
Structural-Dynamics and Binding Analysis of RBD from SARS-CoV-2 Variants of Concern (VOCs) and GRP78 Receptor Revealed Basis for Higher Infectivity

 , , , ,
, , , ,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Variants Modeling and Preparation
2.2. Docking of Variants with the Host GRP78 Receptor
2.3. Estimation of Dissociation Constant (KD)
2.4. Dynamics Features of Wild-Type RBD and Variants-GRP78 Complexes
2.5. Estimation of Binding Free Energy
3. Results and Discussion
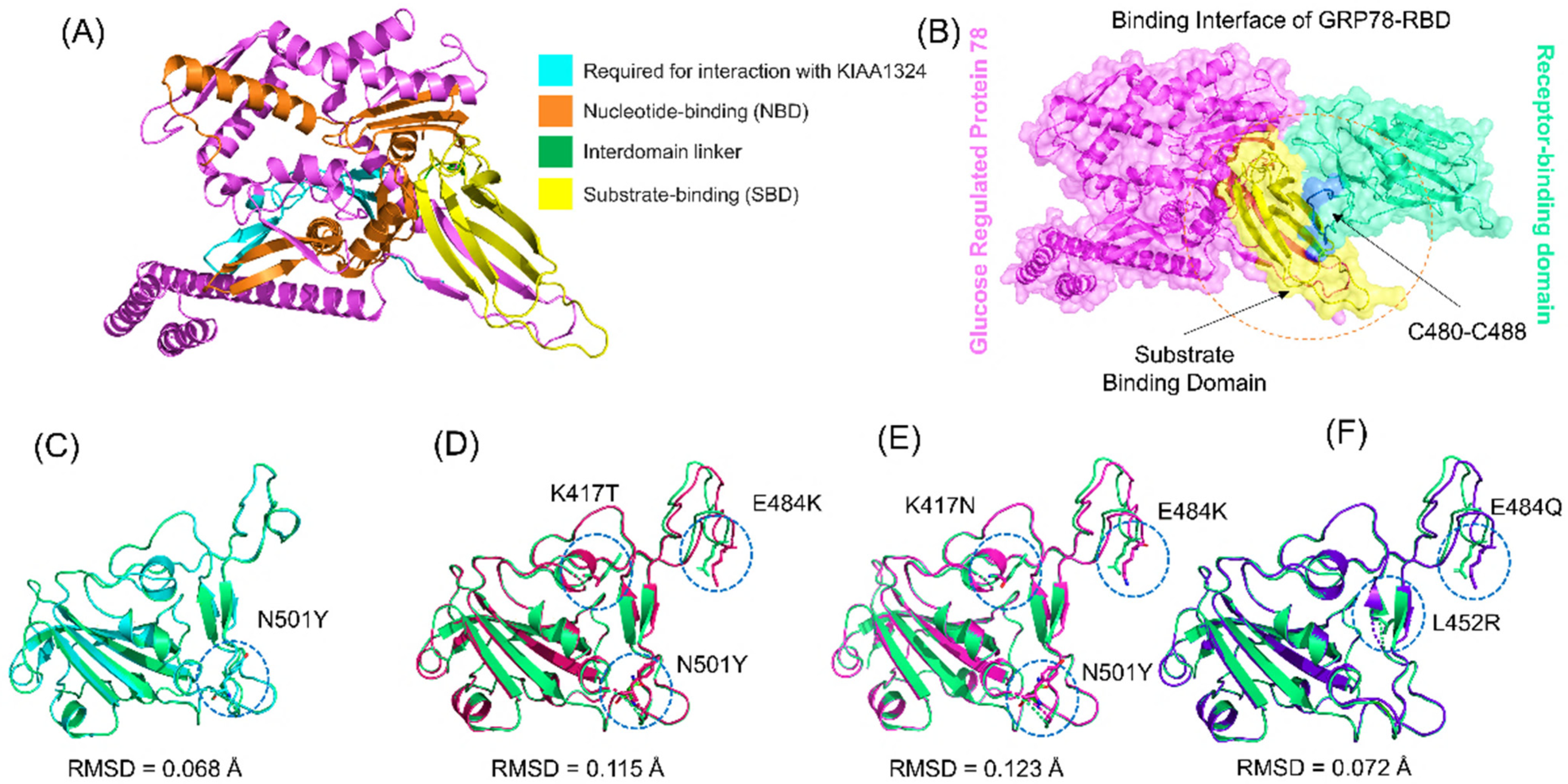
3.1. Structural Modeling and Analysis
3.2. Docking of Spike RBDs and GRP78
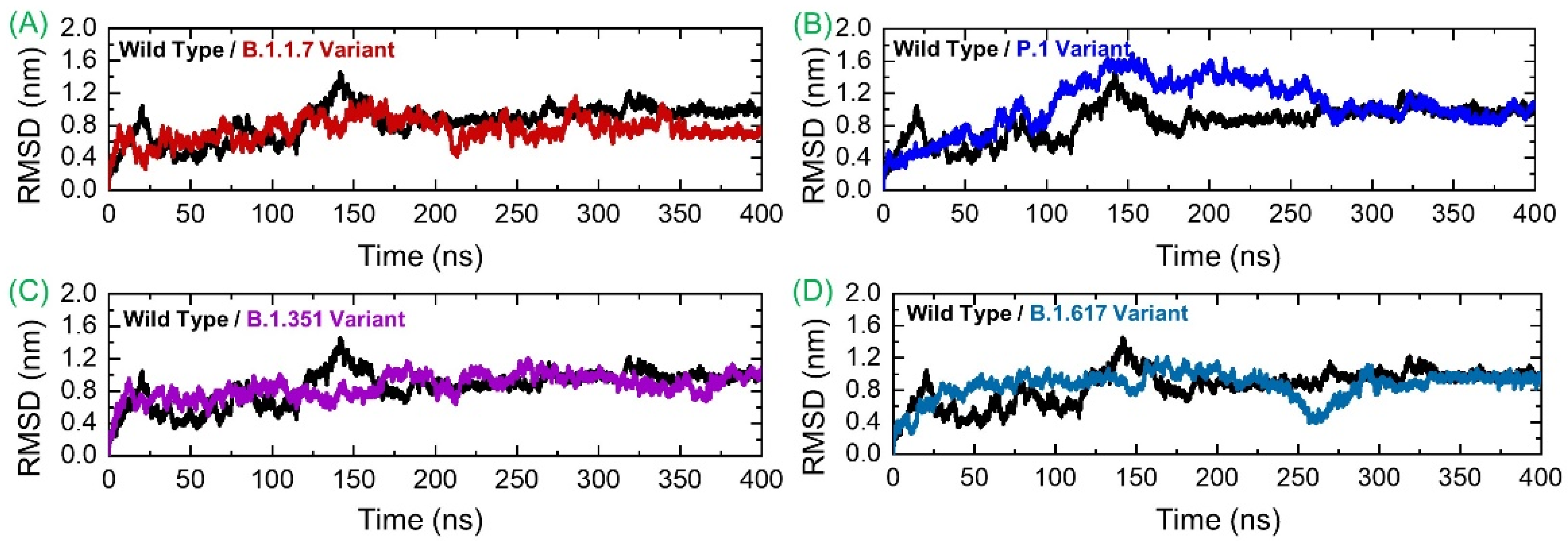
3.3. Conformational Dynamics Analysis by RMSD
3.4. Investigating System Compactness
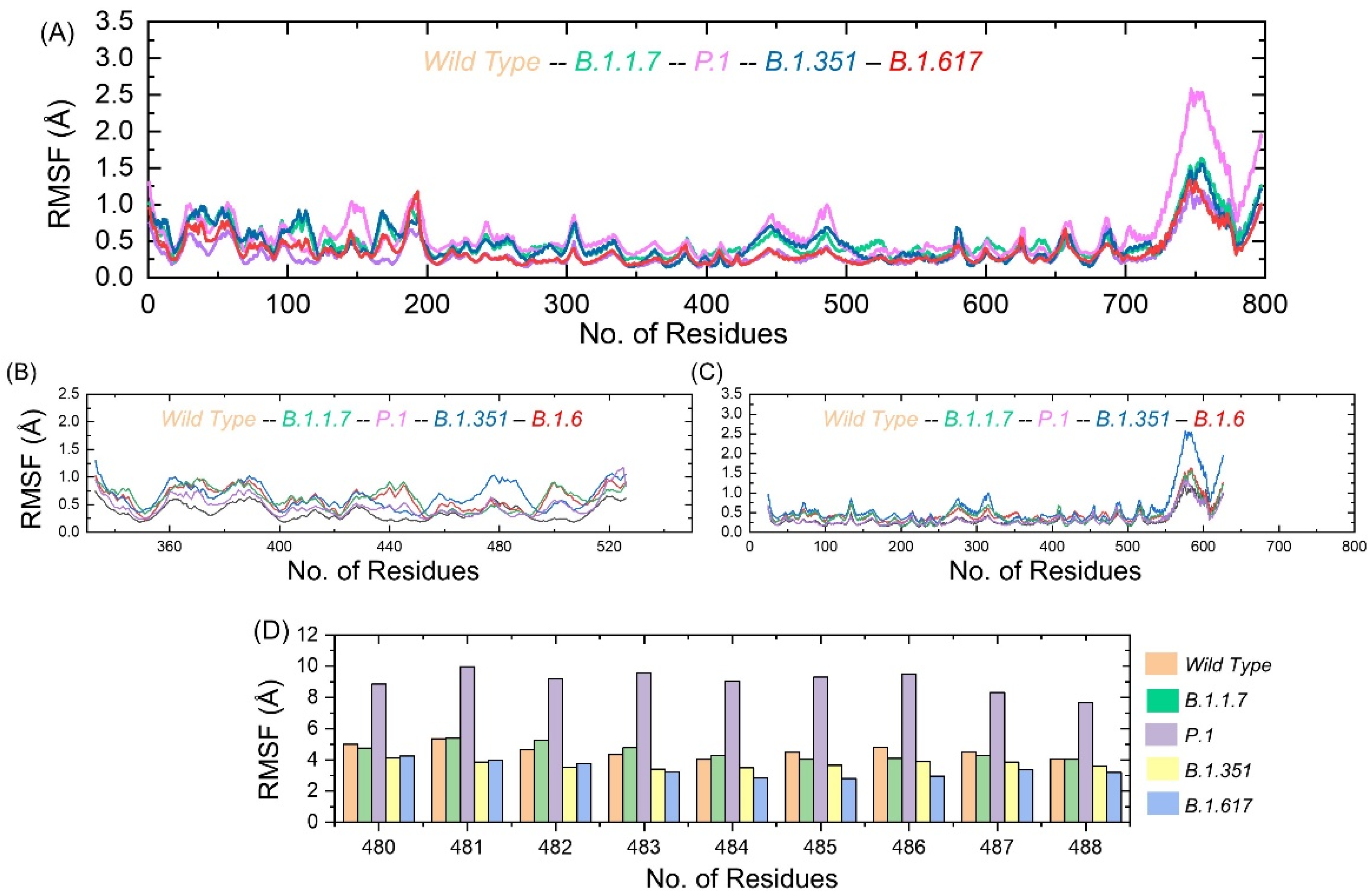
3.5. Residue Level Flexibility Analysis
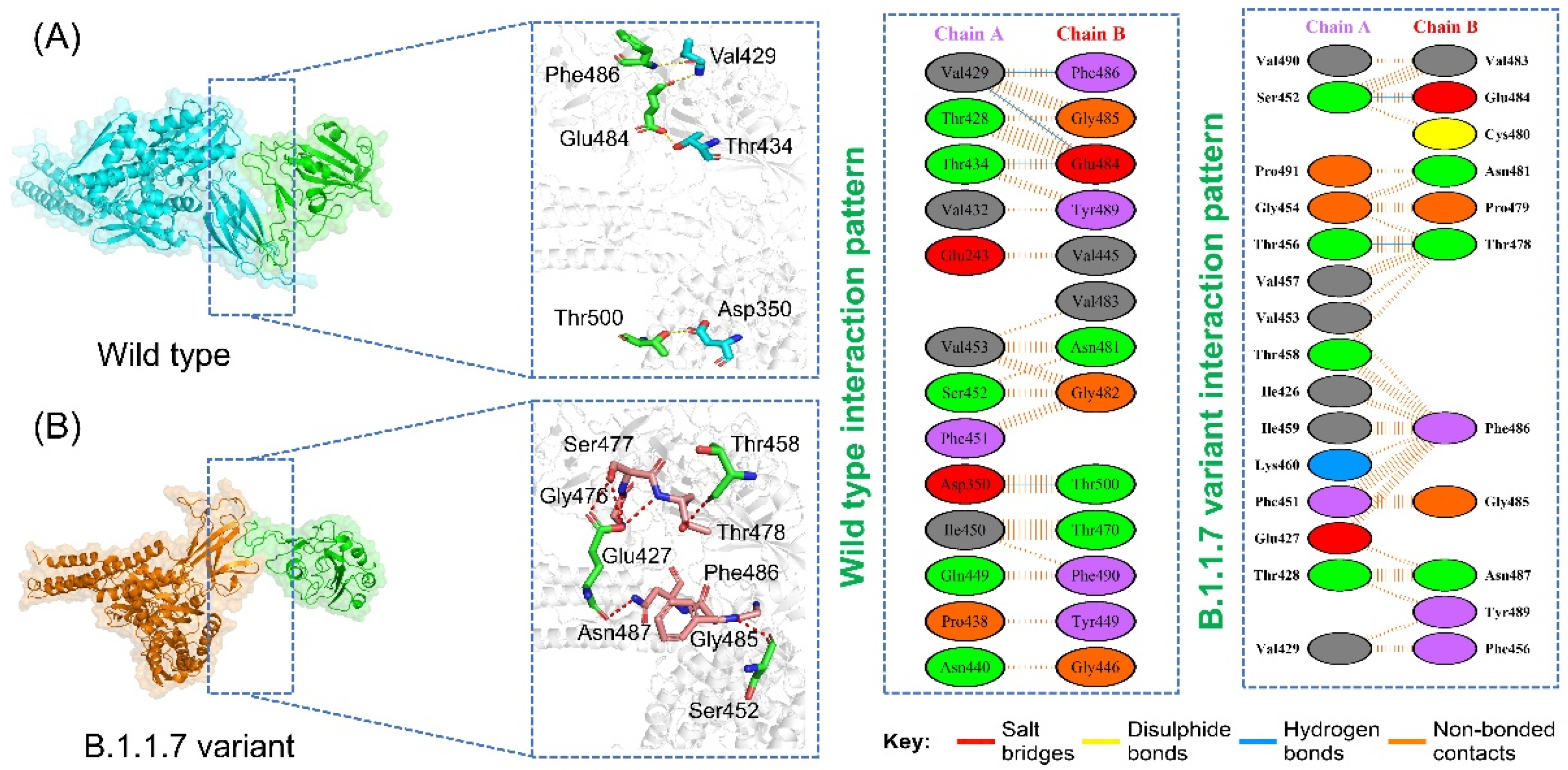
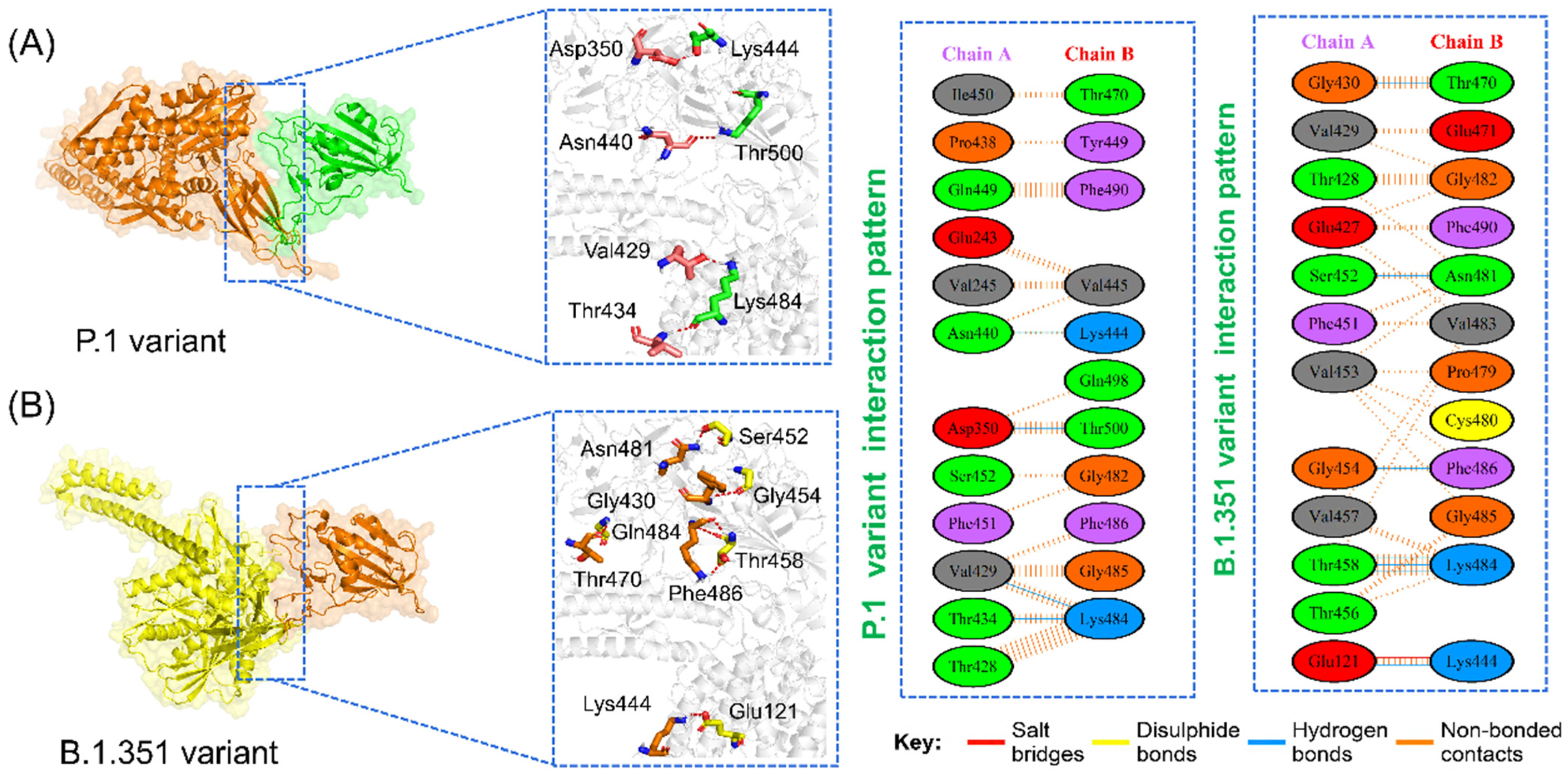
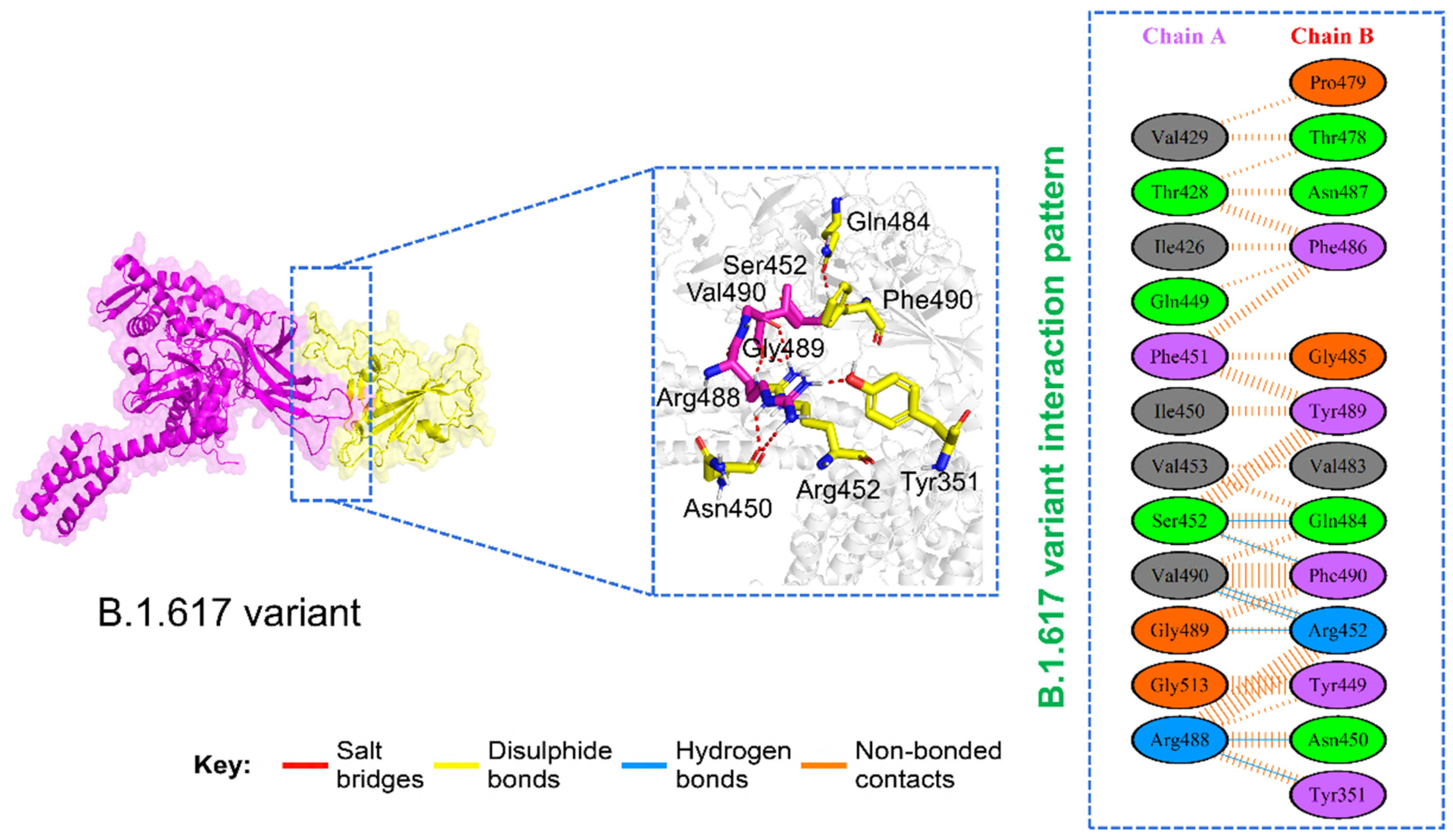
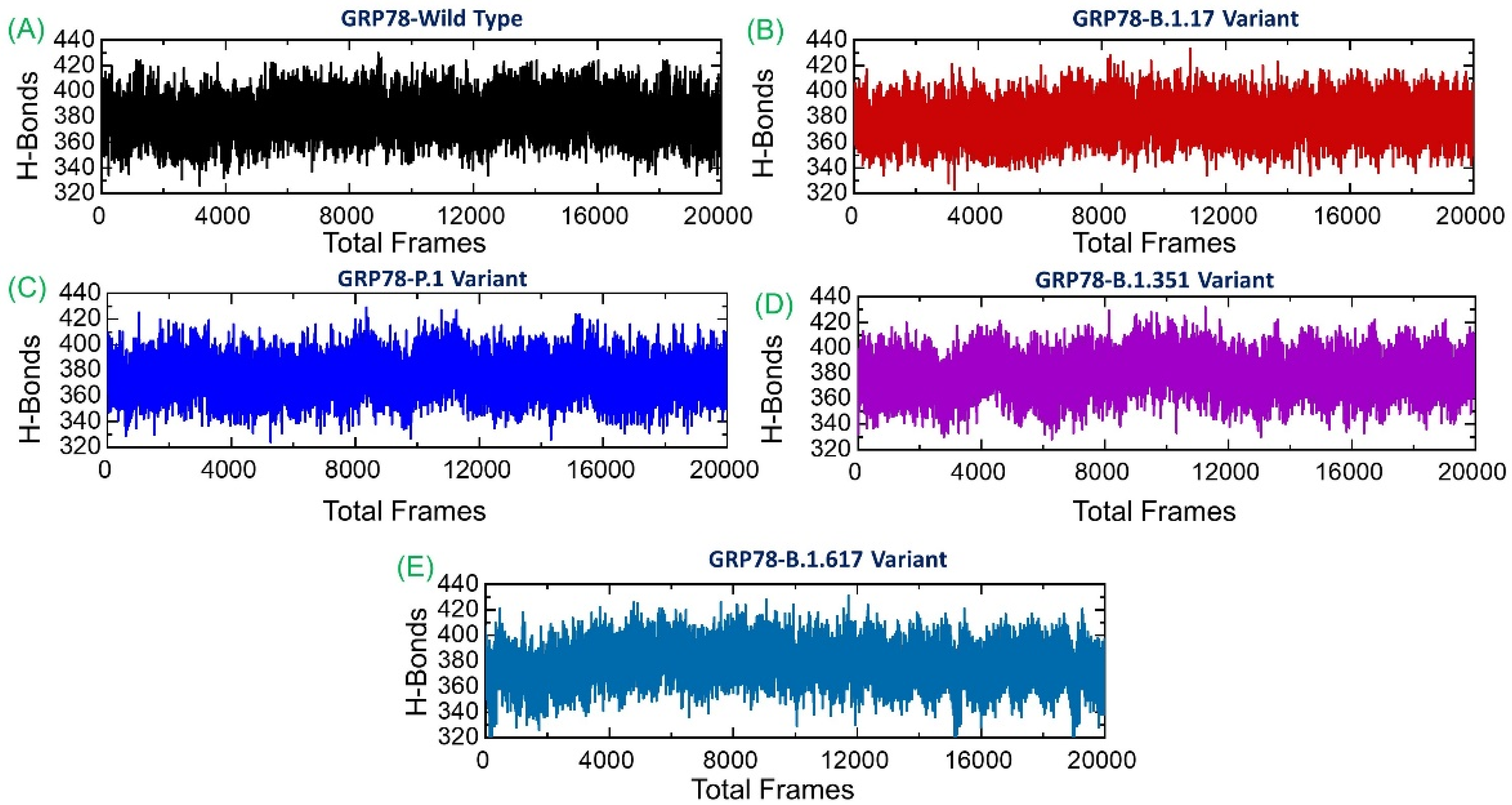
3.6. Analysis of Intermolecular Hydrogen Bonding
3.7. Estimation of Binding Free Energy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the sars-cov-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Abd El-Aziz, T.M.; Al-Sabi, A.; Stockand, J.D. Human recombinant soluble ace2 (hrsace2) shows promise for treating severe covid19. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C. Human recombinant soluble ace2 in severe covid-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of sars-cov-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Carlos, A.J.; Ha, D.P.; Yeh, D.-W.; Van Krieken, R.; Tseng, C.-C.; Zhang, P.; Gill, P.; Machida, K.; Lee, A.S. The chaperone grp78 is a host auxiliary factor for sars-cov-2 and grp78 depleting antibody blocks viral entry and infection. J. Biol. Chem. 2021, 296, 100759. [Google Scholar] [CrossRef]
- Lee, A.S. The er chaperone and signaling regulator grp78/bip as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Aguiar, J.A.; Tremblay, B.J.; Mansfield, M.J.; Woody, O.; Lobb, B.; Banerjee, A.; Chandiramohan, A.; Tiessen, N.; Cao, Q.; Dvorkin-Gheva, A. Gene expression and in situ protein profiling of candidate sars-cov-2 receptors in human airway epithelial cells and lung tissue. Eur. Respir. J. 2020, 56, 2001123. [Google Scholar] [CrossRef]
- Felordi, M.S.; Memarnejadian, A.; Najimi, M.; Vosough, M. Is there any alternative receptor for sars-cov-2? Cell J. 2021, 23, 247–250. [Google Scholar]
- Elfiky, A.A. Sars-cov-2 spike-heat shock protein a5 (grp78) recognition may be related to the immersed human coronaviruses. Front. Pharmacol. 2020, 11, 1997. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, C.M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.; Sze, K.H.; Yang, D.; Shuai, H.; et al. Middle east respiratory syndrome coronavirus and bat coronavirus hku9 both can utilize grp78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. Covid-19 spike-host cell receptor grp78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Elfiky, A.A.; Elgohary, A.M. Recognition through grp78 is enhanced in the uk, south african, and brazilian variants of sars-cov-2; an in silico perspective. Biochem. Biophys. Res. Commun. 2021, 562, 89–93. [Google Scholar] [CrossRef]
- Carlos, A.J.; Ha, D.P.; Yeh, D.-W.; Van Krieken, R.; Gill, P.; Machida, K.; Lee, A. Grp78 binds sars-cov-2 spike protein and ace2 and grp78 depleting antibody blocks viral entry and infection in vitro. bioRxiv 2021. [Google Scholar] [CrossRef]
- Khan, A.; Zia, T.; Suleman, M.; Khan, T.; Ali, S.S.; Abbasi, A.A.; Mohammad, A.; Wei, D.-Q. Higher infectivity of the sars-cov-2 new variants is associated with k417n/t, e484k, and n501y mutants: An insight from structural data. J. Cell. Physiol. 2021, 236, 7045–7057. [Google Scholar] [CrossRef]
