
Abstract
Following the detection of the first imported case of COVID-19 in the northern sector of Ghana, we molecularly characterized and phylogenetically analysed sequences, including three complete genome sequences, of severe acute respiratory syndrome coronavirus 2 obtained from nine patients in Ghana. We performed high-throughput sequencing on nine samples that were found to have a high concentration of viral RNA. We also assessed the potential impact that long-distance transport of samples to testing centres may have on sequencing results. Here, two samples that were similar in terms of viral RNA concentration but were transported from sites that are over 400 km apart were analyzed. All sequences were compared to previous sequences from Ghana and representative sequences from regions where our patients had previously travelled. Three complete genome sequences and another nearly complete genome sequence with 95.6% coverage were obtained. Sequences with coverage in excess of 80% were found to belong to three lineages, namely A, B.1 and B.2. Our sequences clustered in two different clades, with the majority falling within a clade composed of sequences from sub-Saharan Africa. Less RNA fragmentation was seen in sample KATH23, which was collected 9 km from the testing site, than in sample TTH6, which was collected and transported over a distance of 400 km to the testing site. The clustering of several sequences from sub-Saharan Africa suggests regional circulation of the viruses in the subregion. Importantly, there may be a need to decentralize testing sites and build more capacity across Africa to boost the sequencing output of the subregion.
Similar content being viewed by others


Introduction
Coronaviruses (CoVs) are minute in size (65–125 nm in diameter) and contain a single-stranded RNA ranging in length from 26 to 32 kb. The subfamily Orthocoronavirinae includes the genera Alphacoronavirus, Betacoronavirus, Gammacoronavirus, and Deltacoronavirus. Until 2002, when the world witnessed a severe acute respiratory syndrome (SARS) outbreak caused by SARS-CoV in Guangdong, China, coronaviruses, with the exception of 229E and OC43, were thought to infect only animals [1]. Only a decade later, another pathogenic coronavirus, known as Middle East respiratory syndrome coronavirus (MERS-CoV), caused an endemic outbreak in Middle Eastern countries [2].
In December 2019, the World Health Organization (WHO) was informed about a cluster of patients who presented with pneumonia of unknown aetiology in the city of Wuhan (Hubei province) in China [3]. Shortly afterwards, a new type of coronavirus, now termed severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was isolated and identified by scientists from China [4]. Sequencing results revealed that it belongs to the genus Betacoronavirus of the family Coronaviridae and has 96% genome sequence identity to a previously detected SARS‐like bat coronavirus [5, 6]. The genetic sequence of this new virus was shared with the international community on January 10, 2020 [7, 8]. Infections caused by this virus have spread to all WHO regions and had an enormous adverse global impact. On March 11, 2020, WHO declared a global pandemic [9], which prompted increased and sustained international action and response. By August 27, 2020, over 24 million cases were confirmed, with more than 830,000 deaths worldwide (https://www.worldometers.info/coronavirus/).
The first confirmed case of COVID-19 in Africa was reported in Egypt on February 14, 2020 [10], and the first case in sub-Saharan Africa was in Nigeria on February 28, 2020 [11]. Ghana recorded its first confirmed case of COVID-19 on March 12, 2020 [12]. After this, all suspected cases of COVID-19 were confirmed by reverse transcription polymerase chain reaction (RT-PCR) as recommended by WHO [13]. While RT-PCR still remains the gold standard, issues relating to transport of samples from primary healthcare facilities to centralised testing laboratories, laboratory infrastructure, human resources, supply chain management, and stockpiling laboratory consumables and reagents have remained key challenges [14,15,16]. Beyond using RT-PCR to establish the presence of the virus, one big shortfall in several African countries is the inability to sequence the genome of the virus and to study its biology. Thankfully, the Africa Centres for Disease Control and Prevention (Africa CDC) is making a massive effort to establish genomic sequencing centres in Africa [17]. However, until this becomes a reality, the inability of several African countries to sequence viral isolates may result in deficits in our understanding of the distribution of the various SARS-CoV-2 strains circulating on the African continent as well as a lack of knowledge about its transmission dynamics [18].
