
Multiple SARS-CoV-2 Introductions Shaped the Early Outbreak in Central Eastern Europe: Comparing Hungarian Data to a Worldwide Sequence Data-Matrix

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Direct Sequencing and Primary Data Analysis from Patient Samples
2.3. Genome Data Analysis
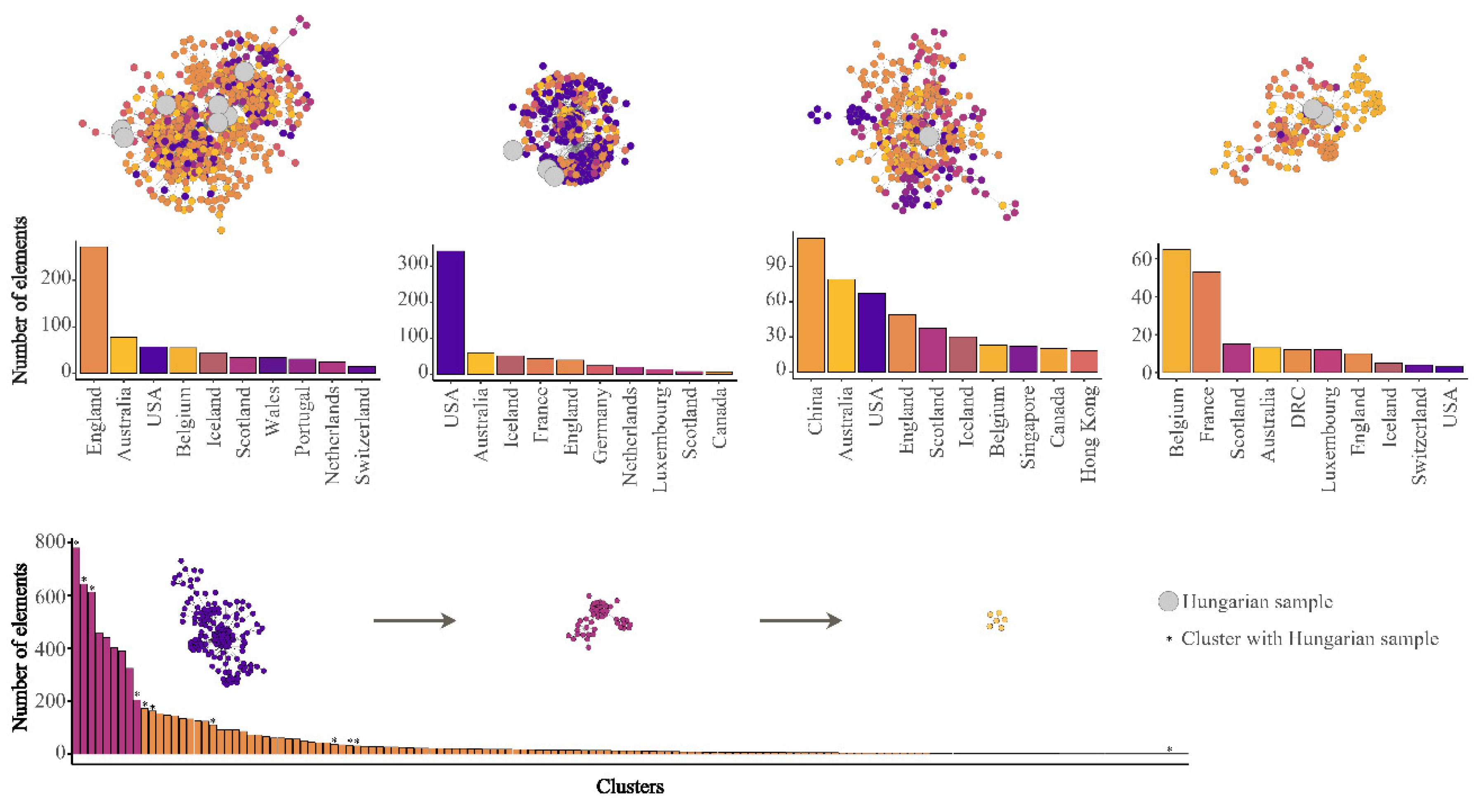
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19. 13 March 2020. Available online: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-mission-briefing-on-covid-19 (accessed on 28 April 2020).
