
Hereditary Thrombophilia in the Era of COVID-19

 , , , , , and
, , , , , and
Abstract
:1. Introduction
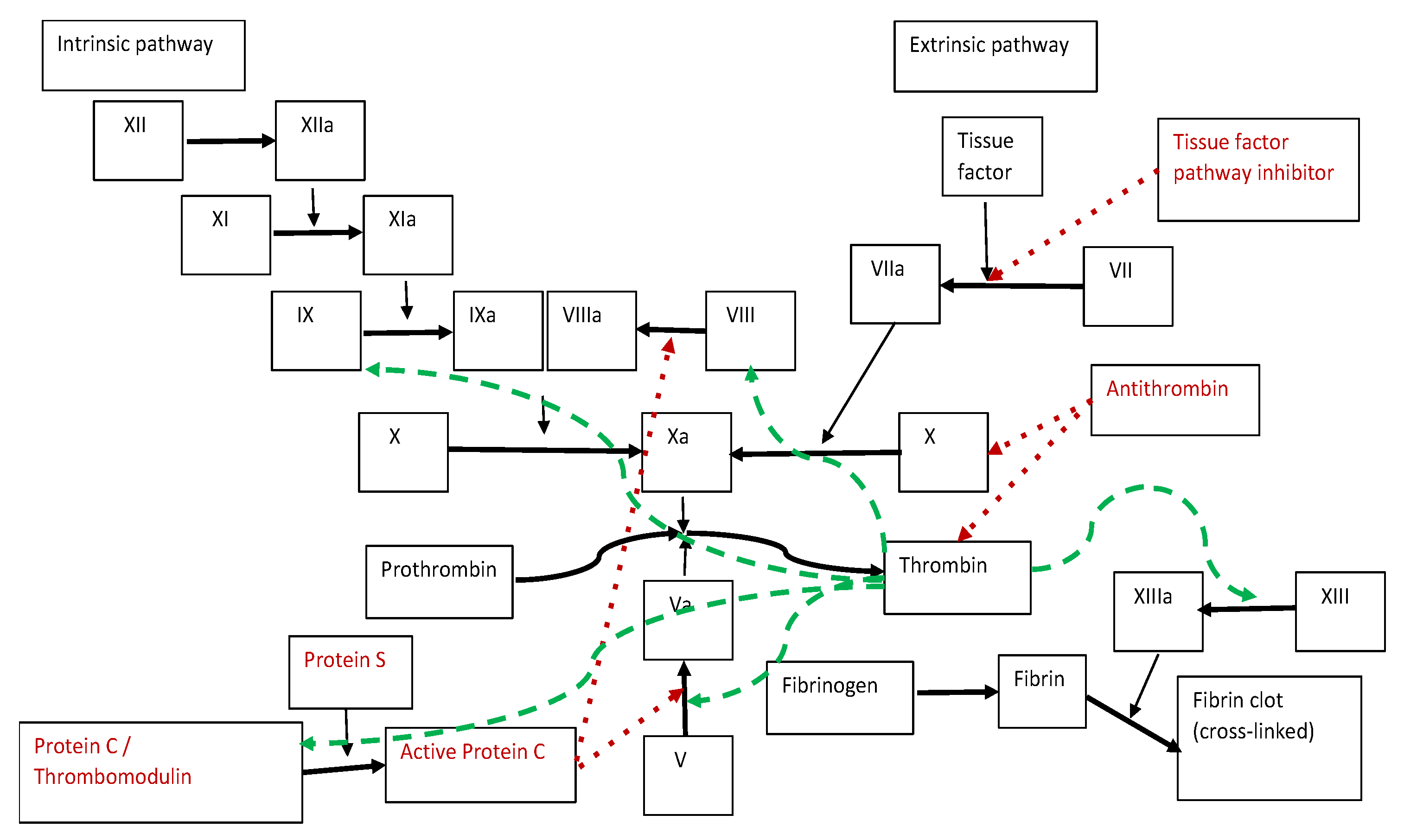
2. Blood Clotting Process
3. Overview of COVID-19
4. Inherited Thrombophilia
4.1. Antithrombin Deficiency
- -
- Type I deficiency: reduced synthesis of the molecule (both antigenic and functional activity of AT in the blood are reduced);
- -
- Type II deficiency: molecular defect (AT immunological activity is normal but functional activity is reduced);
- -
- Type III: affected interaction between AT and heparin [71].
4.2. Protein C Deficiency
4.3. Protein S Deficiency
- -
- Type I: characterized by decreased activated protein C cofactor activity, low values of total S protein and free S protein;
- -
- Type II: characterized by decreased activated protein C cofactor activity, normal values of total S protein and free S protein;
- -
- Type III: characterized by decreased activated protein C cofactor activity, normal values of total S protein and low values of free S protein [73].
