
Machine Learning Techniques for the Prediction of B-Cell and T-Cell Epitopes as Potential Vaccine Targets with a Specific Focus on SARS-CoV-2 Pathogen: A Review

 ,
,  , , and
, , and
Abstract
:1. Introduction
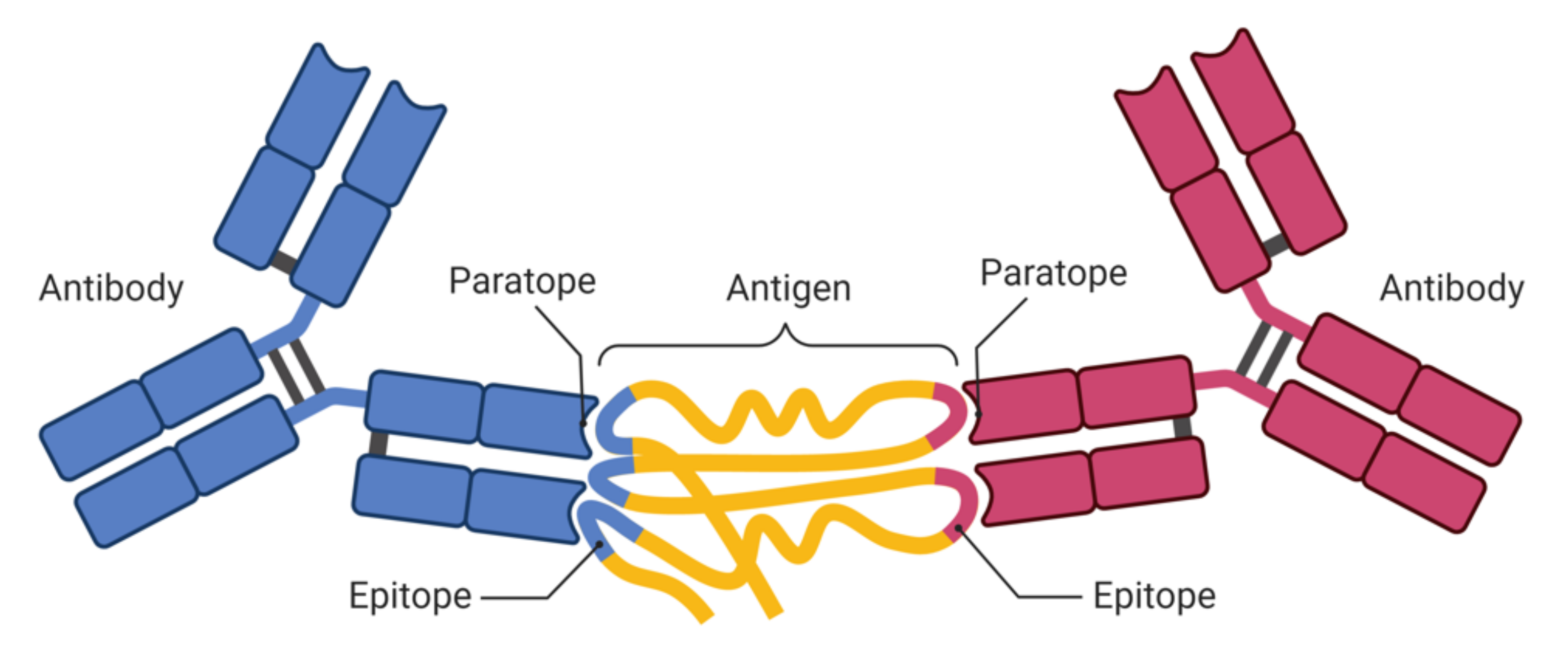
1.1. Epitopes and Paratopes
1.2. Need for T- and B-Cell Epitope Prediction
1.3. Motivations behind This Study
- To highlight the work done in T- and B-cell epitope prediction using ML, along with the strengths and limitations of the existing ML methods and tools, with the aim of promoting the EBPV design approach as this approach has received less attention so far. This will also stimulate continuing research efforts for designing an EBPV.
- With the increase in data related to antigenic determinants (TCEs and BCEs) and advances in immunoinformatics, the scientific community is overwhelmed.
- To provide future directions in terms of taking advantage of ensemble ML and exploring additional physicochemical properties of amino acids, and to use other confusion matrix-based performance metrics apart from accuracy and area under the curve (AUC) for designing an effective EBPV.
2. Existing ML-Based Studies for the Prediction of T- and B-Cell Epitopes
3. Existing Tools for T- and B-Cell Epitope Prediction
3.1. Tools for T-Cell Epitope Prediction
3.2. Tools for B-Cell Epitope Prediction
4. Studies Conducted for Predicting SARS-CoV-2 Epitopes
5. Future Research Directions in T- and B-Cell Epitope Prediction
- 1.
- The majority of current state-of-the-art approaches estimate a peptide’s binding capability. These approaches struggle to predict deterministically whether a given peptide is an epitope or not. CTLpred [41], one of the servers, operates in this category; however, it is limited to peptides that are up to 9 mers in length. To circumvent the limitations of the previous approaches, a direct method of predicting epitopes is sought. Furthermore, the technique should be capable of predicting variable-length peptides with a length greater than 9 mers.
- 2.
- Current state-of-the-art ML epitope prediction approaches rely heavily on just a few classifiers, including ANNs, SVMs, and Hidden Markov models (HMM) [121]. There are other robust classifiers available that can be utilized to achieve even more promising results, including decision trees (DT), random forest (RF), convolutional neural networks (CNNs), and AdaBoost [122]. In the literature surveyed, ANN-based models constitute the majority of the epitope prediction methods. However, relying on ANNs only is not safe. ANNs suffer from a hardware dependency as they require processors with parallel processing power in accordance with their structure [123]. Because epitope prediction is such a delicate task, the ANN’s behavior is occasionally unexplainable. When an ANN generates a probing solution, it does not explain why or how it was generated, which reduces the trust in the network [123]. However, to have high-performing models and robust models for applications such as the healthcare domain, explainable ML can be explored, which is in its initial stage and remains an open issue [124]. Gagniuc et al. have proposed a spectral-based forecast model as an alternative to the classical ANN. In their experiment, the ANN categorized the collection of data fairly but failed to reveal any useful information about the evolution of a subject over time. In this regard, forecasts based on Markov chains or traditional statistical methodologies have produced more trustworthy outcomes in the biology and medicine domains. The proposed novel method of analysis based on spectral forecasts outperformed the classical ANNs [125].
- 3.
