
Genetic Characterisation and Comparison of Three Human Coronaviruses (HKU1, OC43, 229E) from Patients and Bovine Coronavirus (BCoV) from Cattle with Respiratory Disease in Slovenia


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Specimens
2.2. Sample Preparation, Nucleic-Acid Extraction, and Real-Time RT-PCR
2.3. RT-PCR for Coronaviruses and DNA Sequencing
2.4. Data Analysis
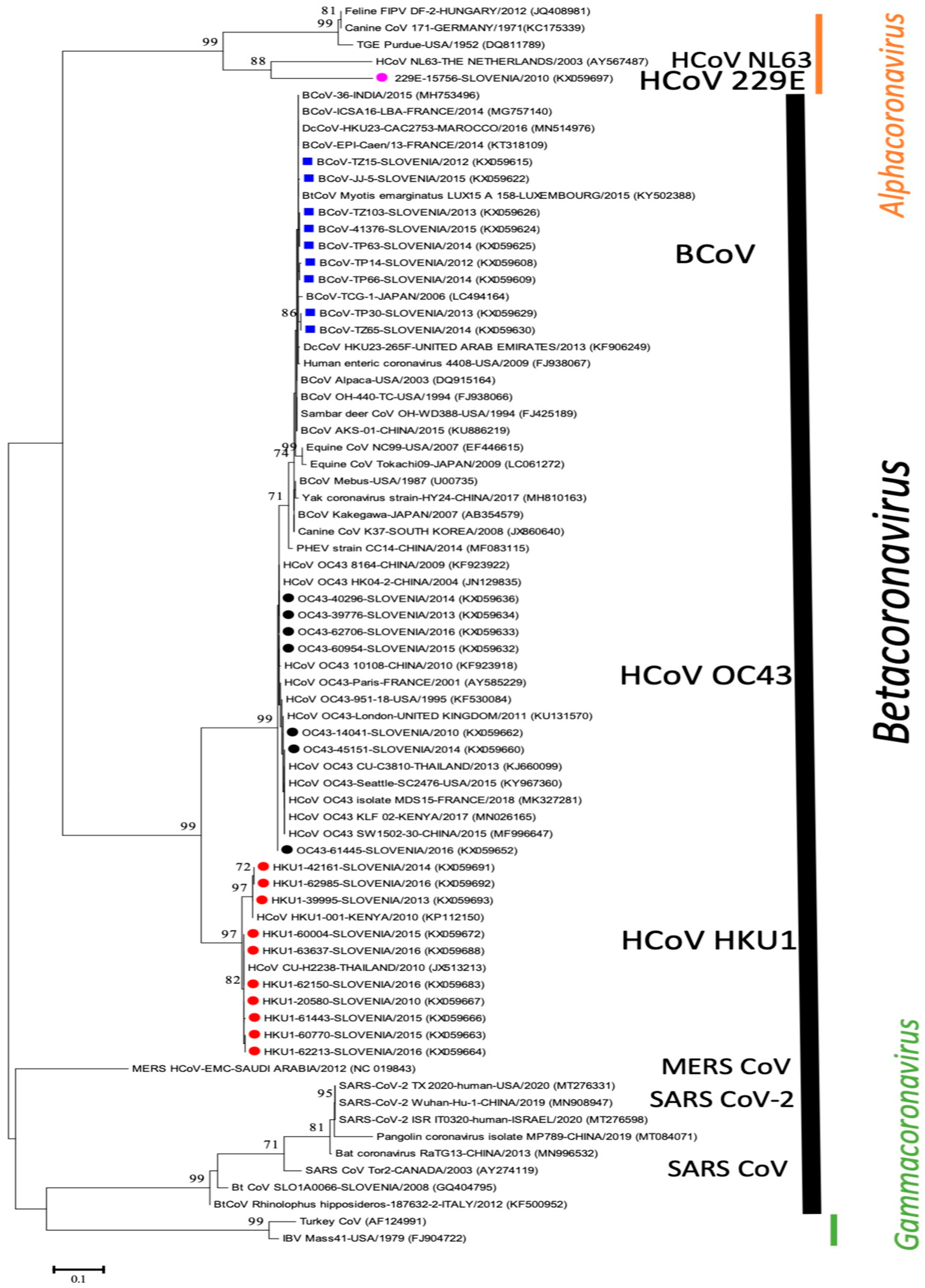
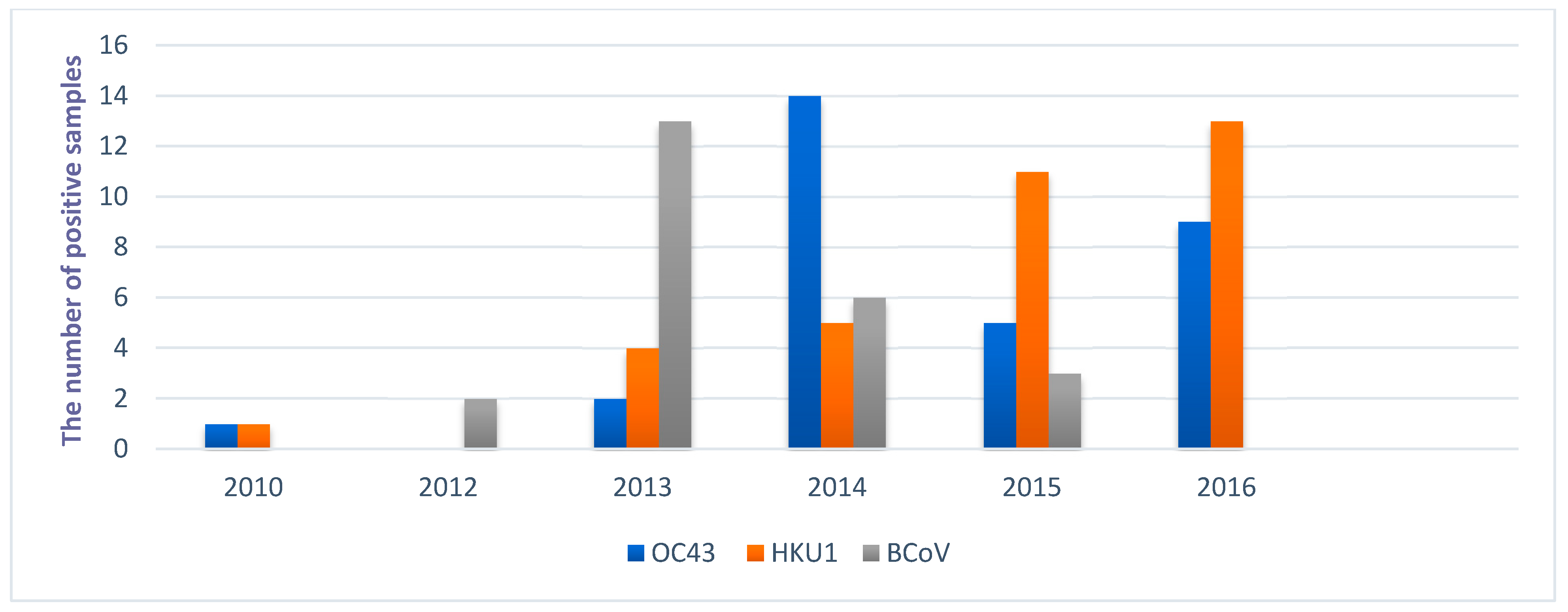
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Navas-Martin, S.; Weiss, S.R. SARS: Lessons Learned from Other Coronaviruses. Viral Immunol. 2003, 16, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Enjuanes, L.; Ziebuhr, J.; Snijder, E.J. Nidovirales: Evolving the largest RNA virus genome. Virus Res. 2006, 117, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Carstens, E.B. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2012). Arch. Virol. 2012, 157, 1411–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [Green Version]
- ICTV Virus Taxonomy: 2019 Release. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/223/coronaviridae-figures (accessed on 23 March 2021).