4.4. Disturbances in Fibrinogen Levels
4.5. Elevated Homocysteine Levels
4.6. Factor II Mutation
4.7. Factor V Leiden
5. A Closer Look at the Correlation with COVID-19
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Montagnana, M.; Lippi, G.; Danese, E. An Overview of Thrombophilia and Associated Laboratory Testing. Hemostasis and Thrombosis. In Methods in Molecular Biology; Favaloro, E., Lippi, G., Eds.; Humana Press: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Tripodi, A.; Mannucci, P.M. Laboratory investigation of thrombophilia. Clin. Chem. 2001, 47, 1597–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siriez, R.; Dogné, J.M.; Gosselin, R.; Laloy, J.; Mullier, F.; Douxfils, J. Comprehensive review of the impact of direct oral anticoagulants on thrombophilia diagnostic tests: Practical recommendations for the laboratory. Int. J. Lab. Hematol. 2021, 43, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Khider, L.; Gendron, N.; Mauge, L. Inherited thrombophilia in the era of direct oral anticoagulants. Int. J. Mol. Sci. 2022, 23, 1821. [Google Scholar] [CrossRef] [PubMed]
- Badulescu, O.V.; Filip, N.; Sirbu, P.D.; Bararu-Bojan, I.; Vladeanu, M.; Bojan, A.; Ciocoiu, M. Current practices in haemophilic patients undergoing orthopedic surgery—A systematic review. Exp. Ther. Med. 2020, 20, 207. [Google Scholar] [CrossRef]
- Badulescu, O.V.; Sirbu, P.D.; Ungureanu, C.; Pȋnzariu, A.; Cojocaru, E.; Filip, N.; Bararu-Bojan, I.; Vladeanu, M.; Ciocoiu, M. Orthopedic surgery in hemophilic patients with musculoskeletal disorders: A systematic review. Exp. Ther. Med. 2021, 22, 995. [Google Scholar] [CrossRef]
- Badulescu, O.V.; Ciocoiu, M.; Filip, N.; Veringa, V. Tranexamic acid-major antifibrinolytic agent used to achieve hemostasis in hemophilic patients with anti-factor VIII antibodies who must undergo total joint replacement. Rev. Chim. Buchar. 2019, 70, 638–641. [Google Scholar] [CrossRef]
- Goldhaber, S.Z. Risk factors for venous thromboembolism. J. Am. Coll. Cardiol. 2010, 29, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Engbers, M.J.; Van Hylckama Vlieg, A.; Rosendaal, F.R. Venous thrombosis in the elderly: Incidence, risk factors and risk groups. J. Thromb. Haemost. 2010, 8, 2105–2112. [Google Scholar] [CrossRef]
- Cushman, M. Epidemiology and risk factors for venous thrombosis. Semin. Hematol. 2007, 44, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Li, W.; Xie, J.; Hou, Y.; You, C. Cytokine storm induced by SARS-CoV-2. Clin. Chim. Acta 2020, 509, 280–287. [Google Scholar] [CrossRef]
- Merrill, J.T.; Erkan, D.; Winakur, J.; James, J.A. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat. Rev. Rheumatol. 2020, 16, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Klok, F.A.; Kruip, M.J.H.A.; Van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.A.M.P.J.; Kant, K.M.; Kaptein, F.H.J.; van Passen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Gąsecka, A.; Borovac, J.A.; Guerreiro, R.A.; Giustozzi, M.; Parker, W.; Caldeira, D.; Chiva-Blanch, G. Thrombotic complications in patients with COVID-19: Pathophysiological mechanisms, diagnosis, and treatment. Cardiovasc. Drugs Ther. 2021, 35, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Long, B.; Holladay, D.; Gottlieb, M. Thrombotic complications of COVID-19. Am. J. Emerg. Med. 2021, 39, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Losso, J.N. The potential of dietary bioactive compounds against SARS-CoV-2 and COVID-19-induced endothelial dysfunction. Molecules 2022, 27, 1623. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Stevens, H.; Peter, K. The emerging threat of (micro) thrombosis in COVID-19 and its therapeutic implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef]
- Barnes, G.D.; Burnett, A.; Allen, A.; Blumenstein, M.; Clark, N.P.; Cuker, A.; Dager, W.E.; Deitelzweig, S.B.; Ellsworth, S.; Garcia, D.; et al. Thromboembolism and anticoagulant therapy during the COVID-19 pandemic: Interim clinical guidance from the anticoagulation forum. J. Thromb. Thrombolysis. 2020, 50, 72–81. [Google Scholar] [CrossRef]
