenced in Wuhan, the epicentre of the epidemic in China. This virus mutates relatively little, once a month on average, so there’s no point in sequencing small pieces of the genome. on February 29, the researchers isolated the sequence of whole genome of the corona virus from the second Brazilian patient diagnosed with COVID-19. Completed only 24 hours after confirmation of the case in Brazil, the study shows that there are differences between the genomes of the viruses isolated from the first and second patients. “The second genomic sequence of Brazilian is more similar to the genome sequenced in the United Kingdom, and it both differ from the Chinese sequences. This show that the COVID-19 epidemic is maturing in Europe in the sense that internal transmission is already occurring in European countries.
The main advantage of real-time epidemic monitoring. It is used to exactly identifying,where the virus came from. This information is useful to guide containment actions and help reduce dissemination of the disease. on February 26, the first case of COVID-19 was diagnosed in Brazil by molecular biology.99,100
References
- Weiss SR, Navas-Martin S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol Mol Bio Rev 2005; 69: 635-64.
CrossRef - Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbial 2019; 17: 181-92.
CrossRef - Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. A novel coronavirus from patients with pneumonia in China, 2019. New Engl J Med 2020; 382: 727-33.
CrossRef - Lu H, Stratton CW, Tang YW. Outbreak of pneumonia of unknown etiology in Wuhan, China: The mystery and the miracle. J Med Virol2020; 92: 401-2.
CrossRef - Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020; 395: 507-13.
CrossRef - Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020; 395: 565-74.
CrossRef - Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology Nature Publishing Group 2020; 5: 536-44.
CrossRef - Paraskevis D, Kostaki EG, Magiorkinis G,Panayiotopoulos G, Sourvinos G, Tsiodras S. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect Genet Evol2020; 79:
CrossRef
- Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B beta coronaviruses. Nat Microbiol doi: 10.1038/s41564-020-0688-y.
CrossRef - Li F, Li W, Farzan M, Harrison SC. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science2005; 309: 1864-8.
CrossRef - Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS. J Virol2020; 94. pi: e00127-20.
CrossRef - World Health Organization. Coronavirus disease(COVID-2019) situation reports. WHO; 2020? February 29, 2020.
- World Health Organization. Coronavirus disease (COVID-19) technical guidance: Laboratory testing for 2019-nCoV in humans. February 29, 2020.
- Ministry of Health & Family Welfare, Government of India; 2020.
- Yadav PD, Albariño CG, Nyayanit DA, Guerrero L, Jenks MH, Sarkale P, et al. Equine Encephalosis Virus in India, 2008. Emerg Infect Dis 2018; 24: 898-901.
CrossRef - Yadav PD, Whitmer SLM, Sarkale P, Ng TFF, Goldsmith CS, Nyayanit DA, et al. Characterization of novel reoviruses [Wad Medani virus (Orbivirus) and Kundal (Coltivirus)] collected from hyalommaantolicum ticks in India during surveillance for Crimean Congo Haemorrhagic fever. J Virol doi:10.1128/JVI.00106-19.
CrossRef - Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob Chall2017; 1: 33-46.
CrossRef - Kumar S, Stecher G, Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol BiolEvol2016; 33: 1870-4.
CrossRef - Andersen K, Rambaut A, Lipkin I, Holmes EC, Garry R. The proximal origin of SARS-CoV2.
- Pan Y, Zhang D, Yang P, Poon LLM, Wang Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect Dis pi: S1473-3099(20)30113-4.
CrossRef - To KK, Tsang OT, Chik-Yan Yip C, Chan KH, Wu TC, Chan JMC, et al. Consistent detection of 2019 novel coronavirus in saliva. Clin Infect Dis pii: ciaa149.
CrossRef - Volz E, Baguelin M, Bhatia S, Boonyasiri A, Cori A, Cucunuba Z, et al. Report 5: Phylogenetic analysis of SARS-CoV-2.
- Kirchdoerfer RN, Ward AB. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat Commun2019; 10:
CrossRef
- Buchholz UJ, Bukreyev A, Yang L, Lamirande EW, Murphy BR, Subbarao K, et al. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc Natl Acad Sci U S A 2004; 101: 98049.
CrossRef - O’Neil D, Swanton C, Jones A, Medd PG, Rayment N, Chain B. IFN-γ down-regulates MHC expression and antigen processing in a human B cell line. J Immunol 1999; 162: 791-8.