- Garbino, J.; Crespo, S.; Aubert, J.D.; Rochat, T.; Ninet, B.; Deffernez, C.; Wunderli, W.; Pache, J.C.; Soccal, P.M.; Kaiser, L. A prospective hospital-based study of the clinical impact of non-severe acute respiratory syndrome (Non-SARS)-related human coronavirus infection. Clin. Infect. Dis. 2006, 43, 1009–1015. [Google Scholar] [CrossRef]
- Esposito, S.; Bosis, S.; Niesters, H.G.; Tremolati, E.; Begliatti, E.; Rognoni, A.; Tagliabue, C.; Principi, N.; Osterhaus, A.D. Impact of human coronavirus infections in otherwise healthy children who attended an emergency department. J. Med. Virol. 2006, 78, 1609–1615. [Google Scholar] [CrossRef]
- Principi, N.; Bosis, S.; Esposito, S. Effects of Coronavirus Infections in Children. Emerg. Infect. Dis. 2010, 16, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Khan, G. A novel coronavirus capable of lethal human infections: An emerging picture. Virol. J. 2013, 10, 66. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Gossner, C.; Danielson, N.; Gervelmeyer, A.; Berthe, F.; Faye, B.; Kaasik Aaslav, K.; Adlhoch, C.; Zeller, H.; Penttinen, P.; Coulombier, D. Human-Dromedary Camel Interactions and the Risk of Acquiring Zoonotic Middle East Respiratory Syn-drome Coronavirus Infection. Zoonoses Public Health 2016, 63, 1–9. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jevšnik, M.; Uršič, T.; Žigon, N.; Lusa, L.; Krivec, U.; Petrovec, M. Coronavirus infections in hospitalized pediatric patients with acute respiratory tract disease. BMC Infect. Dis. 2012, 12, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mebus, C.A.; Stair, E.L.; Rhodes, M.B.; Twiehaus, M.J. Neonatal calf diarrhea: Propagation, attenuation, and characteristics of a coronavirus-like agent. Am. J. Veter. Res. 1973, 34, 145–150. [Google Scholar]
- Saif, L.J.; Redman, D.R.; Brock, K.V.; Kohler, E.M.; Heckert, R.A. Winter dysentery in adult dairy cattle: Detection of coronavirus in the faeces. Veter. Rec. 1988, 123, 300–301. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.-O.; Hoet, A.E.; Loerch, S.C.; Wittum, T.E.; Saif, L.J. Evaluation of concurrent shedding of bovine coronavirus via the respiratory tract and enteric route in feedlot cattle. Am. J. Veter. Res. 2001, 62, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Hasoksuz, M.; Vlasova, A.; Saif, L.J. Detection of group 2a coronaviruses with emphasis on bovine and wild ruminant strains. Virus isolation and detection of antibody, antigen, and nucleic acid. Methods Mol. Biol. 2008, 454, 43–59. [Google Scholar]
- Barros, I.N.; Silva, S.O.; Nogueira Neto, F.S.; Asano, K.M.; Souza, S.P.; Richtzenhain, L.J.; Brandao, P.E. A multigene approach for comparing genealogy of Betacoronavirus from cattle and horses. Sci. World J. 2013, 2013, 349702. [Google Scholar] [CrossRef] [PubMed]
- Saif, L.J. Bovine Respiratory Coronavirus. Veter. Clin. N. Am. Food Anim. Pr. 2010, 26, 349–364. [Google Scholar] [CrossRef]
- Suzuki, T.; Otake, Y.; Uchimoto, S.; Hasebe, A.; Goto, Y. Genomic Characterization and Phylogenetic Classification of Bovine Coronaviruses Through Whole Genome Sequence Analysis. Viruses 2020, 12, 183. [Google Scholar] [CrossRef] [Green Version]
- Fulton, R.W.; Herd, H.R.; Sorensen, N.J.; Confer, A.W.; Ritchey, J.W.; Ridpath, J.F.; Burge, L.J. Enteric disease in postweaned beef calves associated with Bovine coronavirus clade 2. J. Veter. Diagn. Investig. 2015, 27, 97–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Byrum, B.; Zhang, Y. Porcine Coronavirus HKU15 Detected in 9 US States, 2014. Emerg. Infect. Dis. 2014, 20, 1594–1595. [Google Scholar] [CrossRef]