- Hill, J.; Oderberg, D.S.; Gibbins, J.M.; Bojak, I. Mistake-making: A theoretical framework for generating research questions in biology, with illustrative application to blood clotting. Q. Rev. Biol. 2022, 97, 1–13. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Pieters, M.; De Lange-Loots, Z.; Weisel, J.W. Fibrinogen and Fibrin. In Macromolecular Protein Complexes III: Structure and Function; Harris, J.R., Marles-Wright, J., Eds.; Springer: Cham, Switzerland, 2021; Volume 94, pp. 471–501. [Google Scholar] [CrossRef]
- Kumar, R. Physiology, coagulation cascade: Inherited disorders, and the molecular phenomenon of alterations in hemostasis. J. Clin. Haematol. 2021, 2, 62–64. [Google Scholar]
- Gailani, D.; Renné, T. Intrinsic pathway of coagulation and arterial thrombosis. Arter. Thromb Vasc. Biol. 2007, 27, 2507–2513. [Google Scholar] [CrossRef]
- Chaudhry, R.; Usama, S.M.; Babiker, H.M. Physiology, Coagulation Pathways; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Hamed, M.A. An overview on COVID-19: Reality and expectation. Bull. Natl. Res. Cent. 2020, 44, 86. [Google Scholar] [CrossRef]
- Yang, L.; Liu, S.; Liu, J.; Zhang, Z.; Wan, X.; Huang, B.; Chen, Y.; Zhang, Y. COVID-19: Immunopathogenesis and immunotherapeutics. Signal Transduct. Target Ther. 2020, 5, 128. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Xu, S. Comprehensive overview of COVID-19 based on current evidence. Dermatol. Ther. 2020, 33, e13525. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Chen, C.S.; Chan, Y.J. The outbreak of COVID-19: An overview. J. Chin. Med. Assoc. 2020, 83, 217–220. [Google Scholar] [CrossRef]
- Zeng, J.H.; Liu, Y.X.; Yuan, J.; Wang, F.X.; Wu, W.B.; Li, J.X.; Wang, L.F.; Gao, H.; Wang, Y.; Dong, C.F.; et al. First case of COVID-19 complicated with fulminant myocarditis: A case report and insights. Infection 2020, 48, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Alhogbani, T. Acute myocarditis associated with novel Middle east respiratory syndrome coronavirus. Ann. Saudi Med. 2016, 36, 78–80. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Wu, D.; Chen, H.; Yan, W.; Yang, D.; Chen, G.; Ma, K.; Xu, D.; Yu, H.; Wang, H.; et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ 2020, 368, m1091. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Driggin, E.; Madhavan, M.V.; Bikdeli, B.; Chuich, T.; Laracy, J.; Biondi-Zoccai, G.; Brown, T.S.; Der Nigoghossian, C.; Zidar, D.A.; Haythe, J.; et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. J. Am. Coll. Cardiol. 2020, 75, 2352–2371. [Google Scholar] [CrossRef]
- Sedaghat, Z.; Karimi, N. Guillain Barre syndrome associated with COVID-19 infection: A case report. J. Clin. Neurosci. 2020, 76, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Paybast, S.; Gorji, R.; Mavandadi, S. Guillain-Barré Syndrome as a neurological complication of novel COVID-19 infection: A case report and review of the literature. Neurologist 2020, 25, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Diez-Porras, L.; Vergés, E.; Gil, F.; Vidal, M.J.; Massons, J.; Arboix, A. Guillain-Barré-Strohl syndrome and COVID-19: Case report and literature review. Neuromuscul. Disord. 2020, 30, 859–861. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Lima, A.A.; Chandra, R.; Valeriano, J.; Desai, T.; Freiberg, W.; Small, G. Novel coronavirus (COVID-19)-Associated Guillain-Barré syndrome: Case Report. J. Clin. Neuromuscul. Dis. 2020, 21, 240–242. [Google Scholar] [CrossRef]
- Zito, A.; Alfonsi, E.; Franciotta, D.; Todisco, M.; Gastaldi, M.; Cotta Ramusino, M.; Ceroni, M.; Costa, A. COVID-19 and Guillain-Barré syndrome: A case report and review of literature. Front. Neurol. 2020, 11, 909. [Google Scholar] [CrossRef]
- Ottaviani, D.; Boso, F.; Tranquillini, E.; Gapeni, I.; Pedrotti, G.; Cozzio, S.; Guarrera, G.M.; Giometto, B. Early Guillain-Barré syndrome in coronavirus disease 2019 (COVID-19): A case report from an Italian COVID-hospital. Neurol. Sci. 2020, 41, 1351–1354. [Google Scholar] [CrossRef]
- Ye, M.; Ren, Y.; Lv, T. Encephalitis as a clinical manifestation of COVID-19. Brain Behav. Immun. 2020, 88, 945–946. [Google Scholar] [CrossRef]