- Khan, A.; Wei, D.Q.; Kousar, K.; Abubaker, J.; Ahmad, S.; Ali, J.; Al-Mulla, F.; Ali, S.S.; Nizam-Uddin, N.; Mohammad Sayaf, A.; et al. Preliminary structural data revealed that the sars-cov-2 b.1.617 variant’s rbd binds to ace2 receptor stronger than the wild type to enhance the infectivity. Chembiochem. A Eur. J. Chem. Biol. 2021, 22, 2641–2649. [Google Scholar] [CrossRef] [PubMed]
- Challen, R.; Brooks-Pollock, E.; Read, J.M.; Dyson, L.; Tsaneva-Atanasova, K.; Danon, L. Risk of mortality in patients infected with sars-cov-2 variant of concern 202012/1: Matched cohort study. BMJ 2021, 372, n579. [Google Scholar] [CrossRef]
- Khan, A.; Khan, T.; Ali, S.; Aftab, S.; Wang, Y.; Qiankun, W.; Khan, M.; Suleman, M.; Ali, S.; Heng, W. Sars-cov-2 new variants: Characteristic features and impact on the efficacy of different vaccines. Biomed. Pharmacother. 2021, 143, 112176. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium, UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [PubMed] [Green Version]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C. Structural and functional analysis of the d614g sars-cov-2 spike protein variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Ferrin, T.E. Software extensions to ucsf chimera for