- Moreover, instead of relying on predictions by a single model, we can combine several robust classifiers, called an ensemble model. Ensemble learning (EL) is a powerful technique for boosting the model accuracy by combining a number of base classifiers [126]. Such a technique has considerably better generalization capability than its individual counterparts. Indeed, EL is appealing because it can elevate weak learners (also known as base classifiers), which are marginally better than random guesses, to strong learners, which can make accurate forecasts [127]. The base classifiers vote for a new data instance, and, based on the majority of votes, a class label is returned. An ensemble model can be created by training homogeneous base models on different subsets of the training set or heterogeneous base models using the same training dataset. The main three types of ensembling techniques are bagging, boosting, and stacking. Multiple base learners (homogenous) can be integrated in bagging using different sub-samples from the same dataset [128]. The final prediction is obtained by taking the average prediction from multiple base learners. In boosting, base learners are added sequentially, and the predictions reported by previous learners are corrected. The final output is decided by taking the weighted average of all the predictions [128]. On the other hand, stacking involves fitting heterogeneous base learners on the same dataset [128] and then using another learner to learn how to best combine all the predictions. Moreover, while dealing with complex data, such as high-dimensional, imbalanced, noisy data, etc., traditional ML algorithms may fail to produce satisfactory results. The reason for this is that, for these methods, it is difficult to capture various attributes and the underlying layout of the data. Ensemble learning aims to combine data modeling, data fusion, and data mining into a cohesive framework [129] To conclude, the main reasons for employing ensemble learning in epitope prediction are as follows:
- 4.
- 5.
- The existing ML-based methods for epitope prediction have been assessed using metrics such as accuracy and area under the curve (AUC). However, other confusion matrix-based performance metrics such as Gini, specificity, sensitivity, F-score, kappa, Matthews correlation coefficient (MCC), and precision, etc., can be utilized to analyze the performance of the model in a better way.
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Immunology Guidebook|ScienceDirect. Available online: https://www.sciencedirect.com/book/9780121983826/immunology-guidebook (accessed on 25 September 2021).
- COVID Live Update: 270,426,226 Cases and 5,321,864 Deaths from the Coronavirus—Worldometer. Available online: https://www.worldometers.info/coronavirus/ (accessed on 10 December 2021).
- Centers for Disease Control and Prevention (CDC). SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 7 August 2021).
- WHO Director-General’s opening remarks at the 8th meeting of the IHR Emergency Committee on COVID-19—14 July 2021. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-8th-meeting-of-the-ihr-emergency-committee-on-covid-19-14-july-2021 (accessed on 10 December 2021).
- Coronavirus Disease 2019 (COVID-19)|CDC. Available online: https://www.cdc.gov/coronavirus/2019-ncov/index.html (accessed on 7 August 2021).
- Callaway, E. Delta coronavirus variant: Scientists brace for impact. Nature 2021, 595, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Deng, A.; Li, K.; Hu, Y.; Li, Z.; Xiong, Q.; Liu, Z.; Guo, Q.; Zou, L.; Zhang, H.; et al. Viral infection and transmission in a large, well-traced outbreak caused by the SARS-CoV-2 Delta variant. MedRxiv 2021. [Google Scholar] [CrossRef]
- COVID-19: What Is the Mu Variant? United Nations Western Europe. Available online: https://unric.org/en/covid-19-what-is-the-mu-variant/ (accessed on 25 September 2021).
- Guruprasad, L. Human SARS CoV-2 spike protein mutations. Proteins Struct. Funct. Bioinform. 2021, 89, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Cellular and Molecular Immunology; Saunders Elsevier: Philadelphia, PA, USA, 2007; p. 566. [Google Scholar]
- Doan, T.; Melvold, R.; Viselli, S. Lippincott’s Illustrated Reviews, Immunology, 2nd ed.; Wolter Kluwel: Alphen aan den Rijn, The Netherlands, 2012; ISBN 9781451109375. [Google Scholar]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Basic Immunology: Functions and Disorders of the Immune System; Elsevier Slanders Publishing: Amsterdam, The Netherlands, 2015; ISBN 9780323400152. [Google Scholar]
- Barlow, D.J.; Edwards, M.S.; Thornton, J. Continuous and discontinuous protein antigenic determinants. Nature 1986, 322, 747–748. [Google Scholar] [CrossRef]
- BioRender Templates. Available online: https://app.biorender.com/biorender-templates (accessed on 26 September 2021).
- Mix, E.; Goertsches, R.; Zettl, U.K. Immunoglobulins—Basic considerations. J. Neurol. 2006, 253 (Suppl. 5), V9–V17, Erratum in J. Neurol. 2008, 255, 308. [Google Scholar] [CrossRef] [PubMed]
- A Compact Vocabulary of Paratope-Epitope Interactions Enables Predictability of Antibody-Antigen Binding|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S2211124721001704?token=74748F25258D74599D0802A9AFA03C34793008C315DF289599AE40FDBA0AF1A482C4B92C75ADC47372988E9FABB4A34B&originRegion=eu-west-1&originCreation=20210904091233 (accessed on 4 September 2021).
- Ravetch, J.V.; Bolland, S. IgG Fc Receptors. Annu. Rev. Immunol. 2001, 19, 275–290. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. Immunobiology: The Immune System in Health and Disease, 5th ed.; Garland Science: New York, NY, USA, 2001; Available online: https://www.ncbi.nlm.nih.gov/books/NBK10757/ (accessed on 12 October 2021).
- Al Qaraghuli, M.M.; Kubiak-Ossowska, K.; Ferro, V.A.; Mulheran, P.A. Antibody-protein binding and conformational changes: Identifying allosteric signalling pathways to engineer a better effector response. Sci. Rep. 2020, 10, 13696. [Google Scholar] [CrossRef]
- Introduction to Antigen-Antibody Reactions. Available online: https://microbenotes.com/introduction-to-antigen-antibody-reactions/ (accessed on 4 September 2021).
- An Introduction to Antibodies: Antibody-Antigen Interaction. Available online: https://www.sigmaaldrich.com/IN/en/technical-documents/technical-article/protein-biology/elisa/antibody-antigen-interaction (accessed on 4 September 2021).