- Filatov, A.; Sharma, P.; Hindi, F.; Espinosa, P.S. Neurological complications of coronavirus disease (COVID-19): Encephalopathy. Cureus 2020, 12, e7352. [Google Scholar] [CrossRef] [Green Version]
- Bridwell, R.; Long, B.; Gottlieb, M. Neurologic complications of COVID-19. Am. J. Emerg. Med. 2020, 38, e3–e1549. [Google Scholar] [CrossRef]
- Ucpinar, B.A.; Sahin, C.; Yanc, U. Spontaneous pneumothorax and subcutaneous emphysema in COVID-19 patient: Case report. J. Infect. Public Health 2020, 13, 887–889. [Google Scholar] [CrossRef]
- Al-Shokri, S.D.; Ahmed, A.O.E.; Saleh, A.O.; AbouKamar, M.; Ahmed, K.; Mohamed, M.F.H. Case report: COVID-19-related pneumothorax-case series highlighting a significant complication. Am. J. Trop. Med. Hyg. 2020, 103, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Alhakeem, A.; Khan, M.M.; Al Soub, H.; Yousaf, Z. Case report: COVID-19-Associated bilateral spontaneous pneumothorax-a literature review. Am. J. Trop. Med. Hyg. 2020, 103, 1162–1165. [Google Scholar] [CrossRef] [PubMed]
- Hazariwala, V.; Hadid, H.; Kirsch, D.; Big, C. Spontaneous pneumomediastinum, pneumopericardium, pneumothorax and subcutaneous emphysema in patients with COVID-19 pneumonia, a case report. J. Cardiothorac. Surg. 2020, 15, 301. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Guangming, L.; Wei, L.; Xuedong, Y. Spontaneous pneumomediastinum, pneumothorax and subcutaneous emphysema in COVID-19: Case report and literature review. Rev. Inst. Med. Trop. Sao Paulo 2020, 62, e76. [Google Scholar] [CrossRef]
- Volpi, S.; Ali, J.M.; Suleman, A.; Ahmed, R.N. Pneumomediastinum in COVID-19 patients: A case series of a rare complication. Eur. J. Cardiothorac. Surg. 2020, 58, 646–647. [Google Scholar] [CrossRef] [PubMed]
- Quincho-Lopez, A.; Quincho-Lopez, D.L.; Hurtado-Medina, F.D. Case report: Pneumothorax and pneumomediastinum as uncommon complications of COVID-19 pneumonia-literature review. Am. J. Trop. Med. Hyg. 2020, 103, 1170–1176. [Google Scholar] [CrossRef]
- Garg, D.; Muthu, V.; Sehgal, I.S.; Ramachandran, R.; Kaur, H.; Bhalla, A.; Puri, G.D.; Chakrabarti, A.; Agarwal, R. Coronavirus disease (COVID-19) associated mucormycosis (CAM): Case report and systematic review of literature. Mycopathologia 2021, 186, 289–298. [Google Scholar] [CrossRef]
- Akel, T.; Qaqa, F.; Abuarqoub, A.; Shamoon, F. Pulmonary embolism: A complication of COVID 19 infection. Thromb. Res. 2020, 193, 79–82. [Google Scholar] [CrossRef]
- Gasparotto, M.; Framba, V.; Piovella, C.; Doria, A.; Iaccarino, L. Post-COVID-19 arthritis: A case report and literature review. Clin. Rheumatol. 2021, 40, 3357–3362. [Google Scholar] [CrossRef]
- Melquist, S.; Estepp, K.; Aleksandrovich, Y.; Lee, A.; Beiseker, A.; Hamedani, F.S.; Bassett, J. COVID-19 presenting as fulminant hepatic failure: A case report. Medicine 2020, 99, e22818. [Google Scholar] [CrossRef]
- Chacko, M.; Job, A.; Caston, F.; George, P.; Yacoub, A.; Cáceda, R. COVID-19-Induced psychosis and suicidal behavior: Case report. SN Compr. Clin. Med. 2020, 2, 2391–2395. [Google Scholar] [CrossRef] [PubMed]
- Huarcaya-Victoria, J.; Herrera, D.; Castillo, C. Psychosis in a patient with anxiety related to COVID-19: A case report. Psychiatry Res. 2020, 289, 113052. [Google Scholar] [CrossRef] [PubMed]
- Chandra, P.S.; Shiva, L.; Nagendrappa, S.; Ganjekar, S.; Thippeswamy, H. COVID 19 related Psychosis as an interface of fears, socio-cultural issues and vulnerability—Case report of two women from India. Psychiatry Res. 2020, 290, 113136. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Florido, M.J.; López-Díaz, Á.; Palermo-Zeballos, F.J.; Martínez-Molina, I.; Martín-Gil, V.E.; Crespo-Facorro, B.; Ruiz-Veguilla, M. Reactive psychoses in the context of the COVID-19 pandemic: Clinical perspectives from a case series. Rev. Psiquiatr. Salud Ment. 2020, 13, 90–94. [Google Scholar] [CrossRef]