The paucity of data is evidenced by the measure and proportion of sequences and genome data deposited in the Global Initiative on Sharing Avian Flu Data (GISAID) repository originating from Africa institutions (https://www.gisaid.org/). This means that Africa needs to build more capacity for genome sequencing in order to understand the distribution of the various strains circulating on the continent and their transmission dynamics. This could eventually assist in the development of vaccines against COVID-19. It is likely that the African sequences are already relatively widely distributed globally [19]. In an effort to contribute to this initiative and following the detection of the first imported case in the northern sector of Ghana on March 13, 2020, we have now molecularly characterized and phylogenetically analyzed sequences, including three complete genome sequences, of SARS‐CoV‐2 obtained from nine of the first 200 patients observed in Ghana. Eight of these patients had a recent history of foreign travel, and one did not.
Materials and methods
Sample collection and transport
Between February 29 and March 28, 2020, samples were obtained from patients with suspected COVID-19 from the Tamale Teaching Hospital (TTH) in the Northern Region, and Komfo Anokye Teaching Hospital (KATH) and Kumasi South Hospital (KSH) in the Ashanti Region of Ghana (Fig. 1). Recruitment of cases at these facilities was done according to the Ghana National Surveillance Strategy protocol [20]. Suspected COVID-19 cases were defined as individuals presenting with fever (>38 °C) or a history of fever and symptoms of respiratory tract illness such as cough or shortness of breath, or individuals who were in close contact with a person who was suspected or confirmed to have COVID-19.

Map of Ghana showing sample collection sites. The map was generated using Quantum GIS version 3.6.2 and data freely available from www.openstreetmaps.org. Samples were collected from the Northern Region (orange) and the Ashanti Region (green) and tested at the KCCR, also in Kumasi in the Ashanti Region of Ghana
Nasopharyngeal and or oropharyngeal swabs were obtained using flocked swabs (Copan Group, Brescia, Italy) and kept in 500 µl of RNAlater (QIAGEN, Hilden, Germany) in 1.5-ml tubes (Eppendorf, Regensburg, Germany) and transported immediately at ambient temperature for confirmation at the Kumasi Centre for Collaborative Research in Tropical Medicine (KCCR), Kumasi, in the Ashanti Region of Ghana.
The KCCR is one of the two main research laboratories in Ghana designated for COVID-19 testing. This is because of its longstanding experience and the expertise of its scientists in studies related to coronaviruses (http://kccr.org/). The Centre is currently Ghana’s second largest testing site, which serves the northern sector of the country. During the early phase of the pandemic, the Centre received samples from 12 out of 16 regions in Ghana and tested approximately 1,200 samples daily.
Viral RNA extraction and PCR detection
Using a starting volume of 140 µl, both nasopharyngeal and oropharyngeal swabs from each patient were extracted together as a single sample using a QIAGEN Viral RNA Mini Kit (QIAGEN, Hilden, Germany) according to manufacturer’s instructions. Samples were eluted in a 100-µl volume, and SARS-CoV-2 RNA was detected using a RealStar® SARS-CoV-2 RT-PCR Kit (Altona, Germany) according to the manufacturer’s instructions. Sample quantification was done using an externally generated standard curve based on serially diluted SARS-CoV-2 in vitro transcripts. All samples with a cycle threshold (Ct) of 40 or above were considered negative. All amplification runs were validated by including positive and negative controls.
Whole-genome sequencing
High-throughput sequencing for samples with sufficiently high RNA concentrations as determined by quantitative real-time PCR were sequenced using an Illumina NextSeq platform (Illumina, San Diego, California, U.S.) and a KAPA RNA Hyper Prep kit (Roche Molecular Diagnostics, Basel, Switzerland) according to manufacturer’s instructions. In order to estimate the potential impact that long-distance transport of samples to testing centers may have on sequencing results, the two samples that were closest in terms of viral RNA concentration were tested using an Agilent 4200 TapeStation system (Agilent Technologies, CA, USA). These samples comprised one from TTH in the north, which is approximately 400 kilometers from the testing site, and another from KATH in Kumasi (in the same city as the testing site, approximately 9 km away).
Phylogenetic analysis
Sequences from this study, with the exception of three that had less than 80% of the genome sequenced, were compared to previous sequences from Ghana and representative sequences from regions where patients had previously travelled. These included sequences from the USA, Japan, France, and Guinea and available sequences from sub-Saharan Africa. All sequences from the region of interest as of April 2020 were obtained from GISAID (https://www.gisaid.org/), and duplicates were removed. The non-redundant sequences were then clustered at a minimum threshold of 99.9% using CD-HIT (http://weizhongli-lab.org/cd-hit/), and representative sequences from each cluster were selected. Multiple sequence alignments were done using the MAFFT plugin in Geneious prime (http://www.geneious.com). Phylogenetic analysis was done by Bayesian inference using the MrBayes [21] plugin in Geneious prime with a chain length of 1.1 million and a subsampling frequency of 200. A general time-reversible substitution model with a gamma distribution and proportion of invariable sites (GTR+G+I) was used for the analysis. All sequences with genome coverage greater than 80% were analyzed using the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGOLIN) online resource (https://pangolin.cog-uk.io/) for lineage assignment.