interactive visualization of large molecular assemblies. Structure 2005, 13, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. Ucsf chimerax: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlić, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D. The rcsb protein data bank: Redesigned web site and web services. Nucleic Acids Res. 2010, 39, D392–D401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. Haddock: A protein− protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. Prodigy: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with amber on gpus. 2. Explicit solvent particle mesh ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. Ptraj and cpptraj: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the mm/pbsa and mm/gbsa methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Modeling 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Gui, J.; Ahmad, W.; Haq, I.; Shahid, M.; Khan, A.A.; Shah, A.; Khan, A.; Ali, L.; Anwar, Z. The sars-cov-2 b. 1.618 variant slightly alters the spike rbd–ace2 binding affinity and is an antibody escaping variant: A computational structural perspective. RSC Adv. 2021, 11, 30132–30147. [Google Scholar] [CrossRef]
- Sun, Z.; Yan, Y.N.; Yang, M.; Zhang, J.Z. Interaction entropy for protein-protein binding. J. Chem. Phys. 2017, 146, 124124. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C. Deep mutational scanning of sars-cov-2 receptor binding domain reveals constraints on folding and ace2 binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Davenport, T.M.; Gorman, J.; Joyce, M.G.; Zhou, T.; Soto, C.; Guttman, M.; Moquin, S.; Yang, Y.; Zhang, B.; Doria-Rose, N.A.; et al. Somatic hypermutation-induced changes in the structure and dynamics of hiv-1 broadly neutralizing antibodies. Structure 2016, 24, 1346–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovchinnikov, V.; Louveau, J.E.; Barton, J.P.; Karplus, M.; Chakraborty, A.K. Role of framework mutations and antibody flexibility in the evolution of broadly neutralizing antibodies. Elife 2018, 7, e33038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warwicker, J. A model for ph coupling of the sars-cov-2 spike protein open/closed equilibrium. Brief. Bioinform. 2021, 22, 1499–1507. [Google Scholar] [CrossRef]
- Chen, X.; Geiger, J.D. Janus sword actions of chloroquine and hydroxychloroquine against covid-19. Cell. Signal. 