- Roper, R.L.; Rehm, K.E. SARS vaccines: Where are we? Expert Rev. Vaccines 2009, 8, 887–898. [Google Scholar] [CrossRef]
- Shang, W.; Yang, Y.; Rao, Y.; Rao, X. The outbreak of SARS-CoV-2 pneumonia calls for viral vaccines. NPJ Vaccines 2020, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. iBCE-EL: A New Ensemble Learning Framework for Improved Linear B-Cell Epitope Prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larrañaga, P.; Calvo, B.; Santana, R.; Bielza, C.; Galdiano, J.; Inza, I.; Lozano, J.A.; Armañanzas, R.; Santafé, G.; Pérez, A.; et al. Machine learning in bioinformatics. Brief. Bioinform. 2006, 7, 86–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha-Neto, E.; Rosa, D.; Harris, P.; Olson, T.; Morrow, A.; Ciotlos, S.; Herst, C.V.; Rubsamen, R.M. An Approach for a Synthetic CTL Vaccine Design against Zika Flavivirus Using Class I and Class II Epitopes Identified by Computer Modeling. Front. Immunol. 2017, 8, 640. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Shi, K.; Li, W. Deep learning methods improve linear B-cell epitope prediction. BioData Min. 2020, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Fatoba, A.J.; Maharaj, L.; Adeleke, V.T.; Okpeku, M.; Adeniyi, A.A.; Adeleke, M.A. Immunoinformatics prediction of overlapping CD8+ T-cell, IFN-γ and IL-4 inducer CD4+ T-cell and linear B-cell epitopes based vaccines against COVID-19 (SARS-CoV-2). Vaccine 2021, 39, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Moody, R.; Wilson, K.L.; Boer, J.C.; Holien, J.K.; Flanagan, K.L.; Jaworowski, A.; Plebanski, M. Predicted B Cell Epitopes Highlight the Potential for COVID-19 to Drive Self-Reactive Immunity. Front. Bioinform. 2021, 1, 31. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Mahajan, S.; Peters, B.; Nielsen, M.; Marcatili, P. Antibody Specific B-Cell Epitope Predictions: Leveraging Information from Antibody-Antigen Protein Complexes. Front. Immunol. 2019, 10, 298. [Google Scholar] [CrossRef]
- Liu, L.-Y.; Yang, H.-G.; Cheng, B. Prediction of Linear B-cell Epitopes Based on PCA and RNN Network. In Proceedings of the 2019 IEEE 7th International Conference on Bioinformatics and Computational Biology (ICBCB), Hangzhou, China, 21–23 March 2019. [Google Scholar]
- Cheng, B.; Liu, L.-Y.; Qi, Z.-H.; Yang, H.-G. Prediction of Continuous B-cell Epitopes Using Long Short Term Memory Networks. In Proceedings of the 2018 6th International Conference on Bioinformatics and Computational Biology, Chengdu, China, 12–14 March 2018; pp. 55–59. [Google Scholar] [CrossRef]
- Hu, Y.-J.; You, S.-N.; Ko, C.-L. Computational Ensemble Approach for Immune System Study: Conformational B-cell Epitope Prediction. Eur. J. Biomed. Inform. 2017, 14, 4–15. [Google Scholar] [CrossRef]
- Ren, J.; Song, J.; Ellis, J.; Li, J. Staged heterogeneity learning to identify conformational B-cell epitopes from antigen sequences. BMC Genom. 2017, 18, 113. [Google Scholar] [CrossRef] [Green Version]
- Georgios, A.; Rooman, D.M. SEPIa, a knowledge-driven algorithm for predicting conformational B-cell epitopes from the amino acid sequence. BMC Bioinform. 2017, 18, 95. [Google Scholar]
- Sher, G.; Zhi, D.; Zhang, S. DRREP: Deep ridge regressed epitope predictor. BMC Genom. 2017, 18, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Niu, Y.; Zou, H.; Luo, L.; Liu, Q.; Wu, W. Accurate Prediction of Immunogenic T-Cell Epitopes from Epitope Sequences Using the Genetic Algorithm-Based Ensemble Learning. PLoS ONE 2015, 10, e0128194. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhang, C.; Hanlon, M.; Ruan, J.; Gao, J. An ensemble method for prediction of con-formational B-cell epitopes from antigen sequences. Comput. Biol. Chem. 2014, 49, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, X.; Sun, P.; Gao, B.; Ma, Z. Conformational B-Cell Epitopes Prediction from Sequences Using Cost-Sensitive Ensemble Classifiers and Spatial Clustering. BioMed Res. Int. 2014, 2014, 689219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O.; Nielsen, M. An integrative approach to CTL epitope prediction: A combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur. J. Immunol. 2005, 35, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Reche, P.; Reinherz, E.L. Sequence Variability Analysis of Human Class I and Class II MHC Molecules: Functional and Structural Correlates of Amino Acid Polymorphisms. J. Mol. Biol. 2003, 331, 623–641. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Udaka, K.; Mamitsuka, H.; Zhu, S. Toward more accurate pan-specific MHC-peptide binding prediction: A review of current methods and tools. Brief. Bioinform. 2012, 13, 350–364. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Sidney, J.; Dow, C.; Mothe, B.; Sette, A.; Peters, B. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 2008, 4, e1000048. [Google Scholar] [CrossRef] [Green Version]
- Atanasova, M.; Patronov, A.; Dimitrov, I.; Flower, D.R.; Doytchinova, I. EpiDOCK: A molecular docking-based tool for MHC class II binding prediction. Protein Eng. Des. Sel. 2013, 26, 631–634. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Xiang, Z.; Mobley, H.L.T. Vaxign: The first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J. Biomed. Biotechnol. 2010, 2010, 297505. [Google Scholar] [CrossRef]
- Reche, P.A.; Reinherz, E.L. PEPVAC: A web server for multi-epitope vaccine development based on the prediction of supertypic MHC ligands. Nucleic Acids Res. 2005, 33, W138–W142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.M.M.; Lafuente, E.M.; Flower, D.R.; Reche, P.A. Selection of Conserved Epitopes from Hepatitis C Virus for Pan-Populational Stimulation of T-Cell Responses. Clin. Dev. Immunol. 2013, 2013, 601943. [Google Scholar] [CrossRef]
- Hakenberg, J.; Nussbaum, A.K.; Schild, H.; Rammensee, H.-G.; Kuttler, C.; Holzhütter, H.-G.; Kloetzel, P.-M.; Kaufmann, S.H.; Mollenkopf, H.-J. MAPPP: MHC class I antigenic peptide processing prediction. Appl. Bioinform. 2003, 2, 155–158. [Google Scholar]
- Oyarzún, P.; Ellis, J.J.; Bodén, M.; Kobe, B. PREDIVAC: CD4+ T-cell epitope prediction for vaccine design that covers 95% of HLA class II DR protein diversity. BMC Bioinform. 2013, 14, 52. [Google Scholar] [CrossRef]
- Rammensee, H.-G.; Bachmann, J.; Emmerich, N.P.N.; Bachor, O.A.; Stevanović, S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics 1999, 50, 213–219. [Google Scholar] [CrossRef]
- Reche, P.A.; Glutting, J.P.; Zhang, H.; Reinherz, E.L. Enhancement to the RANKPEP resource for the prediction of pep-tide binding to MHC molecules using profiles. Immunogenetics 2004, 56, 405–419. [Google Scholar] [CrossRef] [Green Version]
