The Phylogeography of MERS-CoV in Hospital Outbreak-Associated Cases Compared to Sporadic Cases in Saudi Arabia

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Data Collection and Data Linkage
2.1.1. MERS-CoV Human Cases Epidemiological Dataset
2.1.2. MERS-CoV Complete Genome Sequences Dataset
2.1.3. Matching Sequences with Human Cases
- Demographic characteristics: age, gender, healthcare worker, contact history;
- Location: county or town, city, province, region;
- Date: collection date of isolate, lab confirmation date (0–10 days after collection date), symptoms onset date (14 days before collection date), death date (after collection date).
- Unique isolates had at least one criterion from group 1 (and) group 2 (and) group 3 matched with a human case;
- Likely unique isolates had at least one criterion from group 2 (and) 3 matched with a human case;
- Possible unique isolates had at least one criterion from group 2 (or) 3 matched with a human case.
2.2. Compilation of MERS-CoV Genetic Sequences
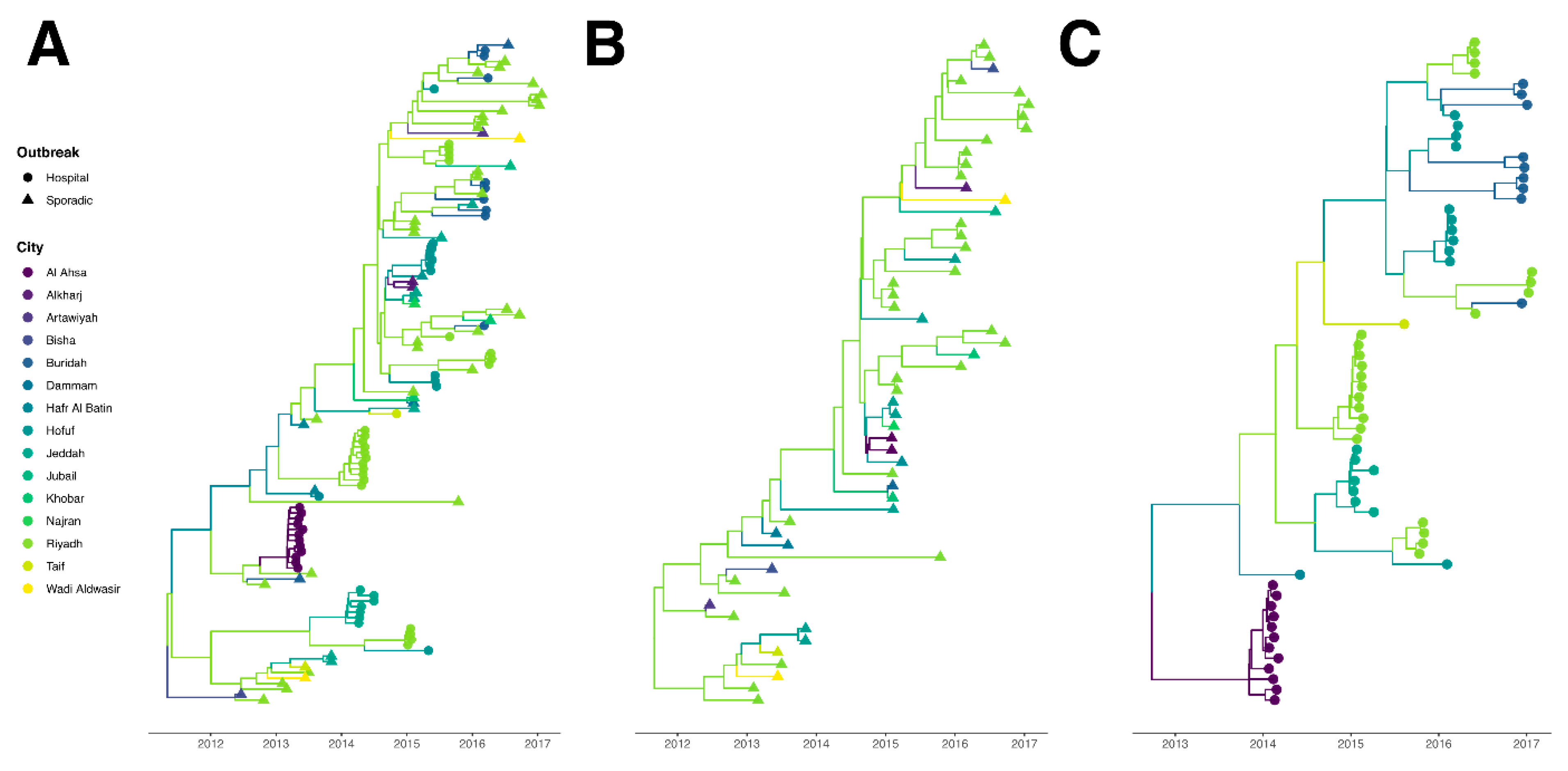
2.3. Comparison of the Phylogeography between Hospital Outbreak-Associated Cases and Sporadic Cases
2.3.1. Temporal Analysis
2.3.2. Discrete Trait Analysis
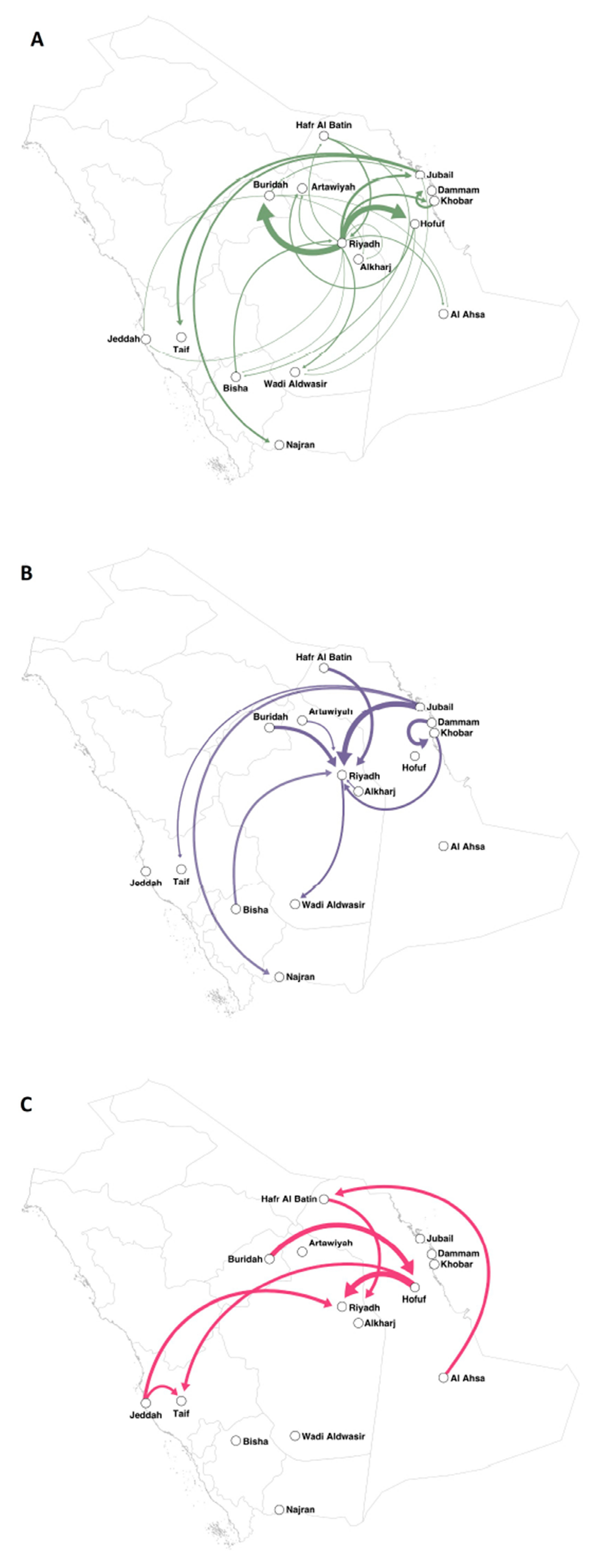
2.3.3. Mapping the Transmission Routes
3. Results
3.1. Data Collection and Data Linkage
3.2. Evolutionary Analysis and Evaluation of Transmission Routes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shehata, M.M.; Gomaa, M.R.; Ali, M.A.; Kayali, G. Middle East respiratory syndrome coronavirus: A comprehensive review. Front. Med. 2016, 10, 120–136. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.-W.; Lau, S.K.-P.; Woo, P.C.-Y. The emerging novel Middle East respiratory syndrome coronavirus: The “knowns” and “unknowns”. J. Med. Assoc. 