- Paul, T.; Joy, A.R.; Alsoub, H.A.R.S.; Parambil, J.V. Case Report: Ischemic colitis in severe COVID-19 pneumonia: An unforeseen gastrointestinal complication. Am. J. Trop. Med. Hyg. 2021, 104, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.; Wolf, J.M. Digital ischemia in COVID-19 patients: Case report. J. Hand Surg. Am. 2020, 45, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.; Bunker, C.B.; Ciurtin, C.; Porter, J.C.; Chambers, R.C.; Papdopoulou, C.; Garthwaite, H.; Hillman, T.; Heightman, M.; Howell, K.J.; et al. Chilblain-like acral lesions in long COVID-19: Management and implications for understanding microangiopathy. Lancet Infect. Dis. 2021, 21, 912. [Google Scholar] [CrossRef]
- Ciotti, M.; Ciccozzi, M.; Terrinoni, A.; Jiang, W.C.; Wang, C.B.; Bernardini, S. The COVID-19 pandemic. Crit. Rev. Clin. Lab. Sci. 2020, 57, 365–388. [Google Scholar] [CrossRef]
- Zimmermann, P.; Pittet, L.F.; Curtis, N. How common is long COVID in children and adolescents? Pediatr. Infect. Dis. J. 2021, 40, e482–e487. [Google Scholar] [CrossRef]
- Zimmermann, P.; Pittet, L.F.; Curtis, N. Long COVID in children and adolescents. BMJ 2022, 376, o143. [Google Scholar] [CrossRef]
- Siebach, M.K.; Piedimonte, G.; Ley, S.H. COVID-19 in childhood: Transmission, clinical presentation, complications and risk factors. Pediatr. Pulmonol. 2021, 56, 1342–1356. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.C.; Chow, V.C.; Lee, M.K.; Lai, R.W. Deep throat saliva as an alternative diagnostic specimen type for the detection of SARS-CoV-2. J. Med. Virol. 2021, 93, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, E.; Bamford, A.; Kenny, J.; Kaforou, M.; Jones, C.E.; Shah, P.; Ramnarayan, P.; Fraisse, A.; Miller, O.; Davies, P.; et al. PIMS-TS study group and EUCLIDS and PERFORM consortia. clinical characteristics of 58 children with a pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2. JAMA 2020, 324, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020, 395, 1771–1778. [Google Scholar] [CrossRef]
- Swann, O.V.; Holden, K.A.; Turtle, L.; Pollock, L.; Fairfield, C.J.; Drake, T.M.; Seth, S.; Egan, C.; Hardwick, H.E.; Halpin, S.; et al. ISARIC4C investigators. Clinical characteristics of children and young people admitted to hospital with COVID-19 in United Kingdom: Prospective multicentre observational cohort study. BMJ 2020, 370, m3249. [Google Scholar] [CrossRef]
- Zhang, F.; Xiong, Y.; Wei, Y.; Hu, Y.; Wang, F.; Li, G.; Liu, K.; Du, R.; Wang, C.Y.; Zhu, W. Obesity predisposes to the risk of higher mortality in young COVID-19 patients. J. Med. Virol. 2020, 92, 2536–2542. [Google Scholar] [CrossRef]
- Leon-Abarca, J.A. Obesity and immunodeficiencies are the main pre-existing conditions associated with mild to moderate COVID-19 in children. Pediatr. Obes. 2020, 15, e12713. [Google Scholar] [CrossRef]
- Khan, S.; Dickerman, J.D. Hereditary thrombophilia. Thromb. J. 2006, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Aydin, S.; Ugur, K.; Yalcin, H.; Sahin, İ.; Akkoc, R.F.; Yakar, B.; Yucel, D.; Aydin, S. Overview of COVID-19’s relationship with thrombophilia proteins. Turk. J. Biochem. 2021, 46, 609–622. [Google Scholar] [CrossRef]
- Ten Kate, M.K.; Van der Meer, J. Protein S deficiency: A clinical perspective. Haemophilia 2008, 14, 1222–1228. [Google Scholar] [CrossRef]
- Juhl, D.; Kuta, P.; Shneyder, M.; Wünsche, F.; Nowak-Göttl, U. Two novel variants in the protein S Gene PROS1 are associated with protein S deficiency and thrombophilia. Acta Haematol. 2021, 144, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, J.L.; Lijfering, W.M.; Ten Kate, M.K.; Kluin-Nelemans, H.C.; Veeger, N.J.; Van der Meer, J. High long-term absolute risk of recurrent venous thromboembolism in patients with hereditary deficiencies of protein S, protein C or antithrombin. Thromb. Haemostasis. 2009, 101, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Verhovsek, M.; Moffat, K.A.; Hayward, C.P. Laboratory testing for fibrinogen abnormalities. Am. J. Hematol. 2008, 83, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Haverkate, F.; Samama, M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen. Thromb. Haemost. 1995, 73, 151–161. [Google Scholar] [PubMed]
- Filip, A.; Badulescu, O.V.; Sirbu, P.D.; Cojocaru, E.; Filip, N.; Puha, G.; Trandafir, L.; Iancu, C.; Trandafirescu, M.F.; Alexa, O. Serum homocysteine and reactive species levels in fragility fractures of the pelvis. Revista de Chimie 2019, 70, 3216–3219. [Google Scholar] [CrossRef]