- Yusim, K.; Korber, B.T.; Brander, C.; Barouch, D.; de Boer, R.; Haynes, B.F.; Koup, R.; Moore, J.P.; Walker, B.D.; Watkins, D. HIV Molecular Immunology; Los Alamos National Lab: Los Alamos, NM, USA, 2015. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Guan, P.; Flower, D.R. EpiJen: A server for multistep T cell epitope prediction. BMC Bioinform. 2006, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Raghava, G.P.S. ProPred: Prediction of HLA-DR binding sites. Bioinformatics 2001, 17, 1236–1237. [Google Scholar] [CrossRef] [Green Version]
- Sturniolo, T.; Bono, E.; Ding, J.; Raddrizzani, L.; Tuereci, O.; Sahin, U.; Braxenthaler, M.; Gallazzi, F.; Protti, M.P.; Sinigaglia, F.; et al. Generation of tissue specific and promiscuous HLA ligand databases using DNA mi-croarrays and virtual HLA class II matrices. Nat. Biotechnol. 1999, 17, 555–561. [Google Scholar] [CrossRef]
- Singh, H.; Raghava, G. ProPred1: Prediction of promiscuous MHC Class-I binding sites. Bioinformatics 2003, 19, 1009–1014. [Google Scholar] [CrossRef]
- Parker, K.C.; Bednarek, M.A.; Coligan, J.E. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J. Immunol. 1994, 152, 163–175. [Google Scholar] [PubMed]
- Dimitrov, I.; Garnev, P.; Flower, D.R.; Doytchinova, I. EpiTOP—A proteochemometric tool for MHC class II binding prediction. Bioinformatics 2010, 26, 2066–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, P.; Doytchinova, I.; Zygouri, C.; Flower, D.R. MHCPred: A server for quantitative prediction of peptide-MHC binding. Nucleic Acids Res. 2003, 31, 3621–3624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.L.; DeLuca, D.S.; Keskin, D.B.; Chitkushev, L.; Zlateva, T.; Lund, O.; Reinherz, E.L.; Brusic, V. MULTIPRED2: A computational system for large-scale identification of peptides predicted to bind to HLA supertypes and alleles. J. Immunol. Methods 2011, 374, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Worning, P.; Lauemøller, S.L.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003, 12, 1007–1017. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Lamberth, K.; Harndahl, M.; Justesen, S.; Røder, G.; Peters, B.; Sette, A.; Lund, O.; et al. NetMHCpan, a Method for Quantitative Predictions of Peptide Binding to Any HLA-A and -B Locus Protein of Known Sequence. PLoS ONE 2007, 2, e796. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Lund, O. Prediction of MHC class II binding affinity using SMM-align, a novel stabilization matrix alignment method. BMC Bioinform. 2007, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Peters, B.; Sette, A.; Justesen, S.; Buus, S.; Lund, O. Quantitative Predictions of Peptide Binding to Any HLA-DR Molecule of Known Sequence: NetMHCIIpan. PLoS Comput. Biol. 2008, 4, e1000107. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.; Flower, D.R. In Silico Identification of Supertypes for Class II MHCs. J. Immunol. 2005, 174, 7085–7095. [Google Scholar] [CrossRef] [Green Version]
- Dhanda, S.K.; Gupta, S.; Vir, P.; Raghava, G.P.S. Prediction of IL4 Inducing Peptides. Clin. Dev. Immunol. 2013, 2013, 263952. [Google Scholar] [CrossRef]
- Dönnes, P.; Kohlbacher, O. Integrated modeling of the major events in the MHC class I antigen processing pathway. Protein Sci. 2005, 14, 2132–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Meng, X.; Xu, Q.; Flower, D.R.; Li, T. Quantitative prediction of mouse class I MHC peptide binding affinity using support vector machine regression (SVR) models. BMC Bioinform. 2006, 7, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dönnes, P.; Elofsson, A. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinform. 2002, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, M.; Raghava, G.P.S. SVM based method for predicting HLA-DRB1*0401 binding peptides in an antigen sequence. Bioinformatics 2004, 20, 421–423. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, P.; Kim, Y.; Haste-Andersen, P.; Beaver, J.; Bourne, P.E.; Bui, H.-H.; Buus, S.; Frankild, S.; Greenbaum, J.; et al. Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 2008, 36, W513–W518. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni-Kale, U.; Bhosle, S.; Kolaskar, A.S. CEP: A conformational epitope prediction server. Nucleic Acids Res. 2005, 33, W168–W171. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Alix, A.J.P. Predictive estimation of protein linear epitopes by using the program PEOPLE. Vaccine 1999, 18, 311–314. [Google Scholar] [CrossRef]
- Singh, H.; Ansari, H.R.; Raghava, G.P.S. Improved Method for Linear B-Cell Epitope Prediction Using Antigen’s Primary Sequence. PLoS ONE 2013, 8, e62216. [Google Scholar] [CrossRef] [Green Version]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A Method to Predict Antigenic Epitopes Using Support Vector Machine to Integrate Tri-Peptide Similarity and Propensity. PLoS ONE 2012, 7, e45152. [Google Scholar] [CrossRef] [Green Version]
- El-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. 2008, 21, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.H.; Nielsen, M.; Lund, O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006, 15, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Sweredoski, M.J.; Baldi, P. PEPITO: Improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics 2008, 24, 1459–1460. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, J.V.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, N.D.; Mayrose, I.; Martz, E.; Pupko, T. Epitopia: A web-server for predicting B-cell epitopes. BMC Bioinform. 2009, 10, 287. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, K.; Liu, X.; Baker, T.; Shi, J.; Deane, C.M. Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics 2014, 30, 2288–2294. [Google Scholar] [CrossRef]
- Liang, S.; Zheng, D.; Standley, D.M.; Yao, B.; Zacharias, M.; Zhang, C. EPSVR and EPMeta: Prediction of antigenic epitopes using support vector regression and multiple server results. BMC Bioinform. 2010, 11, 381. [Google Scholar] [CrossRef] [Green Version]
- Mayrose, I.; Penn, O.; Erez, E.; Rubinstein, N.D.; Shlomi, T.; Freund, N.T.; Bublil, E.M.; Ruppin, E.; Sharan, R.; Gershoni, J.M.; et al. Pepitope: Epitope mapping from affinity-selected peptides. Bioinformatics 2007, 23, 3244–3246. [Google Scholar] [CrossRef] [Green Version]
- CBTOPE- Prediction of Conformational B-cell Epitopes. Retrieved 26 October 2021. Available online: https://webs.iiitd.edu.in/raghava/cbtope/ (accessed on 10 December 2021).