2013, 112, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. New Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Cotten, M.; Watson, S.J.; Zumla, A.I.; Makhdoom, H.Q.; Palser, A.L.; Ong, S.H.; Al Rabeeah, A.A.; Alhakeem, R.F.; Assiri, A.; Al-Tawfiq, J.A.; et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio 2014, 5, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemi, M.; Fitch, W.M.; Ciccozzi, M.; Ruiz-Alvarez, M.J.; Rezza, G.; Lewis, M.J. Severe acute respiratory syndrome coronavirus sequence characteristics and evolutionary rate estimate from maximum likelihood analysis. J. Virol. 2004, 78, 1602–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobusawa, E.; Sato, K. Comparison of the Mutation Rates of Human Influenza A and B Viruses. J. Virol. 2006, 80, 3675–3678. [Google Scholar] [CrossRef] [Green Version]
- Raj, V.S.; Farag, E.A.; Reusken, C.B.; Lamers, M.M.; Pas, S.D.; Voermans, J.; Smits, S.L.; Osterhaus, A.D.; Al-Mawlawi, N.; Al-Romaihi, H.E.; et al. Isolation of MERS coronavirus from a dromedary camel, Qatar, 2014. Emerg. Infect. Dis. 2014, 20, 1339–1342. [Google Scholar] [CrossRef]
- Chen, X.; Chughtai, A.A.; Dyda, A.; MacIntyre, C.R. Comparative epidemiology of Middle East respiratory syndrome coronavirus (MERS-CoV) in Saudi Arabia and South Korea. Emerg. Microbes Infect. 2017, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- MacIntyre, C.R.; Engells, T.E.; Scotch, M.; Heslop, D.J.; Gumel, A.B.; Poste, G.; Chen, X.; Herche, W.; Steinhöfel, K.; Lim, S. Converging and emerging threats to health security. Env. Syst. Decis. 2018, 38, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Al-Tawfiq, J.A.; Auwaerter, P.G. Healthcare-associated infections: The hallmark of Middle East respiratory syndrome coronavirus with review of the literature. J. Hosp. Infect. 2019, 101, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Assiri, A.; McGeer, A.; Perl, T.M.; Price, C.S.; Al Rabeeah, A.A.; Cummings, D.A.; Alabdullatif, Z.N.; Assad, M.; Almulhim, A.; Makhdoom, H.; et al. Hospital outbreak of Middle East respiratory syndrome coronavirus. New Engl. J. Med. 2013, 369, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Memish, Z.A.; Al-Tawfiq, J.A.; Alhakeem, R.F.; Assiri, A.; Alharby, K.D.; Almahallawi, M.S.; Alkhallawi, M. Middle East respiratory syndrome coronavirus (MERS-CoV): A cluster analysis with implications for global management of suspected cases. Travel Med. Infect. Dis. 2015, 13, 311–314. [Google Scholar] [CrossRef] [PubMed]
- Fagbo, S.F.; Skakni, L.; Chu, D.K.; Garbati, M.A.; Joseph, M.; Peiris, M.; Hakawi, A.M. Molecular Epidemiology of Hospital Outbreak of Middle East Respiratory Syndrome, Riyadh, Saudi Arabia, 2014. Emerg. Infect. Dis. 2015, 21, 1981–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, D.L.; Tokars, J.I.; Abdel Aziz, I.Z.; Alkhaldi, K.Z.; Bensadek, A.T.; Alraddadi, B.M.; Jokhdar, H.; Jernigan, J.A.; Garout, M.A.; Tomczyk, S.M.; et al. Outbreak of Middle East Respiratory Syndrome at Tertiary Care Hospital, Jeddah, Saudi Arabia, 2014. Emerg. Infect. Dis. 2016, 22, 794–801. [Google Scholar] [CrossRef] [Green Version]
- Assiri, A.; Abedi, G.R.; Bin Saeed, A.A.; Abdalla, M.A.; al-Masry, M.; Choudhry, A.J.; Lu, X.; Erdman, D.D.; Tatti, K.; Binder, A.M.; et al. Multifacility Outbreak of Middle East Respiratory Syndrome in Taif, Saudi Arabia. Emerg. Infect Dis. 2016, 22, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Balkhy, H.H.; Alenazi, T.H.; Alshamrani, M.M.; Baffoe-Bonnie, H.; Al-Abdely, H.M.; El-Saed, A.; Al Arbash, H.A.; Al Mayahi, Z.K.; Assiri, A.M.; Bin Saeed, A. Notes from the Field: Nosocomial Outbreak of Middle East Respiratory Syndrome in a Large Tertiary Care Hospital--Riyadh, Saudi Arabia, 2015. Mmwr. -Morb. Mortal. Wkly. Rep. 2016, 65, 163–164. [Google Scholar] [CrossRef] [Green Version]
- MacIntyre, C.R. Biopreparedness in the Age of Genetically Engineered Pathogens and Open Access Science: An Urgent Need for a Paradigm Shift. Mil. Med. 2015, 180, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. Plos Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- FluTrackers. FluTrackers. 20122–018 Case List of MoH/WHO Novel Coronavirus MERS nCoV Announced Cases. Available online: https://flutrackers.com/forum/forum/novel-coronavirus-ncov-mers-2012-2014/146270-2012-2018-case-list-of-moh-who-novel-coronavirus-mers-ncov-announced-cases (accessed on 22 February 2018).