Ethical approval
We obtained ethical approval from the Committee on Human Research Publications and Ethics of the School of Medicine and Dentistry at the Kwame Nkrumah University of Science and Technology (CHPRE/AP/462/19) and the Institutional Review Board of the Ghana Health Service (GHS-ERC087/03/20).
Results
Sample description and comparison
A total of nine samples obtained from nine patients that tested positive for SARS-CoV-2 were analyzed. Six of the samples were from the Northern Region, while three were from the Ashanti Region of Ghana. Six patients were asymptomatic, and three were symptomatic, presenting with cough, headache, general weakness, sore throat, shortness of breath, and diarrhea. All but one of the patients had a history of travel to the USA, Japan, France or Guinea (Table 1).
Sample TTH6 was transported over 400 km by road and was presumably exposed to ambient temperature for a longer time than sample KATH23, which was transported over a distance of 9 km to the testing centre, where both samples underwent similar processing procedures.
Despite a difference of approximately only 576 copies/µL between samples KATH23 and TTH6 (Table 1), TapeStation analysis showed less RNA fragmentation in KATH23 (Supplementary Figs. S1 and S2), which yielded a complete genome sequence on the first attempt, whereas TTH6 yielded a sequence with approximately 82.8% genome coverage, which was increased to 95.6% with resequencing efforts.
Sequence description and phylogenetic analysis
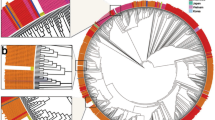
The percent coverage of the available genome sequences in this study ranged from as low as 23.2% to complete coverage. Complete genome sequences were obtained for three samples, and another nearly complete genome sequence had a coverage of 95.6%. Sequences with coverage in excess of 80% were found to belong to three lineages, namely A, B.1, and B.2. The least number of nucleotide substitutions in comparison to the original SARS-CoV-2 isolate from China (NC_045512) was the one from a patient with no known travel history (KATH23). The one with the most nucleotide substitutions was from a patient known to have travelled to at least two locations in Asia and Europe before arriving in Ghana (KSH61). The D614G amino acid substitution in the spike protein (A23403G), which is purported to enhance viral infectivity [22], was found in three sequences from this study (Table 2). Our sequences clustered in two different clades, with the majority falling within a clade composed solely of sequences from sub-Saharan Africa (Fig. 2). All seven sequences that had greater than 50% coverage were submitted to the SARS-CoV-2 repository on the GISAID platform and assigned the accession numbers EPI_ISL_515181-515184 and EPI_ISL_515247-515249.

Phylogenetic analysis of SARS-CoV-2 genome sequences Phylogenetic analysis was performed on 76 representative genome sequences by Bayesian inference using the GTR+G+I substitution model. Sequences in the tree are designated by location, GISAID accession numbers and date of collection. Sequences from this study are highlighted in red with sequence-specific names. The tree was rooted with randomly selected sequences from England collected in June 2020
Discussion
Despite the impressive drive from different regions of the globe, including Africa, to sequence SARS-CoV-2 genomes for studying the epidemiology of the virus, Africa still lags behind in terms of its genome sequencing output, as was seen by searching the SARS-CoV-2 repository on GISAID in August 2020. Apart from a lack of availability of sequencing technology and expertise, which could account for this shortfall [23], contributory factors may be degradation of genetic material during transport to available diagnostic centres due to the mostly centralized testing system prevalent on the African continent [24]. Our study shows that although two samples had similar viral loads (difference = 576 copies/µL), the samples obtained from Kumasi with a viral load of 1.20 × 103 copies/µL (approximately 9 km from the testing laboratory) showed less fragmentation than the one from Tamale in the northern part of Ghana, with a viral load of 6.24 × 102 copies/µL, that had to be transported over a distance of approximately 400 km across the country before being processed. This highlights the importance of correct sample storage and transport conditions. Although some level of viral RNA fragmentation can be tolerated when diagnostic real-time PCR is used due to short amplicon sizes, the same cannot be said for downstream whole-genome sequencing [25]. Therefore, decentralization of diagnostic centres and proper sample storage are important for boosting the sequencing output of the subregion. In addition, as the world races towards development of a vaccine, the importance of sequence data from the African region cannot be overemphasized. Capacity building in Africa for this purpose is paramount.