- Negi, S.S.; Braun, W. Automated Detection of Conformational Epitopes Using Phage Display Peptide Sequences. Bioinform. Biol. Insights 2009, 3, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Pellequer, J.-L.; Westhof, E. PREDITOP: A program for antigenicity prediction. J. Mol. Graph. 1993, 11, 204–210. [Google Scholar] [CrossRef]
- Blythe, M.J.; Flower, D.R. Benchmarking B cell epitope prediction: Underperformance of existing methods. Protein Sci. 2005, 14, 246–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenbaum, J.A.; Andersen, P.H.; Blythe, M.; Bui, H.-H.; Cachau, R.E.; Crowe, J.; Davies, M.; Kolaskar, A.S.; Lund, O.; Morrison, S.; et al. Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J. Mol. Recognit. 2007, 20, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Levitt, M. Nature of the protein universe. Proc. Natl. Acad. Sci. USA 2009, 106, 11079–11084. [Google Scholar] [CrossRef] [Green Version]
- Sela-Culang, I.; Ofran, Y.; Peters, B. Antibody specific epitope prediction—Emergence of a new paradigm. Curr. Opin. Virol. 2015, 11, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Ansari, H.R.; Raghava, G.P. Identification of conformational B-cell Epitopes in an antigen from its primary sequence. Immunome Res. 2010, 6, 6–9. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Lineburg, K.E.; Grant, E.J.; Swaminathan, S.; Chatzileontiadou, D.S.; Szeto, C.; Sloane, H.; Panikkar, A.; Raju, J.; Crooks, P.; Rehan, S.; et al. CD8+ T cells specific for an immunodominant SARS-CoV-2 nucleocapsid epitope cross-react with selective seasonal coronaviruses. Immunity 2021, 54, 1055–1065.e5. [Google Scholar] [CrossRef]
- Zhang, X.; Tan, Y.; Ling, Y.; Lu, G.; Liu, F.; Yi, Z.; Jia, X.; Wu, M.; Shi, B.; Xu, S.; et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 2020, 583, 437–440. [Google Scholar] [CrossRef]
- Schmidt, M.E.; Varga, S.M. The CD8 T Cell Response to Respiratory Virus Infections. Front. Immunol. 2018, 9, 678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, O.-W.; Chia, A.; Tan, A.T.; Jadi, R.S.; Leong, H.N.; Bertoletti, A.; Tan, Y.-J. Memory T cell responses targeting the SARS coronavirus persist up to 11 years post-infection. Vaccine 2016, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.E.; Beek, J.E.; de Jonge, J.; Eluytjes, W.; Baarle, D.E. T Cell Responses to Viral Infections—Opportunities for Peptide Vaccination. Front. Immunol. 2014, 5, 171. [Google Scholar] [CrossRef]
- Seder, R.A.; Darrah, P.A.; Roederer, M. T-cell quality in memory and protection: Implications for vaccine design. Nat. Rev. Immunol. 2008, 8, 247–258. [Google Scholar] [CrossRef]
- Saqib, M.; Faraz, S.; Abdul, A.; Mckay, M.R. In silico T cell epitope identification for SARS-CoV-2: Progress and perspectives. Adv. Drug Deliv. Rev. 2021, 171, 29–47. [Google Scholar]
- Stranzl, T.; Larsen, M.V.; Lundegaard, C.; Nielsen, M. NetCTLpan: Pan-specific MHC class I pathway epitope predictions. Immunogenetics 2010, 62, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Croft, N.P.; Purcell, A.W.; Tscharke, D.C.; Sette, A.; Nielsen, M.; Peters, B. Benchmarking predictions of MHC class I restricted T cell epitopes in a comprehensively studied model system. PLoS Comput. Biol. 2020, 16, e1007757. [Google Scholar] [CrossRef]
- Abelin, J.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst. 2018, 7, 129–132.e4. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.K.; Andreatta, M.; Marcatili, P.; Buus, S.; Greenbaum, J.A.; Yan, Z.; Sette, A.; Peters, B.; Nielsen, M. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 2018, 154, 394–406. [Google Scholar] [CrossRef]
- Karosiene, E.; Rasmussen, M.; Blicher, T.; Lund, O.; Buus, S.; Nielsen, M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 2013, 65, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef]
- Abelin, J.; Harjanto, D.; Malloy, M.; Suri, P.; Colson, T.; Goulding, S.P.; Creech, A.L.; Serrano, L.R.; Nasir, G.; Nasrullah, Y.; et al. Defining HLA-II Ligand Processing and Binding Rules with Mass Spectrometry Enhances Cancer Epitope Prediction. Immunity 2019, 51, 766–779.e17. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Olsson, N.; Wagar, L.; Fast, E.; Liu, C.L.; Muftuoglu, Y.; Sworder, B.; Diehn, M.; Levy, R.; et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019, 37, 1332–1343. [Google Scholar] [CrossRef]
- Nielsen, M.; Lundegaard, C.; Lund, O.; Keşmir, C. The role of the proteasome in generating cytotoxic T-cell epitopes: Insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005, 57, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 2007, 8, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reche, P.A.; Glutting, J.P.; Reinherz, E.L. Prediction of MHC class I binding peptides using profile motifs. Hum. Immunol. 2002, 63, 701–709. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef]
- Crooke, S.; Ovsyannikova, I.G.; Kennedy, R.B.; Poland, G.A. Immunoinformatic identification of B cell and T cell epitopes in the SARS-CoV-2 proteome. Sci. Rep. 2020, 10, 14179. [Google Scholar] [CrossRef]
- Bukhari, S.N.H.; Jain, A.; Haq, E. A Novel Ensemble Machine Learning Model for Prediction of Zika Virus T-Cell Epitopes. In Lecture Notes on Data Engineering and Communications Technologies; Gupta, D., Polkowski, Z., Khanna, A., Bhattacharyya, S., Castillo, O., Eds.; Springer: Singapore, 2022; Volume 91, pp. 275–292. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- The Effects of Virus Variants on COVID-19 Vaccines. Available online: https://www.who.int/news-room/feature-stories/detail/the-effects-of-virus-variants-on-covid-19-vaccines (accessed on 7 August 2021).
- Wee, L.J.; Simarmata, D.; Kam, Y.W.; Ng, L.F.; Tong, J.C. SVM-based prediction of linear B-cell epitopes using Bayes Feature Extraction. BMC Genom. 2010, 11, S21. [Google Scholar] [CrossRef] [Green Version]
- Nisar, S.; Bukhari, H.; Dar, M.A. Using Random Forest to Predict T -Cell Epitopes of Dengue Virus. Dengue Virus 2021, 20, 2543–2547. [Google Scholar]
- Artificial Neural Network Disadvantages. Retrieved 4 September 2021. Available online: https://www.datascienceexamples.com/artificial-neural-network-disadvantages/ (accessed on 10 December 2021).