2020, 73, 109706. [Google Scholar] [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-em structure of the sars coronavirus spike glycoprotein in complex with its host cell receptor ace2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of sars-cov-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Mobley, D.L. Entropy-enthalpy compensation: Role and ramifications in biomolecular ligand recognition and design. Annu. Rev. Biophys. 2013, 42, 121–142. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | Wild-Type | B.1.1.7 Variant | P.1 Variant | B.1.351 Variant | B.1.617 Variant |
|---|---|---|---|---|---|
| Interface residues | 13,13 | 14,16 | 12,11 | 12,12 | 12,13 |
| Salt bridges | *** | 1 | 2 | *** | *** |
| Disulfide bonds | *** | *** | *** | *** | *** |
| Hydrogen bonds | 4 | 7 | 7 | 7 | 4 |
| Non-bonded contact | 67 | 75 | 50 | 83 | 70 |
| Docking scores | −205.36 | −289.67 | −279.59 | −265.66 | −224.32 |
| Dissociation constant (KD) | 4.02 × 10−8 | 3.00 × 10−4 | 3.00 × 10−7 | 3.65 × 10−7 | 4.01 × 10−5 |
| Wild-Type | B.1.1.7 Variant | P.1 Variant | B.1.351 Variant | B.1.617 Variant | |
|---|---|---|---|---|---|
| Van der Waals | −76.54 | −68.88 | −84.05 | −72.32 | −68.15 |
| Electrostatic Interactions | −453.55 | −180.57 | −280.96 | −436.79 | −560.29 |
| Generalized Born | 495.75 | 201.03 | 315.11 | 472.59 | 601.11 |