- Cavanagh, D. A nomenclature for avian coronavirus isolates and the question of species status. Avian Pathol. 2001, 30, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paller, T.; Hostnik, P.; Pogačnik, M.; Toplak, I. The prevalence of ten pathogens detected by a real-time PCR method in nasal swab samples collected from live cattle with respiratory disease. Slov. Vet. Res. 2017, 54, 101–107. [Google Scholar]
- Vijgen, L.; Keyaerts, E.; Moes, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.M.; Van Ranst, M. Complete genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.-Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.K.P.; Lee, P.; Tsang, A.K.L.; Yip, C.C.Y.; Tse, H.; Lee, R.A.; So, L.-Y.; Lau, Y.-L.; Chan, K.-H.; Woo, P.C.Y.; et al. Molecular Epidemiology of Human Coronavirus OC43 Reveals Evolution of Different Genotypes over Time and Recent Emergence of a Novel Genotype due to Natural Recombination. J. Virol. 2011, 85, 11325–11337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, J.; Xiao, Y.; Zhang, J.; Wang, Y.; Chen, L.; Paranhos-Baccalà, G.; Ren, L.; Wang, J. Genotype shift in human coronavirus OC43 and emergence of a novel genotype by natural recombination. J. Infect. 2015, 70, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diallo, I.S.; Hewitson, G.; Wright, L.; Rodwell, B.J.; Corney, B.G. Detection of equine herpesvirus type 1 using a real-time polymerase chain reaction. J. Virol. Methods 2006, 131, 92–98. [Google Scholar] [CrossRef]
- Mankoč, S.; Hostnik, P.; Grom, J.; Toplak, I.; Klobučar, I.; Kosec, M.; Barlic-Maganja, D. Comparison of different molecular methods for assessment of equine arteritis virus (EAV) infection: A novel one-step MGB real-time RT-PCR assay, PCR-ELISA and classical RT-PCR for detection of highly diverse sequences of Slovenian EAV variants. J. Virol. Methods 2007, 146, 341–354. [Google Scholar] [CrossRef]
- Kuypers, J.; Wright, N.; Morrow, R. Evaluation of quantitative and type-specific real-time RT-PCR assays for detection of respiratory syncytial virus in respiratory specimens from children. J. Clin. Virol. 2004, 31, 123–129. [Google Scholar] [CrossRef]
- Kuypers, J.; Wright, N.; Ferrenberg, J.; Huang, M.-L.; Cent, A.; Corey, L.; Morrow, R. Comparison of Real-Time PCR Assays with Fluorescent-Antibody Assays for Diagnosis of Respiratory Virus Infections in Children. J. Clin. Microbiol. 2006, 44, 2382–2388. [Google Scholar] [CrossRef] [Green Version]
- Daum, L.T.; Canas, L.C.; Arulanandam, B.P.; Niemeyer, D.; Valdes, J.J.; Chambers, J.P. Real-time RT-PCR assays for type and subtype detection of influenza A and B viruses. Influ. Other Respir. Viruses 2007, 1, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Maertzdorf, J.; Wang, C.K.; Brown, J.B.; Quinto, J.D.; Chu, M.; De Graaf, M.; Hoogen, B.G.V.D.; Spaete, R.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Real-Time Reverse Transcriptase PCR Assay for Detection of Human Metapneumoviruses from All Known Genetic Lineages. J. Clin. Microbiol. 2004, 42, 981–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Chittaganpitch, M.; Olsen, S.J.; Mackay, I.M.; Sloots, T.P.; Fry, A.M.; Erdman, D.D. Real-Time PCR Assays for Detection of Bocavirus in Human Specimens. J. Clin. Microbiol. 