- Janice Oh HL, Ken-En Gan S, Bertoletti A, Tan YJ.Understanding the T cell immune response in SARScoronavirus infection. Emerg Microbes Infect 2012; 1:
CrossRef
- Baruah V, Bose S. Immunoinformatic-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J Med Virol2020; 92: 495-500.
CrossRef - Gao GF. From “A” IV to “Z” IKV: attacksfrom emerging and re-emerging pathogens. Cell 2018; 172:1157-9.
CrossRef - Weiss SR, Leibowitz JL. Coronaviruspathogenesis. Adv Virus Res 2011; 81:85-164.
CrossRef - Masters PS, Perlman S. Coronaviridae.In: Knipe DM, Howley PM, eds. Fields virology. 6th ed. Lippincott Williams & Wilkins, 2013:825-58.
- Su S, Wong G, Shi W, et al. Epidemiology, genetic recombination, and pathogenesis ofcoronaviruses. Trends Microbiol 2016; 24:490-502.
CrossRef - Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 2019; 17:181-92.
CrossRef - Zhong NS, Zheng BJ, Li YM, et al. Epidemiology and cause of severe acute respiratorysyndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 2003; 362:1353-8.
CrossRef - Ksiazek TG, Erdman D, GoldsmithCS, et al. A novel coronavirus associatedwith severe acute respiratory syndrome.N Engl J Med2003; 348:1953-66.
- Drosten C, Günther S, Preiser W, etal. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 2003; 348:1967-76.
CrossRef - Zaki AM, van Boheemen S, BestebroerTM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012; 367:1814-20.
CrossRef - Wong G, Liu W, Liu Y, Zhou B, Bi Y,Gao GF. MERS, SARS, and Ebola: the roleof super-spreaders in infectious disease. Cell Host Microbe 2015; 18:398-401.
CrossRef - Report of clustering pneumonia ofunknown etiology in Wuhan City. WuhanMunicipal Health Commission,
- Liu GS, Li H, Zhao SC, Lu RJ, Niu PH,Tan WJ. Viral and bacterial etiology ofacute febrile respiratory syndrome amongpatients in Qinghai, China. Biomed Environ Sci 2019; 32:438-45.
- Palacios G, Druce J, Du L, et al. A newarenavirus in a cluster of fatal transplants associated diseases. N Engl J Med 2008; 358:991-8.
CrossRef - Tan WJ, Zhao X, Ma XJ, et al. A novelcoronavirus genome identified in a cluster of pneumonia cases — Wuhan, China 2019-2020. China CDC Weekly 2020;2: 61-2.
CrossRef - Armstrong GL, MacCannell DR, Taylor J, et al. Pathogen genomics in public health. N Engl J Med 2019; 381:2569-80.
CrossRef - Report of novel coronavirus-infectedpneumonia in Wuhan City. Wuhan Municipal Health Commission, 2020.
- S. M. Peiris, Y. Guan, and K. Y. Yuen, “Severe acute respiratory syndrome,” NatureMedicine, vol. 10, no. 12, pp. S88–S97, 2004.
CrossRef - A. Rota, M. S. Oberste, S. S. Monroe et al., “Characterization of a novel coronavirusassociated with severe acute respiratory syndrome,” Science, vol. 300, no. 5624, pp. 1394–1399, 2003.
CrossRef - Shi and Z. Hu, “A review of studies on animal reservoirs of the SARS coronavirus,” VirusResearch, vol. 133, no. 1, pp. 74–87, 2008.
CrossRef - Balboni, A. Palladini, G. Bogliani, and M. Battilani, “Detection of a virus related to beta coronaviruses in Italian greater horseshoe bats,” Epidemiology and Infection, vol. 139,
no. 2, pp. 216–219, 2011.
CrossRef - F. Drexler, F. Gloza-Rausch, J. Glende et al., “Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences,” Journal of Virology, vol. 84, no. 21, pp. 11336–11349, 2010.
CrossRef - Li, Z. Shi, M. Yu et al., “Bats are natural reservoirs of SARS like coronaviruses,” Science, vol. 310, no. 5748, pp. 676–679, 2005.
CrossRef - L. Quan, C. Firth, C. Street et al., “Identification of a severe acute respiratory syndrome coronavirus-like virus in a leafnosed bat in Nigeria,” mBio, vol. 1, no. 4, article e00208-10, 2010.