- Linardatos, P.; Papastefanopoulos, V.; Kotsiantis, S. Explainable AI: A Review of Machine Learning Interpretability Methods. Entropy 2021, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Gagniuc, P.A.; Ionescu-Tirgoviste, C.; Gagniuc, E.; Militaru, M.; Nwabudike, L.C.; Pavaloiu, B.I.; Vasilăţeanu, A.; Goga, N.; Drăgoi, G.; Popescu, I.; et al. Spectral forecast: A general purpose prediction model as an alternative to classical neural networks. Chaos Interdiscip. J. Nonlinear Sci. 2020, 30, 033119. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.N.H.; Jain, A.; Haq, E.; Khder, M.A.; Neware, R.; Bhola, J.; Najafi, M.L. Machine Learning-Based Ensemble Model for Zika Virus T-Cell Epitope Prediction. J. Health Eng. 2021, 2021, 9591670. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Xie, G.; Xiao, R. Research on Ensemble Learning. In Proceedings of the 2009 International Conference on Artificial Intelligence and Computational Intelligence, Shanghai, China, 7–8 November 2009; Volume 3, pp. 249–252. [Google Scholar]
- A Gentle Introduction to Ensemble Learning Algorithms. Available online: https://machinelearningmastery.com/tour-of-ensemble-learning-algorithms (accessed on 8 September 2021).
- Dong, X.; Yu, Z.; Cao, W.; Shi, Y.; Ma, Q. A survey on ensemble learning. Front. Comput. Sci. 2020, 14, 241–258. [Google Scholar] [CrossRef]
- Why Use Ensemble Learning? Available online: https://machinelearningmastery.com/why-use-ensemble-learning/ (accessed on 10 July 2021).
- Osorio, D.; Rondón-Villarreal, P.; Torres, R.T.R. Peptides: A Package for Data Mining of Antimicrobial Peptides. Small 2015, 12, 44–444. [Google Scholar] [CrossRef]
- Hofmann, H.; Hare, E.; GGobi Foundation. Peptider: Evaluation of Diversity in Nucleotide Libraries. R Package Version 0.2.2. 2015. Available online: https://CRAN.R-project.org/package=peptider (accessed on 10 September 2021).
- Jain, P.; Chawla, P. A Novel Smart Healthcare System Design for Internet of Health Things. In Proceedings of the 2021 International Conference on Innovative Computing, Intelligent Communication and Smart Electrical Systems (ICSES), Chennai, India, 24–25 September 2021; pp. 1–8. [Google Scholar]
- Bukhari, S.N.H.; Jain, A.; Haq, E.; Mehbodniya, A.; Webber, J. Ensemble Machine Learning Model to Predict SARS-CoV-2 T-Cell Epitopes as Potential Vaccine Targets. Diagnostics 2021, 11, 1990. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Study Conducted | Methodology Adopted | Strengths/Limitations |
|---|---|---|
| T. Liu et al. [23] | A feedforward deep neural network-based ensemble of 11 classifiers was created to predict BCEs. IEDB was used to obtain the BCE peptide dataset. On the test set, the model was evaluated using the AUROC metric. | Model reports peptide as an epitope if classified by all 11 classifiers. It would provide the best results if simple majority voting was used for classification. |
| Fatoba, A. J. et al. [24] | In [24], potential epitope-based vaccine candidates were explored. After retrieving 600 genome sequences of SARS-CoV-2 from the ViPR repository, CD8+ and CD4+ epitopes and B-cell (linear) epitopes were generated and screened for immunogenicity, antigenicity, and non-allergenicity. | The results of [25] reported 19 candidate T-cell epitopes (CD8+), which were found to overlap strongly with 8 B-cell epitopes. The results provide the basis for an experimental design for a suitable peptide vaccine against SARS-CoV-2. |
| R. Moody et al. [26] | Authors used IEDB prediction tools for predicting B-cell epitopes and those with high scores in terms of prediction were selected as candidate epitopes. The epitopes were then matched to human proteins using NCBI Blast technology. | The findings showed eleven (11) novel B-cell epitopes in the host that were capable of explaining key elements of COVID-19 extrapulmonary disease that previous research had not been able to explain. |
| Jespersen MC et al. [27] | The authors employed feedforward neural networks (FFNN) with two hidden layers, each with 25 neurons, an activation function (sigmoid) at all neurons, and an ADAM as an optimizing function to predict antibody-specific epitopes (B cell) or epitope targets of provided cognate antibodies. The dataset was obtained from the IEDB database. PCA was used for dimensionality reduction before the model was trained. | It was shown that a simple set of attributes retrieved from the cognate antibody boosted the rate of accuracy in predicting individual epitopes. Furthermore, sophisticated features such as Zernike Moments can improve the model’s predictive potential. When compared to DiscoTope 2.0, this model performs better in finding patches overlapping with an actual patch of an epitope in cross-validation and on an independent dataset. |
| Ling-yun Liu et al. [28] | The authors used PCA and RNN networks. They converted the physicochemical properties into digital vectors, intending to have high-dimensional feature space, and later PCA was applied to process them. The output from PCA was used as an input to the RNN for predicting epitopes. | Prediction results obtained by this process demonstrated that PCA reduced dimensions, but at the same time, original features of the main component were retained, and the rate of prediction was also improved. |
| Bin Cheng et al. [29] | Authors introduced a novel scale to measure feature importance, called the relevance of amino acid pair (RAAP). RAAP was calculated by decomposing the sequences of amino acids based on their physicochemical properties. | The successful prediction rate was drastically improved here by using LSTM. It does not suffer from gradient instability and is good enough for textual classification sequences. Fivefold cross-validation was used to test and validate the models. |
| Balachandran Manavalan et al. [30] | Here, a non-redundant dataset was constructed containing 5500 BCEs experimentally validated, and 6893 non-B-cell epitopes were retrieved from IEDB. Then, an ensemble model to predict B-cell epitopes based on ERT (extremely randomized tree) and a classifier called GB (gradient boosting) was developed. The model works based on the physicochemical properties, AA composition, and combination of dipeptides and PCP as the input features. | After performing cross-validation on a benchmark dataset, it was shown that this model performed far better than the individual classifiers such as ERT and GB, with an MCC (Matthews correlation coefficient) of 0.454. |