- Situation Update for the EU/EEA and the UK, as of 30 December 2020. Available online: https://www.ecdc.europa.eu/en/cases-2019-ncov-eueea (accessed on 30 April 2020).
- Communicable Disease Threats Report, 1–7 March 2020, Week 10. Available online: https://www.ecdc.europa.eu/en/publications-data/communicable-disease-threats-report-1-7-march-2020-week-10 (accessed on 28 April 2020).
- Quick, J. Forked from Ebola virus sequencing protocol. Protocols.io 2020. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, P.M.; Szalay, D.; Kecskeméti, S.; Molnár, T.; Szabó, I.; Bálint, Á. Investigations on spreading of PRRSV among swine herds by improved minimum spanning network analysis. Sci. Rep. 2020, 10, 19217. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 28 April 2020).
- Csárdi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. Version 0.8.4. Available online: https://igraph.org/c/doc/igraph-docs.pdf (accessed on 15 November 2020).
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GitHub-hCoV-2019/Pangolin: Software Package for Assigning SARS-CoV-2 Genome Sequences to Global Lineages. Available online: https://github.com/hCoV-2019/pangolin (accessed on 30 April 2020).
- Rambaut, A.; Holmes, E.C.; Hill, V.; OToole, A.; McCrone, J.; Ruis, C.; du Plessis, L.; Pybus, O. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Barabási, A.L.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeling, M.J.; Eames, K.T. Networks and epidemic models. J. R. Soc. Interface 2005, 2, 295–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of ‘small-world’ networks. Nature 1998, 393, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA 2002, 99, 7821–7826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SARS-CoV-2 Lineages. Available online: https://cov-lineages.org/lineages/lineage_B.3.html (accessed on 29 November 2020).
- Nextstrain/Narratives/Ncov/Sit-Rep/2020-05-01. Available online: https://nextstrain.org/narratives/ncov/sit-rep/2020-05-01?f_region=Europe (accessed on 2 May 2020).