- Filip, C.; Albu, E.; Lupascu, D.; Filip, N. The influence of a new rutin derivative in an experimental model of induced hyperhomocysteinemia in rats. Farmacia 2017, 65, 596–599. [Google Scholar]
- Albu, E.; Filip, C.; Zamosteanu, N.; Jaba, I.M.; Linic, I.S.; Sosa, I. Hyperhomocysteinemia is an indicator of oxidant stress. Med. Hypotheses 2012, 78, 554–555. [Google Scholar] [CrossRef]
- Filip, C.; Albu, E.; Zamosteanu, N.; Silion, M.; Jerca, L.; Gheroghita, N.; Mungiu, C. Hyperhomocysteinemia’s effect on antioxidant capacity in rats. Open Med. 2010, 5, 620–626. [Google Scholar] [CrossRef]
- Albu, E.; Lupascu, D.; Filip, C.; Jaba, I.M.; Zamosteanu, N. The influence of a new rutin derivative on homocysteine, cholesterol and total antioxidative status in experimental diabetes in rat. Farmacia 2013, 61, 1167–1177. [Google Scholar]
- Filip, N.; Iancu, C.E. Introductory Chapter: General Aspects Regarding Homocysteine. In Non-Proteinogenic Amino Acids; IntechOpen: London, UK, 2018. [Google Scholar]
- Filip, C.; Socolov, D.G.; Albu, E.; Filip, C.; Serban, R.; Popa, R.F. Serological parameters and vascular investigation for a better assessment in dvt during pregnancy—A systematic review. Medicina 2021, 57, 160. [Google Scholar] [CrossRef]
- Trandafir, L.M.; Russu, G.; Moscalu, M.; Miron, I.; Lupu, V.V.; Leon Constantin, M.M.; Cojocaru, E.; Lupu, A.; Frasinariu, O.E. Waist circumference a clinical criterion for prediction of cardio-vascular complications in children and adolescences with overweight and obesity. Medicine 2020, 99, e20923. [Google Scholar] [CrossRef] [PubMed]
- Buca, B.R.; Tartau, L.M.; Rezus, C.; Filip, C.; Pinzariu, A.C.; Rezus, E.; Popa, G.E.; Panainte, A.; Lupusoru, C.E.; Bogdan, M.; et al. The effects of two nitric oxide donors in acute inflammation in rats. Rev. Chim. 2018, 69, 2899–2903. [Google Scholar] [CrossRef]
- Moroşan, E.; Mihailovici, M.S.; Giuşcă, S.E.; Cojocaru, E.; Avădănei, E.R.; Căruntu, I.D.; Teleman, S. Hepatic steatosis background in chronic hepatitis B and C—Significance of similarities and differences. Rom. J. Morphol. Embryol. 2014, 55, 1041–1047. [Google Scholar] [PubMed]
- Ridker, P.M.; Hennekens, C.H.; Selhub, J.; Miletich, J.P.; Malinow, M.R.; Stampfer, M.J. Interrelation of hyperhomocyst(e)inemia, factor V Leiden, and risk of future venous thromboembolism. Circulation 1997, 95, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.A.; Moll, S. Cardiology patient pages. Prothrombin 20210 mutation (factor II mutation). Circulation 2004, 20, e15-18. [Google Scholar] [CrossRef] [Green Version]
- Emmerich, J.; Rosendaal, F.R.; Cattaneo, M.; Margaglione, M.; De Stefano, V.; Cumming, T.; Arruda, V.; Hillarp, A.; Reny, J.L. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism-pooled analysis of 8 case-control studies including 2310 cases and 3204 controls. Study group for pooled-analysis in venous thromboembolism. Thromb. Haemost. 2001, 86, 809–816, Erratum in: Thromb. Haemost. 2001, 86, 1598. [Google Scholar]
- De Stefano, V.; Chiusolo, P.; Paciaroni, K.; Leone, G. Epidemiology of factor V Leiden: Clinical implications. Semin. Thromb. Hemost. 1998, 24, 367–379. [Google Scholar] [CrossRef]
- Gazzaruso, C.; Paolozzi, E.; Valenti, C.; Brocchetta, M.; Naldani, D.; Grignani, C.; Salvucci, F.; Marino, F.; Coppola, A.; Gallotti, P. Association between antithrombin and mortality in patients with COVID-19. A possible link with obesity. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1914–1919. [Google Scholar] [CrossRef]
- Kujovich, J.L. Factor V Leiden Thrombophilia. In GeneReviews; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gardner, A.J.; Kirkin, D.J.; Rodriguez-Villar, S.; Leoz Abellanas, G.; Tee, A.; Valentin, A. Antithrombin III deficiency-induced coagulopathy in the context of COVID-19: A case series. Br. J. Haematol. 2021, 194, 1007–1009. [Google Scholar] [CrossRef]