| Non-polar Solvation Energy | −9.11 | −9.06 | −9.84 | −10.35 | −10.36 |
| Total Binding Energy | −43.45 | −57.48 | −59.74 | −46.87 | −37.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.; Mohammad, A.; Haq, I.; Nasar, M.; Ahmad, W.; Yousafi, Q.; Suleman, M.; Ahmad, S.; Albutti, A.; Khan, T.; et al. Structural-Dynamics and Binding Analysis of RBD from SARS-CoV-2 Variants of Concern (VOCs) and GRP78 Receptor Revealed Basis for Higher Infectivity. Microorganisms 2021, 9, 2331. https://doi.org/10.3390/microorganisms9112331
Khan A, Mohammad A, Haq I, Nasar M, Ahmad W, Yousafi Q, Suleman M, Ahmad S, Albutti A, Khan T, et al. Structural-Dynamics and Binding Analysis of RBD from SARS-CoV-2 Variants of Concern (VOCs) and GRP78 Receptor Revealed Basis for Higher Infectivity. Microorganisms. 2021; 9(11):2331. https://doi.org/10.3390/microorganisms9112331
Chicago/Turabian StyleKhan, Abbas, Anwar Mohammad, Inamul Haq, Mohammad Nasar, Waqar Ahmad, Qudsia Yousafi, Muhammad Suleman, Sajjad Ahmad, Aqel Albutti, Taimoor Khan, and et al. 2021. "Structural-Dynamics and Binding Analysis of RBD from SARS-CoV-2 Variants of Concern (VOCs) and GRP78 Receptor Revealed Basis for Higher Infectivity" Microorganisms 9, no. 11: 2331. https://doi.org/10.3390/microorganisms9112331