- Nextstrain. Available online: https://nextstrain.org/ (accessed on 29 April 2020).
- Remuzzi, A.; Remuzzi, G. COVID-19 and Italy: What next? Lancet 2020, 395, 1225–1228. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
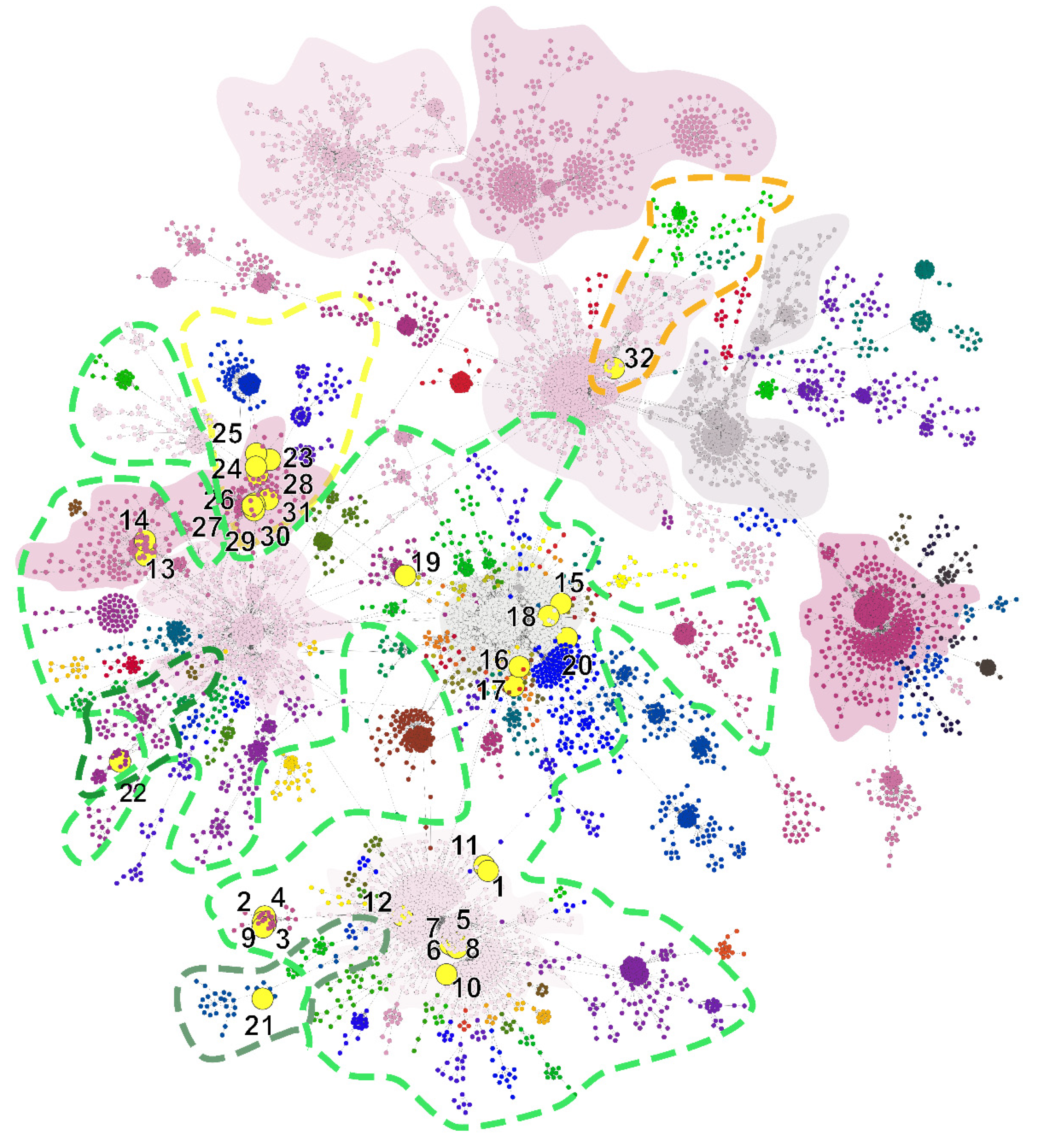
| Taxon Name | Lineage | SH-Alrt | UFbootstrap | Note | Cluster | |
|---|---|---|---|---|---|---|
| 1 | SARS-CoV-2/human/Hungary/49/20_03_2020 | B.1 | 100 | 100 | D | |
| 2 | SARS-CoV-2/human/Hungary/55/20_03_2020 | B.1 | 100 | 100 | E | |
| 3 | SARS-CoV-2/human/Hungary/126/22_03_2020 | B.1 | 100 | 100 | E | |
| 4 | SARS-CoV-2/human/Hungary/186/23_03_2020 | B.1 | 100 | 100 | E | |
| 5 | SARS-CoV-2/human/Hungary/105w/21_03_2020 | B.1 | 100 | 100 | B | |
| 6 | SARS-CoV-2/human/Hungary/278w/25_03_2020 | B.1 | 100 | 100 | A | |
| 7 | SARS-CoV-2/human/Hungary/2801w/25_03_2020 | B.1 | 100 | 100 | A | |
| 8 | SARS-CoV-2/human/Hungary/3670w/29_03_2020 | B.1 | 100 | 100 | B | |
| 9 | SARS-CoV-2/human/Hungary/541/27_03_2020 | B.1 | 100 | 100 | E | |
| 10 | SARS-CoV-2/human/Hungary/777/30_03_2020 | B.1 | 100 | 100 | D | |
| 11 | SARS-CoV-2/human/Hungary/175/23_03_2020 | B.1 | 100 | 100 | D | |
| 12 | SARS-CoV-2/human/Hungary/417/25_03_2020 | B.1 | 100 | 100 | E | |
| 13 | SARS-CoV-2/human/Hungary/3597w/28_03_2020 | B.1 | 100 | 100 | N | |
| 14 | SARS-CoV-2/human/Hungary/MBL-3/25_03_2020 | B.1 | 100 | 100 | Travel-related: France to Hungary | N |
| 15 | SARS-CoV-2/human/Hungary/67/20_03_2020 | B.1 | 100 | 100 | J | |
| 16 | SARS-CoV-2/human/Hungary/183/23_03_2020 | B.1 | 100 | 100 | K | |
| 17 | SARS-CoV-2/human/Hungary/419/26_03_2020 | B.1 | 100 | 100 | K | |
| 18 | SARS-CoV-2/human/Hungary/827/30_03_2020 | B.1 | 100 | 100 | J | |
| 19 | SARS-CoV-2/human/Hungary/836/30_03_2020 | B.1 | 100 | 100 | M | |
| 20 | SARS-CoV-2/human/Hungary/792/30_03_2020 | B.1 | 100 | 100 | L | |
| 21 | SARS-CoV-2/human/Hungary/817/30_03_2020 | B.1.1 | 100 | 93 | C | |
| 22 | SARS-CoV-2/human/Hungary/572w/29_03_2020 | B.1.11 | 100 | 99 | O | |