CrossRef - Rihtaric, P. Hostnik, A. Steyer, J. Grom, and I. Toplak, “Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia,” Archives of Virology,vol. 155, no. 4, pp. 507–514, 2010.
CrossRef - Tong, C. Conrardy, S. Ruone et al., “Detection of novel SARS-like and other coronaviruses in bats from Kenya,” Emerging Infectious Diseases, vol. 15, no. 3, pp. 482–485, 2009.
CrossRef - K. P. Lau, P. C. Y. Woo, K. S. M. Li et al., “Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats,” Proceedings of the National Academy of Sciences of the United States of America, vol. 102, no. 39, pp.14040–14045, 2005.
CrossRef - K. P. Lau, P. C. Y. Woo, K. S. M. Li et al., “Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome,” Virology, vol. 367, no. 2, pp. 428–439, 2007.
CrossRef - K. P. Lau, K. S. M. Li, Y. Huang et al., “Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events,” Journalof Virology, vol. 84, no. 6, pp. 2808–2819, 2010.
CrossRef - Watanabe, J. S. Masangkay, N. Nagata et al., “Bat coronaviruses and experimental infection of bats, the Philippines,” Emerging Infectious Diseases, vol. 16, no. 8, pp. 1217–1223, 2010.
CrossRef - C. Y. Woo, S. K. P. Lau, K. S. M. Li et al., “Molecular diversity of coronaviruses in bats,” Virology, vol. 351, no. 1, pp. 180–187, 2006.
CrossRef - C. Y. Woo, M. Wang, S. K. P. Lau et al., “Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features,” Journal of Virology, vol. 81, no. 4, pp. 1574–1585, 2007.
CrossRef - Yuan, C. C. Hon, Y. Li et al., “Intraspecies diversity of SARS like coronaviruses in Rhinolophus sinicusand its implications for the origin of SARS coronaviruses in humans,” Journal of General Virology, vol. 91, no. 4, pp. 1058–1062, 2010.
CrossRef - Escutenaire, N. Mohamed, M. Isaksson et al., “SYBR Green real-time reverse transcription-polymerase chain reaction assay for the generic detection of coronaviruses,” Archives of Virology, vol. 152, no. 1, pp. 41–58, 2007.
CrossRef - US Environmental Protection Agency, Quality Assurance/ Quality Control Guidance for Laboratories Performing PCR Analysis on Environmental Samples, US Environmental Protection Agency, Washington, DC, USA, 2004.
- Battilani, T. Coradin, A. Scagliarini et al., “Quasispecies composition and phylogenetic analysis of feline coronaviruses (FCoVs) in naturally infected cats,” FEMS Immunology and Medical Microbiology, vol. 39, no. 2, pp. 141–147, 2003.
CrossRef - Battilani, A. Balboni, M. Ustulin, M. Giunti, A. Scagliarini, and S. Prosperi, “Genetic complexity and multiple infections with more Parvovirus species in naturally infected cats,” Veterinary Research, vol. 42, no. 1, article 43, 2011.
CrossRef - M. Poutanenet al., N. Engl. J. Med., available 17 April 2003 athttp://nejm.org/earlyrelease/sars.asp#4-2.
- Centers for Disease Control and Prevention, Mortal. Wkly. Rep. 52, 357 (2003).
- G. Ksiazeket al., N. Engl. J. Med. 348, 1947 (2003).
CrossRef - S. Peiris et al., Lancet 361, 1319 (2003).
CrossRef - M. C. Lai, K. V. Holmes, in Fields Virology, D. M. Knipe, P. M. Howley, Eds. (Lippincott Williams &Wilkins, New York, ed. 4, 2001), chap. 35.9. L. Enjuaneset al., in Virus Taxonomy, M. H. V. vanRegenmortalet al., Eds. (Academic Press, New York,2000), pp. 835–849.
- V. Holmes, in Fields Virology, D. M. Knipe, P. M. Howley, Eds. (Lippincott Williams & Wilkins, New York, ed. 4, 2001), chap. 36.
- Materials and methods are available as supportingmaterial on Science
- Although the match was not statistically significant, the C half of potential protein X1 contains a region of similarity with calcium-transporting adenosine triphosphatases.
- S. Sawicki, D. L. Sawicki. Adv. Exp. Med. Biol. 440,215 (1998).