2006, 44, 3231–3235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vos, N.; Vankeerberghen, A.; Vaeyens, F.; Van Vaerenbergh, K.; Boel, A.; De Beenhouwer, H. Simultaneous detection of human bocavirus and adenovirus by multiplex real-time PCR in a Belgian paediatric population. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Scheltinga, S.; Templeton, K.; Beersma, M.; Claas, E. Diagnosis of human metapneumovirus and rhinovirus in patients with respiratory tract infections by an internally controlled multiplex real-time RNA PCR. J. Clin. Virol. 2005, 33, 306–311. [Google Scholar] [CrossRef]
- Kuypers, J.; Martin, E.T.; Heugel, J.; Wright, N.; Morrow, R.; Englund, J.A. Clinical Disease in Children Associated with Newly Described Coronavirus Subtypes. Pediatrics 2007, 119, e70–e76. [Google Scholar] [CrossRef]
- Stephensen, C.B.; Casebolt, D.B.; Gangopadhyay, N.N. Phylogenetic analysis of a highly conserved region of the polymerase gene from 11 coronaviruses and development of a consensus polymerase chain reaction assay. Virus Res. 1999, 60, 181–189. [Google Scholar] [CrossRef]
- Rihtarič, D.; Hostnik, P.; Steyer, A.; Grom, J.; Toplak, I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol. 2010, 155, 507–514. [Google Scholar] [CrossRef]
- Kin, N.; Miszczak, F.; Diancourt, L.; Caro, V.; Moutou, F.; Vabret, A.; Gouilh, M.A. Comparative molecular epidemiology of two closely related coronaviruses, bovine coronavirus (BCoV) and human coronavirus OC43 (HCoV-OC43), reveals a different evolutionary pattern. Infect. Genet. Evol. 2016, 40, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Beaudeau, F.; Björkman, C.; Alenius, S.; Frössling, J. Spatial patterns of Bovine Corona Virus and Bovine Respiratory Syncytial Virus in the Swedish beef cattle population. Acta Veter. Scand. 2010, 52, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jevšnik, M.; Steyer, A.; Pokorn, M.; Mrvič, T.; Grosek, Š.; Strle, F.; Lusa, L.; Petrovec, M. The Role of Human Coronaviruses in Children Hospitalized for Acute Bronchiolitis, Acute Gastroenteritis, and Febrile Seizures: A 2-Year Prospective Study. PLoS ONE 2016, 11, e0155555. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Yip, C.C.Y.; Huang, Y.; Tsoi, H.-W.; Chan, K.-H.; Yuen, K.-Y. Comparative Analysis of 22 Coronavirus HKU1 Genomes Reveals a Novel Genotype and Evidence of Natural Recombination in Coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jevšnik Virant, M.; Černe, D.; Petrovec, M.; Paller, T.; Toplak, I. Genetic Characterisation and Comparison of Three Human Coronaviruses (HKU1, OC43, 229E) from Patients and Bovine Coronavirus (BCoV) from Cattle with Respiratory Disease in Slovenia. Viruses 2021, 13, 676. https://doi.org/10.3390/v13040676
Jevšnik Virant M, Černe D, Petrovec M, Paller T, Toplak I. Genetic Characterisation and Comparison of Three Human Coronaviruses (HKU1, OC43, 229E) from Patients and Bovine Coronavirus (BCoV) from Cattle with Respiratory Disease in Slovenia. Viruses. 2021; 13(4):676. https://doi.org/10.3390/v13040676
Chicago/Turabian StyleJevšnik Virant, Monika, Danijela Černe, Miroslav Petrovec, Tomislav Paller, and Ivan Toplak. 2021. "Genetic Characterisation and Comparison of Three Human Coronaviruses (HKU1, OC43, 229E) from Patients and Bovine Coronavirus (BCoV) from Cattle with Respiratory Disease in Slovenia" Viruses 13, no. 4: 676. https://doi.org/10.3390/v13040676