| Yuh-Jyh Hu et al. [31] | A cost-sensitive strategy based on bagging MDT was suggested, which integrates two ensemble-based learning algorithms. Without employing the prediction of a pre-trained single predictor, it makes it independent of multiple prediction tools. It can also learn a meta-classification architecture with varied data, without being constrained by a particular hierarchy. | It was demonstrated that the performance of prediction is superior as compared to a single epitope predictor. However, epitope prediction based on meta-learning is purely dependent upon the predictive strength of various other pre-trained linear and conformational epitope prediction tools, which cannot be retained directly by users. Hence, this limits the flexibility and applicability of these meta-classifiers. |
| Jing Ren et al. [32] | The authors proposed a novel staged heterogeneity-based learning model. The model learns both heterogeneity and characteristics of data in a phased manner to identify residue of antigens of conformational B-cell type epitopes that are heterogeneous, purely based on sequences of antigens. In the first stage, the model is made to learn the generic epitope pattern with propensities, and in the second stage, the same model is made to learn the complementarity of the propensities used in the first stage, which is heterogeneous but this time on a small dataset of experimentally verified epitopes. | It was demonstrated that if heterogeneity was learned well, the transferability of the model improved remarkably in handling new data.It was tested and validated on two different datasets: one with epitopes determined experimentally and another with computationally defined. It showed outstanding performance that was around twice that of existing predictors, including CBTOPE. |
| Georgios A. et al. [33] | A novel method, “SEPIa”, has been proposed here to predict B-cell epitopes from protein sequences and is sufficiently faster, and it can also be applied to large-scale datasets. The model is the combination of two classifiers, random forest and naïve Bayes algorithm. | The average prediction accuracy of SEPIa is limited. The AUC score is 0.65 in both 10-fold cross-validation and on the independent test dataset, which is higher than other approaches tested on the same test dataset. |
| Gene Sher et al. [25] | Authors proposed a novel, analytically trained DREEP (Deep Ridge Regressed Epitope Predictor) based on string kernels using a deep neural network tailored to predict continuous epitopes. | The model was tested with input as long sequences of proteins from datasets such as AntiJen, Pellequer, and HIV. The results were compared with epitope predictors such as DMNLBE, LBtope, etc. Using the area under the curve (AUC) metric, the model achieved performance improvements over SARS by 13.7%, HIV by 8.9%, and Pellequer by 1.5%. |
| Wen Zhang et al. [34] | Authors attempted to differentiate immunogenic epitopes from non-immunogenic epitopes based purely on their primary structure. To effectively utilize various features, an ensemble method based on a genetic algorithm was proposed. | The model was tested on two benchmark datasets: IMMA2, PAAQD. The model was compared with methods such as POPI, PAAQD, and POPISK, which are considered state-of-the-art in nature. The model performed better, with an AUC score on IMMA2 of 0.846 and 0.829 on PAAQD. |
| Wei Zheng et al. [35] | The authors used ensemble learning to improve the prediction of BCEs. Their ensemble method combined twelve SVMs. To handle imbalanced datasets, resampling and AdaBoost methods were used. | The proposed ensemble model achieved an AUC score of 0.642–0.672 on the training dataset with five-fold cross-validation and an AUC score of 0.579–0.604 on the test dataset. |
| Jian Zhang et al. [36] | To predict antigenic determinants, the authors devised a cost-sensitive ensemble approach, and a spatial clustering-based algorithm was used to identify probable epitopes. | The model performed admirably in terms of prediction. AUC scores of 0.721 and 0.703 were obtained using leave-one-out cross-validation (LOOCV) on two benchmark datasets: bound and unbound. |
| Kavitha K V et al. [37] | PCA was used to reduce dimensions and to filter out the essential features; for prediction purposes, a random forest algorithm was used. | Experimental results showed that the random forest-based classifier had an improved prediction accuracy rate as compared to BCPred, AAP, etc. |
| Wen Zhang et al. [38] | The authors used sequence-derived features and developed an ensemble model based on random forest to predict epitopes accurately. | The model was evaluated using the leave-one-out cross-validation procedure, and an AUC score of 0.687 and 0.651 on bound and unbound datasets was obtained. |
| Ping Chen et al. [39] | Authors reviewed various prediction models for epitopes, such as models based on SVM, neural network, random forest, etc., to defend computational approaches in the prediction of epitopes as in silico methods require a lot of effort and time. | Apart from defending the computational approaches, it was also concluded that there is a limitation to current models as it is impossible to devise an exact model without having complete knowledge of the immune system, and current models are simply best at approximation. |
| Claus Lundegaard et al. [40] | Here, an artificial neural network was used. The standard feedforward neural network with backpropagation was employed to predict epitopes. The dataset was retrieved from the SYFPEITHI database. | The model efficiently and accurately predicts MHC class I type peptides and outperforms the existing methods. |
| Tool Name | Web URL | MHC Class Prediction Supported (MHC I or MHC II or Both) | S | A | P | T |
|---|---|---|---|---|---|---|
| Structure-based | ||||||
| EpiDOCK [45] | epidock.ddg-pharmfac.net, accessed on 10 December 2021 | II | - | - | - | - |