| 23 | SARS-CoV-2/human/Hungary/2/17_03_2020 | B.1.5 | 100 | 85 | G | |
| 24 | SARS-CoV-2/human/Hungary/MBL-2/23_03_2020 | B.1.5 | 100 | 74 | Household infection | F |
| 25 | SARS-CoV-2/human/Hungary/MBL-1/17_03_2020 | B.1.5 | 100 | 79 | Household infection | F |
| 26 | SARS-CoV-2/human/Hungary/66/20_03_2020 | B.1.5 | 100 | 94 | Travel-related: Spain to Hungary | G |
| 27 | SARS-CoV-2/human/Hungary/1788lc/19_03_2020 | B.1.5 | 100 | 93 | G | |
| 28 | SARS-CoV-2/human/Hungary/MBL-464/27_03_2020 | B.1.5 | 100 | 87 | Hospital cluster | H |
| 29 | SARS-CoV-2/human/Hungary/MBL-465/27_03_2020 | B.1.5 | 100 | 93 | Hospital cluster | H |
| 30 | SARS-CoV-2/human/Hungary/MBL-469/27_03_2020 | B.1.5 | 100 | 93 | Hospital cluster | H |
| 31 | SARS-CoV-2/human/Hungary/1136/02_04_2020 | B.1.5 | 85 | 76 | I | |
| 32 | SARS-CoV-2/human/Hungary/620/27_03_2020 | B.3 | 100 | 87 | P |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kemenesi, G.; Zeghbib, S.; Somogyi, B.A.; Tóth, G.E.; Bányai, K.; Solymosi, N.; Szabo, P.M.; Szabó, I.; Bálint, Á.; Urbán, P.; et al. Multiple SARS-CoV-2 Introductions Shaped the Early Outbreak in Central Eastern Europe: Comparing Hungarian Data to a Worldwide Sequence Data-Matrix. Viruses 2020, 12, 1401. https://doi.org/10.3390/v12121401
Kemenesi G, Zeghbib S, Somogyi BA, Tóth GE, Bányai K, Solymosi N, Szabo PM, Szabó I, Bálint Á, Urbán P, et al. Multiple SARS-CoV-2 Introductions Shaped the Early Outbreak in Central Eastern Europe: Comparing Hungarian Data to a Worldwide Sequence Data-Matrix. Viruses. 2020; 12(12):1401. https://doi.org/10.3390/v12121401
Chicago/Turabian StyleKemenesi, Gábor, Safia Zeghbib, Balázs A Somogyi, Gábor Endre Tóth, Krisztián Bányai, Norbert Solymosi, Peter M Szabo, István Szabó, Ádám Bálint, Péter Urbán, and et al. 2020. "Multiple SARS-CoV-2 Introductions Shaped the Early Outbreak in Central Eastern Europe: Comparing Hungarian Data to a Worldwide Sequence Data-Matrix" Viruses 12, no. 12: 1401. https://doi.org/10.3390/v12121401