The assigned PANGOLIN lineages are reflective of the sampling times of the sequences, as seen with the early circulating A, B.1, and B.2 lineages from January to May 2020 [26]. This supports the reported epidemiological links in terms of travel, which was associated with early introduction of the virus into Ghana, mainly from Asia and Europe. The sequence from the individual with no known travel history had the least number of nucleotide substitutions compared to the original sequence from China. This may hint at earlier introductions and possible community spread than previously believed.
Sequences from sub-Saharan Africa, including previous ones from Ghana, were observed to cluster in different clades across the topology of the phylogenetic tree. However the subset of sequences clustering together only from sub-Saharan Africa may point to the circulation of viruses within the region due to movement through porous land borders [27, 28]. A lot of emphasis is placed on introductions from other regions such as Europe and Asia through air travel, but introductions from within the subregion also appear to play a major role in virus circulation through movement of people. The number of sequences included in the analysis, however, was limited, and interpretation of these data should therefore be done with caution.
Conclusion
Analysis of sequences from the early stages of the outbreak can provide important insights into the viral diversity present in regions where genomic data are lacking. There is the need for further studies on adequate sample storage and transportation when testing facilities are far from sample collection sites. The clustering of several sequences from sub-Saharan Africa suggests regional circulation of the viruses in the subregion.
Availability of data and materials
The datasets obtained and analysed during the current study are available from the corresponding author on reasonable request. All sequences have been deposited on GISAID.
Abbreviations
- CoVs:
-
Coronaviruses
- SARS-CoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- WHO:
-
World Health Organization
- GISAID:
-
Global Initiative on Sharing Avian Influenza Data
- KCCR:
-
Kumasi Centre for Collaborative Research in Tropical Medicine
- KNUST:
-
Kwame Nkrumah University of Science and Technology
- TTH:
-
Tamale Teaching Hospital
- KATH:
-
Komfo Anokye Teaching Hospital
- KSH:
-
Kumasi South Hospital
- GHS:
-
Ghana Health Service
- RT-PCR:
-
Real-time polymerase chain reaction
- PANGOLIN:
-
Phylogenetic Assignment of Named Global Outbreak Lineages
- Ct:
-
Cycle threshold
References
Zhong NS, Zheng BJ, Li YM, Poon LLM, Xie ZH et al (2003) Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet 362:1353–1358
Wang N, Shi X, Jiang L, Zhang S, Wang D et al (2013) Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res 23:986–993
Lu R, Zhao X, Li J, Niu P, Yang B et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565–574
Yang L, Wu Z, Ren X, Yang F, He G, et al (2013) Novel SARS-like Betacoronaviruses in Bats, China, 2011. Emerg Infect Dis. https://doi.org/10.3201/eid1906.121648(Epub ahead of print)
Paraskevis D, Kostaki EG, Magiorkinis G, Panayiotakopoulos G, Sourvinos G et al (2020) Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect Genet Evol 79:104212
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. (Published online February 3)
Zhu N, Zhang D, Wang W, Li X, Yang B, et al (2019) A novel coronavirus from patients with pneumonia in China, N Engl J Med.
Shan C, Yao Y-F, Yang X-L, Zhou Y-W, Gao G, et al (2020) Infection with novel coronavirus (SARS-CoV-2) causes pneumonia in Rhesus macaques. Cell Res:1–8.
Cucinotta D, Vanelli M (2020) WHO declares COVID-19 a pandemic. Acta Bio-medica Atenei Parm 91:157–160
Gilbert M, Pullano G, Pinotti F, Valdano E, Poletto C et al (2020) Preparedness and vulnerability of African countries against importations of COVID-19: a modelling study. Lancet 395:871–877
Hilda AE, Kolawole OE, Olufemi AB, Senbadejo TY, Oyawoye OM, et al (2020) Phyloevolutionary analysis of SARS-CoV-2 in Nigeria. New Microbes New Infect:100717.
Asamoah JKK, Owusu MA, Jin Z, Oduro FT, Abidemi A, et al (2020) Global stability and cost-effectiveness analysis of COVID-19 considering the impact of the environment: using data from Ghana. Chaos Solitons Fractals:110103.