| MM-based | ||||||
| Vaxign [46] | www.violinet.org/vaxign/, accessed on 10 December 2021 | Both | - | - | - | - |
| PEPVAC [47] | imed.med.ucm.es/PEPVAC/, accessed on 10 December 2021 | I | X | - | X | - |
| EPISOPT [48] | bio.med.ucm.es/episopt.htmL, accessed on 10 December 2021 | I | X | - | - | - |
| MAPPP [49] | mpiib-berlin.mpg.de/MAPPP/, accessed on 10 December 2021 | I | X | - | X | - |
| PREDIVAC [50] | predivac.biosci.uq.edu.au/, accessed on 10 December 2021 | II | - | - | - | - |
| SYFPEITHI [51] | syfpeithi.de, accessed on 10 December 2021 | Both | - | - | - | - |
| Rankpep [52] | imed.med.ucm.es/Tools/rankpep.html, accessed on 10 December 2021 | Both | - | - | X | - |
| SM-based | ||||||
| MotifScan [53] | www.hiv.lanl.gov/content/immunology/motif_scan/motif_scan, accessed on 10 December 2021 | Both | X | - | - | - |
| QAM-based | ||||||
| EpiJen [54] | ddg-pharmfac.net/epijen/EpiJen/EpiJen.htm, accessed on 10 December 2021 | I | - | X | X | X |
| Propred [55] | imtech.res.in/raghava/propred/, accessed on 10 December 2021 | II | X | X | - | - |
| TEPITOPE [56] | dataminingiip.fudan.edu.cn/service/TEPITOPEpan/TEPITOPEpan.htm, accessed on 10 December 2021 | II | - | X | - | - |
| Propred 1 [57] | http://www.imtech.res.in/raghava/propred1/, accessed on 10 December 2021 | I | X | X | X | - |
| BIMAS [58] | bimas.cit.nih.gov/molbio/hla_bind/, accessed on 10 December 2021 | I | - | X | - | - |
| QSAR-based | ||||||
| EpiTOP [59] | pharmfac.net/EpiTOP, accessed on 10 December 2021 | II | - | X | - | - |
| MHCPred [60] | ddg-pharmfac.net/mhcpred/MHCPred/, accessed on 10 December 2021 | Both | - | X | - | - |
| ANN-based | ||||||
| NetCTL [41] | cbs.dtu.dk/services/NetCTL/, accessed on 10 December 2021 | I | X | X | X | X |
| MULTIPRED2 [61] | cvc.dfci.harvard.edu/multipred2/index.php, accessed on 10 December 2021 | Both | X | - | - | - |
| NetMHC [62] | cbs.dtu.dk/services/NetMHC/, accessed on 10 December 2021 | I | - | X | - | - |
| NetMHCpan [63] | cbs.dtu.dk/services/NetMHCpan/, accessed on 10 December 2021 | I | - | X | - | - |
| NetMHCII [64] | cbs.dtu.dk/services/NetMHCII/, accessed on 10 December 2021 | II | - | X | - | - |
| NetMHCIIpan [65] | cbs.dtu.dk/services/NetMHCIIpan/, accessed on 10 December 2021 | II | - | X | - | - |
| NHLApred [66] | imtech.res.in/raghava/nhlapred/, accessed on 10 December 2021 | I | - | - | X | - |
| SVM-based | ||||||
| IL4pred [67] | webs.iiitd.edu.in/raghava/il4pred/index.php, accessed on 10 December 2021 | II | - | - | - | - |
| WAPP [68] | abi.inf.uni-tuebingen.de/Services/WAPP/index_html, accessed on 10 December 2021 | I | - | - | X | X |
| SVRMHC [69] | us.accurascience.com/SVRMHCdb/, accessed on 10 December 2021 | Both | - | X | - | - |
| SVMHC [70] | abi.inf.uni-tuebingen.de/Services/SVMHC/, accessed on 10 December 2021 | Both | - | - | - | - |
| MHC2PRED [71] | imtech.res.in/raghava/mhc2pred/index.html, accessed on 10 December 2021 | II | - | - | - | - |
| Combined (QAM and ANN) | ||||||
| IEDB-MHCI [72] | tools.immuneepitope.org/mhci/, accessed on 10 December 2021 | I | - | X | - | - |
| IEDB-MHCII [72] | tools.immuneepitope.org/mhcii/, accessed on 10 December 2021 | II | - | X | - | - |
| Tool Name | Web URL | Methodology Used |
|---|---|---|
| Prediction of Linear B-Cell Epitopes | ||
| BepiPred [74] | cbs.dtu.dk/services/BepiPred/, accessed on 10 December 2021 | Decision tree |
| PEOPLE [75] | iedb.org, accessed on 10 December 2021 | Propensity scale |
| LBtope [76] | imtech.res.in/raghava/lbtope/, accessed on 10 December 2021 | ANN |
| SVMTriP [77] | sysbio.unl.edu/SVMTriP/prediction.php, accessed on 10 December 2021 | SVM |
| BCPREDS [78] | ailab.ist.psu.edu/bcpred/, accessed on 10 December 2021 | SVM |
| ABCpred [79] | imtech.res.in/raghava/abcpred/, accessed on 10 December 2021 | ANN |
| Prediction of Conformational B-Cell Epitopes | ||
| DiscoTope [80] | tools.iedb.org/discotope/, accessed on 10 December 2021 | Structure-based (SM) |
| PEPITO [81] | pepito.proteomics.ics.uci.edu/, accessed on 10 December 2021 | SM |
| ElliPro [82] | tools.iedb.org/ellipro/, accessed on 10 December 2021 | SM |
| CEP [73] | bioinfo.ernet.in/cep.htm, accessed on 10 December 2021 | SM |
| EPITOPIA [83] | epitopia.tau.ac.il/, accessed on 10 December 2021 | SM (Naïve Bayes) |
| EPIPRED [84] | opig.stats.ox.ac.uk/webapps/sabdab-sabpred/EpiPred.php, accessed on 10 December 2021 | SM (Docking, ASEP) |
| EPSVR [85] | sysbio.unl.edu/EPSVR/, accessed on 10 December 2021 | SM |
| PEPITOPE [86] | pepitope.tau.ac.il/, accessed on 10 December 2021 | Mimotope |
| CBTOPE [87] | imtech.res.in/raghava/cbtope/submit.php, accessed on 10 December 2021 | SM (SVM) |
| EpiSearch [88] | curie.utmb.edu/episearch.htm, accessed on 10 December 2021 | Mimotope |
| Sr. No. | Method Name | Usage |
|---|---|---|
| 01 | NetMHC [61] | To predict HLA I class or CD8+ SARS-CoV-2 T-cell epitopes |
| 02 | NetMHCpan [62] | |
| 03 | NetCTLpan_1.1 [104] | |
| 04 | NetMHC_4.0 [105] | |
| 05 | HLAthena [106] | |
| 06 | MHCflurry [107] | |
| 07 | NetHMCII_2.3 [108] | To predict HLA II class or CD4+ SARS-CoV-2 T-cell epitopes |
| 08 | NetMHCIIpan_3.0 [109] | |
| 09 | NetMHCIIpan_4.0 [110] | |
| 10 | NeonMHC2 [111] | |
| 11 | MARIA [112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bukhari, S.N.H.; Jain, A.; Haq, E.; Mehbodniya, A.; Webber, J. Machine Learning Techniques for the Prediction of B-Cell and T-Cell Epitopes as Potential Vaccine Targets with a Specific Focus on SARS-CoV-2 Pathogen: A Review. Pathogens 2022, 11, 146. https://doi.org/10.3390/pathogens11020146
Bukhari SNH, Jain A, Haq E, Mehbodniya A, Webber J. Machine Learning Techniques for the Prediction of B-Cell and T-Cell Epitopes as Potential Vaccine Targets with a Specific Focus on SARS-CoV-2 Pathogen: A Review. Pathogens. 2022; 11(2):146. https://doi.org/10.3390/pathogens11020146
Chicago/Turabian StyleBukhari, Syed Nisar Hussain, Amit Jain, Ehtishamul Haq, Abolfazl Mehbodniya, and Julian Webber. 2022. "Machine Learning Techniques for the Prediction of B-Cell and T-Cell Epitopes as Potential Vaccine Targets with a Specific Focus on SARS-CoV-2 Pathogen: A Review" Pathogens 11, no. 2: 146. https://doi.org/10.3390/pathogens11020146