- World Health Organisation (WHO). Coronavirus Infections. Disease Outbreak News. Available online: http://www.who.int/csr/don/archive/disease/coronavirus_infections/en/ (accessed on 5 February 2018).
- Saudi MoH. No New Corona Cases Recorded. Available online: http://www.moh.gov.sa/en/CCC/PressReleases/Pages/Statistics-2015-12-31-001.aspx (accessed on 15 January 2018).
- Geneious. Available online: http://www.geneious.com/ (accessed on 10 January 2018).
- Middle East Respiratory Syndrome Coronavirus, Complete Genome. Available online: https://www.ncbi.nlm.nih.gov/nuccore/NC_019843 (accessed on 10 January 2018).
- MAFFT—A Multiple Sequence Alignment Program. Available online: https://mafft.cbrc.jp/alignment/software/changelog.html (accessed on 25 January 2018).
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baele, G.; Li, W.L.S.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol. Biol. Evol. 2012, 30, 239–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, M.A.; Suchard, M.A. Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 2008, 36, 355–368. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A. Tracer v1. 6. Program Distributed by the Author. Available online: http://tree.bio.ed.ac.uk/software/tracer (accessed on 10 January 2018).
- Rambaut, A.; Drummond, A. Figtree Version 1.4.3. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 10 January 2018).
- Parker, J.; Rambaut, A.; Pybus, O.G. Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infect. Genet. Evol. 2008, 8, 239–246. [Google Scholar] [CrossRef]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- Adobe Illustrator. Available online: https://www.adobe.com/products/illustrator.html# (accessed on 24 March 2018).