- Dinarvand, P.; Moser, K.A. Protein C deficiency. Arch. Pathol. Lab. Med. 2019, 143, 1281–1285. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Chen, M.; Tang, S.; Yu, W. Association of coagulation disturbances with severity of COVID-19: A longitudinal study. Hematology 2021, 26, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Hardy, M.; Michaux, I.; Lessire, S.; Douxfils, J.; Dogné, J.M.; Bareille, M.; Horlait, G.; Bulpa, P.; Chapelle, C.; Laporte, S.; et al. Prothrombotic disturbances of hemostasis of patients with severe COVID-19: A prospective longitudinal observational study. Thromb. Res. 2021, 197, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, E.; Sartre, B.; Squara, F.; Contenti, J.; Occelli, C.; Lemoel, F.; Levraut, J.; Doyen, D.; Dellamonica, J.; Mondain, V.; et al. High prevalence of acquired thrombophilia without prognosis value in patients with coronavirus disease 2019. J. Am. Heart Assoc. 2020, 9, e017773. [Google Scholar] [CrossRef] [PubMed]
- De la Morena-Barrio, M.E.; Bravo-Pérez, C.; De la Morena-Barrio, B.; Orlando, C.; Cifuentes, R.; Padilla, J.; Miñano, A.; Herrero, S.; Marcellini, S.; Revilla, N.; et al. A pilot study on the impact of congenital thrombophilia in COVID-19. Eur. J. Clin. Investig. 2021, 51, e13546. [Google Scholar] [CrossRef]
- Burlacu, A.; Artene, B.; Crisan-Dabija, R.; Popa, I.V.; Covic, A. Is thrombophilic genetic profile responsible for an acute ischemic stroke in a COVID-19 male patient? Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620967107. [Google Scholar] [CrossRef]
- Bussani, R.; Schneider, E.; Zentilin, L.; Collesi, C.; Ali, H.; Braga, L.; Volpe, M.C.; Colliva, A.; Zanconati, F.; Berlot, G.; et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. EBio Med. 2020, 61, 103104. [Google Scholar] [CrossRef]
- Yuriditsky, E.; Horowitz, J.M.; Merchan, C.; Ahuja, T.; Brosnahan, S.B.; McVoy, L.; Berger, J.S. Thromboelastography profiles of critically Ill patients with coronavirus disease 2019. Crit. Care Med. 2020, 48, 1319–1326. [Google Scholar] [CrossRef]
- Brosnahan, S.B.; Smilowitz, N.R.; Amoroso, N.E.; Barfield, M.; Berger, J.S.; Goldenberg, R.; Ishida, K.; Talmor, N.; Torres, J.; Yaghi, S.; et al. Thrombosis at hospital presentation in patients with and without coronavirus disease 2019. J. Vasc. Surg. Venous Lymphat. Disord. 2021, 9, 845–852. [Google Scholar] [CrossRef]
- Abdullaev, A.; Fevraleva, I.; Odilov, A.; Volkov, A.; Babichenko, I.; Sudarikov, A. Thrombotic events and the profile of hereditary thrombophilia factors in COVID-19 patients. HemaSphere 2021, 5, 641. [Google Scholar]
- Zhang, Y.; Guo, R.; Kim, S.H.; Shah, H.; Zhang, S.; Liang, J.H.; Fang, Y.; Gentili, M.; Leary, C.N.O.; Elledge, S.J.; et al. SARS-CoV-2 hijacks folate and one-carbon metabolism for viral replication. Nat. Commun. 2021, 12, 1676. [Google Scholar] [CrossRef]
- Karst, M.; Hollenhorst, J.; Achenbach, J. Life-threatening course in coronavirus disease 2019 (COVID-19): Is there a link to methylenetetrahydrofolic acid reductase (MTHFR) polymorphism and hyperhomocysteinemia? Med. Hypotheses 2020, 144, 110234. [Google Scholar] [CrossRef] [PubMed]
- Cappadona, C.; Paraboschi, E.M.; Ziliotto, N.; Bottaro, S.; Rimoldi, V.; Gerussi, A.; Azimonti, A.; Brenna, D.; Brunati, A.; Cameroni, C.; et al. MEDTEC Students against Coronavirus: Investigating the role of hemostatic genes in the predisposition to COVID-19 Severity. J. Pers. Med. 2021, 11, 1166. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Roli, L.; Oliva, G.; Manfredini, M.; Trenti, T.; Kaleci, S.; Iannella, R.; Balzano, B.; Coppola, A.; Fiorentino, G.; et al. Homocysteine (Hcy) assessment to predict outcomes of hospitalized COVID-19 patients: A multicenter study on 313 COVID-19 patients. Clin. Chem. Lab. Med. 2021, 59, e354–e357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, W.; Jiang, W.; Xiao, M.; Li, Y.; Tang, N.; Liu, Z.; Yan, X.; Zhao, Y.; Li, T.; et al. Profile of natural anticoagulant, coagulant factor and anti-phospholipid antibody in critically ill COVID-19 patients. J. Thromb. Thrombolysis 2020, 50, 580–586. [Google Scholar] [CrossRef]
- Nogueira, R.G.; Abdalkader, M.; Qureshi, M.M.; Frankel, M.R.; Mansour, O.Y.; Yamagami, H.; Qiu, Z.; Farhoudi, M.; Siegler, J.E.; Yaghi, S.; et al. Global impact of COVID-19 on stroke care. Int J. Stroke 2021, 16, 573–584. [Google Scholar] [CrossRef]