Organization WH (2020) Laboratory testing for coronavirus disease 2019 (COVID-19) in suspected human cases: interim guidance, 2 March 2020. World Health Organization
Senghore M, Savi MK, Gnangnon B, Hanage WP, Okeke IN (2020) Leveraging Africa’s preparedness towards the next phase of the COVID-19 pandemic. Lancet Glob Heal.
Kobia F, Gitaka J (2020) COVID-19: Are Africa’s diagnostic challenges blunting response effectiveness? AAS Open Res 3
Adebisi YA, Oke GI, Ademola PS, Chinemelum IG, Ogunkola IO, et al (2020) SARS-CoV-2 diagnostic testing in Africa: needs and challenges. Pan Afr Med J 35
Shey M, Okeibunor JC, Yahaya AA, Herring BL, Tomori O, et al. (2020) Genome sequencing and the diagnosis of novel coronavirus (SARS-COV-2) in Africa: how far are we? Pan Afr Med J 36
Garba SM, Lubuma JMS, Tsanou B (2020) Modeling the transmission dynamics of the COVID-19 Pandemic in South Africa. Math Biosci: 108441
Allam M, Ismail A, Khumalo ZTH, Kwenda S (2020) Whole-genome sequence of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)
Ghana Health Service. Ghana National Strategy Surveillance. 2020.
Huelsenbeck JP, Ronquist F (2020) MrBayes: a program for the bayesian inference of phylogeny, v. 3.1. 2. Rochester, New York
Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, et al (2020) Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell
Helmy M, Awad M, Mosa KA (2016) Limited resources of genome sequencing in developing countries: challenges and solutions. Appl Transl genomics 9:15–19
Nkengasong J (2020) Let Africa into the market for COVID-19 diagnostics. Nature 580:565
Lewandowski K, Bell A, Miles R, Carne S, Wooldridge D et al (2017) The effect of nucleic acid extraction platforms and sample storage on the integrity of viral RNA for use in whole genome sequencing. J Mol Diagnostics 19:303–312
Rambaut A, Holmes EC, Hill V, OToole A, McCrone J, et al (2020) A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv.
Sarfo AK, Karuppannan S (2020) Application of geospatial technologies in the covid-19 fight of Ghana. Trans Indian Natl Acad Eng:1–12.
Merrill RD, Rogers K, Ward S, Ojo O, Kakaī CG et al (2017) Responding to communicable diseases in internationally mobile populations at points of entry and along porous borders, Nigeria, Benin, and Togo. Emerg Infect Dis 23:S114
Acknowledgements
We thank the hospitals for allowing this study to be undertaken in their various facilities. We also appreciate the support of all healthcare workers who helped with sampling and recruitment. We also thank the Ghana Health Service, Ministry of Health, for all the support. We appreciate the efforts of the technicians at the Kumasi Centre for Collaborative Research in Tropical Medicine and the Institute of Virology, Charite, Universitätsmedizin Berlin, Germany, in supporting this work with their expertise.
Funding
This research received no external funding. However, AAS, P-ED, ROP and CD are on the PANDORA-ID-NET (EDCTP Reg/Grant RIA2016E-1609), funded by the European and Developing Countries Clinical Trials Partnership (EDCTP2) programme, which is supported under Horizon 2020, the European Union’s Framework Programme for Research and Innovation. The views and opinions of the authors expressed herein do not necessarily state or reflect those of EDCTP.
Author information
Authors and Affiliations
Contributions
Conceptualization, AAS, MO and RP; methodology, PE-D, JS, JBS, VMC and CD; formal analysis, AAS, MO, PE-D, RY, NKAB, RG, EA, AK, TB, MF, SA, JAA, YAA and JHA; CD and RP; data curation, JAA; writing—original draft preparation, AAS and PE-D.; writing—review and editing, YAA, EOD, YAS and KOD; funding acquisition, CD and RP. All authors have read and accepted the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics and consent to participate
Ethical approval for this work was sought from the Committee on Human Research Publications and Ethics of the School of Medicine and Dentistry at the Kwame Nkrumah University of Science and Technology (CHPRE/AP/462/19) and the Institutional Review Board of the Ghana Health Service (GHS-ERC087/03/20).
Additional information
Handling Editor: T. K. Frey.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sylverken, A.A., El-Duah, P., Owusu, M. et al. Transmission of SARS-CoV-2 in northern Ghana: insights from whole-genome sequencing. Arch Virol 166, 1385–1393 (2021). https://doi.org/10.1007/s00705-021-04986-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-021-04986-3