- Cotten, M.; Watson, S.J.; Kellam, P.; Al-Rabeeah, A.A.; Makhdoom, H.Q.; Assiri, A.; Al-Tawfiq, J.A.; Alhakeem, R.F.; Madani, H.; AlRabiah, F.A.; et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: A descriptive genomic study. Lancet 2013, 382, 19932–20002. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.K.Y.; Bull, R.A.; Rockman, S.; Scott, G.; Stelzer-Braid, S.; Rawlinson, W. Correlation of polymerase replication fidelity with genetic evolution of influenza A/Fujian/411/02 (H3N2) viruses. J. Med. Virol 2011, 83, 510–516. [Google Scholar] [CrossRef]
- Dudas, G.; Carvalho, L.M.; Rambaut, A.; Bedford, T. MERS-CoV spillover at the camel-human interface. Elife 2018, 7, e31257. [Google Scholar] [CrossRef]
- Abdel-Moneim, A.S. Middle East respiratory syndrome coronavirus (MERS-CoV): Evidence and speculations. Arch. Virol. 2014, 159, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Shen, L.; Gu, X. Evolutionary Dynamics of MERS-CoV: Potential Recombination, Positive Selection and Transmission. Sci. Rep. 2016, 6, 25049. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | Total |
|---|---|---|---|---|---|---|---|
| Al Ahsa | 0 | 12 | 0 | 0 | 0 | 0 | 12 |
| Alkharj | 0 | 0 | 0 | 2 | 0 | 0 | 2 |
| Artawiyah | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Bisha | 1 | 0 | 0 | 0 | 0 | 0 | 1 |
| Buraidah | 0 | 1 | 0 | 0 | 10 | 0 | 11 |
| Dammam | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| Hafr Al Batin | 0 | 3 | 0 | 0 | 0 | 0 | 3 |
| Hofuf | 0 | 0 | 0 | 12 | 0 | 0 | 12 |
| Jeddah | 0 | 2 | 7 | 4 | 1 | 0 | 14 |
| Jubail | 0 | 0 | 0 | 0 | 1 | 0 | 1 |
| Khobar | 0 | 0 | 0 | 1 | 1 | 0 | 2 |
| Najran | 0 | 0 | 0 | 1 | 0 | 0 | 1 |
| Riyadh | 2 | 5 | 11 | 16 | 19 | 2 | 55 |
| Taif | 0 | 1 | 1 | 0 | 0 | 0 | 2 |
| Wadi Aldwasir | 0 | 1 | 0 | 0 | 1 | 0 | 2 |
| Total | 3 | 25 | 19 | 37 | 34 | 2 | 120 |
| Origin | Destination | Bayes Factor |
|---|---|---|
| Riyadh | Buraidah | 119631.3937 |
| Riyadh | Hofuf | 29897.87915 |
| Riyadh | Jubail | 165.8164344 |
| Khobar | Dammam | 109.4201215 |
| Jubail | Taif | 81.58842078 |
| Riyadh | Khobar | 49.91136624 |
| Jubail | Najran | 40.43222377 |
| Hafr Al Batin | Riyadh | 21.5996282 |
| Bisha | Riyadh | 14.21214846 |
| Hofuf | Artawiyah | 12.95700557 |
| Riyadh | Wadi Aldwasir | 11.97577204 |
| Riyadh | Artawiyah | 9.566752858 |
| Riyadh | Al Ahsa | 9.231095454 |
| Buraidah | Jubail | 9.159243505 |
| Riyadh | Hafr Al Batin | 7.418145007 |
| Hafr Al Batin | Bisha | 6.715095512 |
| Hofuf | Wadi Aldwasir | 5.413796481 |
| Bisha | Hafr Al Batin | 4.648046686 |
| Buraidah | Jeddah | 4.456426673 |
| Riyadh | Alkharj | 4.401306164 |
| Buraidah | Alkharj | 4.00981903 |
| Riyadh | Jeddah | 3.994819293 |
| Riyadh | Bisha | 3.925163012 |
| Jubail | Wadi Aldwasir | 3.721941193 |
| Buraidah | Al Ahsa | 3.57086268 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Adam, D.C.; Chughtai, A.A.; Stelzer-Braid, S.; Scotch, M.; MacIntyre, C.R. The Phylogeography of MERS-CoV in Hospital Outbreak-Associated Cases Compared to Sporadic Cases in Saudi Arabia. Viruses 2020, 12, 540. https://doi.org/10.3390/v12050540
Chen X, Adam DC, Chughtai AA, Stelzer-Braid S, Scotch M, MacIntyre CR. The Phylogeography of MERS-CoV in Hospital Outbreak-Associated Cases Compared to Sporadic Cases in Saudi Arabia. Viruses. 2020; 12(5):540. https://doi.org/10.3390/v12050540
Chicago/Turabian StyleChen, Xin, Dillon Charles Adam, Abrar Ahmad Chughtai, Sacha Stelzer-Braid, Matthew Scotch, and Chandini Raina MacIntyre. 2020. "The Phylogeography of MERS-CoV in Hospital Outbreak-Associated Cases Compared to Sporadic Cases in Saudi Arabia" Viruses 12, no. 5: 540. https://doi.org/10.3390/v12050540