- Topcuoglu, M.A.; Pektezel, M.Y.; Oge, D.D.; Bulut Yüksel, N.D.; Ayvacioglu, C.; Demirel, E.; Balci, S.; Arat, A.; Akinci, S.B.; Arsava, E.M. Stroke mechanism in COVID-19 infection: A prospective case-control study. J. Stroke Cerebrovasc. Dis. 2021, 30, 105919. [Google Scholar] [CrossRef]


{kind=link}
| Types of Disease | References |
|---|---|
| Cardiovascular | |
| Myocarditis | [29,30] |
| Acute myocardial infarction | [31] |
| Acute heart failure and cardiomyopathy | [31,32] |
| Dysrhythmias | [33] |
| Neurologic | |
| Guillain-Barré Syndrome | [34,35,36,37,38,39] |
| Encephalitis and encephalopathy | [40,41] |
| Acute cerebrovascular disease | [42] |
| Pulmonary | |
| Pneumothorax | [43,44,45,46,47,48,49] |
| Pneumomediastinum | [43,44,45,46,47,48] |
| Pulmonary mucormycosis | [50] |
| Pulmonary embolism | [51] |
| Rheumatologic | |
| Acute arthritis | [52] |
| Hepatic | |
| Acute liver failure | [53] |
| Neuropsychiatric | |
| Psychosis and suicidal behavior | [54,55,56,57] |
| Gastrointestinal | |
| Ischemic colitis | [58] |
| Dermatologic | |
| Digital ischemia | [59] |
| Chilblains | [60] |
| Evaluated Parameters | Comments | Author (Reference) |
|---|---|---|
| Activated partial thromboplastin time, prothrombin time, D-dimer, fibrinogen, protein C, protein S, antithrombin deficiencies, antiphospholipid antibodies | Although they reported a high prevalence of positive thrombophilia tests in COVID-19 infections, with a 20% prevalence of protein S deficiency, they are not correlated with the severity or prognosis of COVID-19. | Ferrari et al. [98] |
| Genes encoding antithrombin (SERPINC1), protein C and protein S | No thrombotic events were found in most subjects with severe thrombophilia during SARS-CoV-2 infection. | de la Morena-Barrio et al. [99] |
| Factor V Leiden, factor V 4070 A > G (Hr2), factor II G20210A, MTHFR C677 T, MTHFR A1298C, CBS 844ins68, PAI-1 4G/5G, glycoprotein IIIa T1565C (HPA-1a/b), ACE D/I, apolipoprotein E, AGT M235 T, ATR-1 A1166C, fibrinogen 455 G > A and factor XIII Val34Leu | One patient tested positive for apolipoprotein E e3/e4, heterozygous MTHFR A1298C, heterozygous ACE D/I, heterozygous AGT M235 T, and heterozygous FXIII Val34Leu. | Burlacu et al. [100] |
| Histological analysis and immunohistochemistry for a number of 41 patients who died of COVID-19 | Increased alveolar damage and micro/macro vascular pulmonary thrombosis. | Bussani et al. [101] |
| Thromboelastography, D-dimer, fibrinogen, C-reactive protein, ferritin | Changes in fibrinogen, platelets and hypercoagulable thromboelastography profiles. | Yuriditsky et al. [102] |
| D-dimer, Alanine transaminase, C-reactive protein, erythrocyte sedimentation rate, partial thromboplastin time, white blood cell | Changes in the complete blood count, liver function test results, D-dimer levels, C-reactive protein, ferritin and coagulation panels. | Brosnahan et al. [103] |
| FV 506R/Q, MTHFR 223A/V, F2 20210G/A and PAI-1 4G/5G alleles | COVID-19 positive patients who died of thrombotic complications had no signs of predisposition to hereditary thrombophilia. | Abdullaev et al. [104] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badulescu, O.V.; Sirbu, P.D.; Filip, N.; Bordeianu, G.; Cojocaru, E.; Budacu, C.C.; Badescu, M.C.; Bararu-Bojan, I.; Veliceasa, B.; Ciocoiu, M. Hereditary Thrombophilia in the Era of COVID-19. Healthcare 2022, 10, 993. https://doi.org/10.3390/healthcare10060993
Badulescu OV, Sirbu PD, Filip N, Bordeianu G, Cojocaru E, Budacu CC, Badescu MC, Bararu-Bojan I, Veliceasa B, Ciocoiu M. Hereditary Thrombophilia in the Era of COVID-19. Healthcare. 2022; 10(6):993. https://doi.org/10.3390/healthcare10060993
Chicago/Turabian StyleBadulescu, Oana Viola, Paul Dan Sirbu, Nina Filip, Gabriela Bordeianu, Elena Cojocaru, Cristian Constantin Budacu, Minerva Codruta Badescu, Iris Bararu-Bojan, Bogdan Veliceasa, and Manuela Ciocoiu. 2022. "Hereditary Thrombophilia in the Era of COVID-19" Healthcare 10, no. 6: 993. https://doi.org/10.3390/healthcare10060993