CrossRef - The sequence immediately upstream of the ORFcoding for the predicted E protein is GTACGAAC anddiffers from the sequence of the consensus TRS at thefirst two positions.
- X. Liu, S. C. Inglis, J. Virol. 66, 6143 (1992).
CrossRef - Thiel, S. G. Siddell, J. Gen. Virol. 75, 3041 (1994).
CrossRef - S. Loleet al., J. Virol. 73, 152 (1999).
CrossRef - Ziebuhr, E. J. Snijder, A. E. Gorbalenya, J. Gen. Virol. 81,853 (2000).
CrossRef - G. Siddell, Ed., The Coronaviridae(Plenum, New York, 1995).
CrossRef - Escors, J. Ortego, H. Laude, L. Enjuanes, J. Virol. 75, 1312 (2001).
CrossRef - Garoff, R Hewson, D.-J. E. Opstelten, Microbiol. Mol. Biol. Rev. 62, 1171 (1998).
CrossRef - M. Sanchez et al., J. Virol. 73, 7607 (1999).
CrossRef - Leparc-Goffartet al., J. Virol. 72, 9628 (1998).
CrossRef - Cleavage sites in the S proteins of coronaviruses are RRFRR, RRSRR, RRSRR, RSRR, RARS, and RARR (26) in infectious bronchitis virus, bovine coronavirus, human coronavirus OC43, porcine hemagglutinating encephalomyelitis virus, mouse hepatitis virus, and rat coronavirus, respectively.
- Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S,Ser; T, Thr; V,Val; W, Trp; and Y, Tyr.
- A. M. de Haanet al., Virus Res. 82, 77 (2002).
- A. M. de Haan, L. Kuo, P. S. Masters, H. Vennema, P. J. M. Rottier, J. Virol. 72, 6838 (1998).
CrossRef - As of this writing, complete genomic sequences ofthree additional SARS-CoV isolates were available atGenBank (Tor-2 strain, Canada, accession no.ay274119; CUHK-W1 isolate, Hong Kong, accessionno. ay278554; and HKU-39849 isolate, Hong Kong,accession no. ay278491).
- A. Marraet al., Science 300, 1399 (2003); published online 1 May 2003 (10.1126.science.1085953).
CrossRef - Akin A, Lin TL, Wu CC, Bryan TA, Hooper T, Schrader D. Nucleocapsid protein gene sequence analysis reveals close genomic relationshipbetween Turkey coronavirus and avian infectious bronchitis virus. Acta Virol 2001; 45:31–8.
- Drosten C, Gunther S, Preiser W, Van der Werf S, Brodt HR, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New Engl J Med 2003; 348:1967–76.
CrossRef - Hiscox JA, Caavanagh D, Britton P. Quantification of individual subgenemic mRNA species during replication of the coronavirus transmissiblegastroenteritis. Virus Res 1995; 36:119–30.
CrossRef - Keck JG, Hogue BG, Brian DA, Lai MMC. Temporal regulation of bovine coronavirus RNA synthesis. Virus Res 1988; 9:343–56.
CrossRef - Poon LL, Wong OK, Chan KH, Luk W, Yuen KY, Peiris JS, et al. Rapid diagnosis of coronavirus associated with severe acute respiratory syndrome (SARS). Clin Chem 2003; 49:953–5.
CrossRef - Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003; 300:1394–9.
CrossRef - Arbour, N., R. Day, J. Newcombe, and P. J. Talbot. 2000. Neuroinvasion by human respiratory coronaviruses. Virol. 74:8913–8921.
CrossRef - Gorbalenya, A. E., E. V. Koonin, A. P. Donchenko, and V. M. Blinov. 1989. Coronavirus genome: prediction of putative functional domains in the nonstructural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 17:4847–4861.
CrossRef - Kiemer, L., O. Lund, S. Brunak, and N. Blom. 2004. Coronavirus 3CL-pro proteinase cleavage sites: possible relevance to SARS virus pathology. BMCBioinformatics 5:72.
CrossRef - Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680.
CrossRef - Ziebuhr, J., V. Thiel, and A. E. Gorbalenya. 2001. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursorinvolves two paralogous papain-like proteases that cleave the same peptide bond. Biol. Chem. 276:33220–33232.
CrossRef - Jonsdottir HR, Dijkman R. Corona viruses and the human airway: a universal system for virus-host interaction studies. Virol J 2016; 13:24.
CrossRef

