
In Vivo and Ex Vivo Mitochondrial Function in COVID-19 Patients on the Intensive Care Unit

 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Design and Setting
2.2. Variables
2.3. MitoPO2 and mitoVO2 Measurements
2.4. Sample Preparation
2.5. High-Resolution Respirometry
2.6. mtDNA Isolation from Plasma
2.7. Quantative Polymerase Chain Reaction (qPCR)
2.8. Data and Statistical Analysis
3. Results
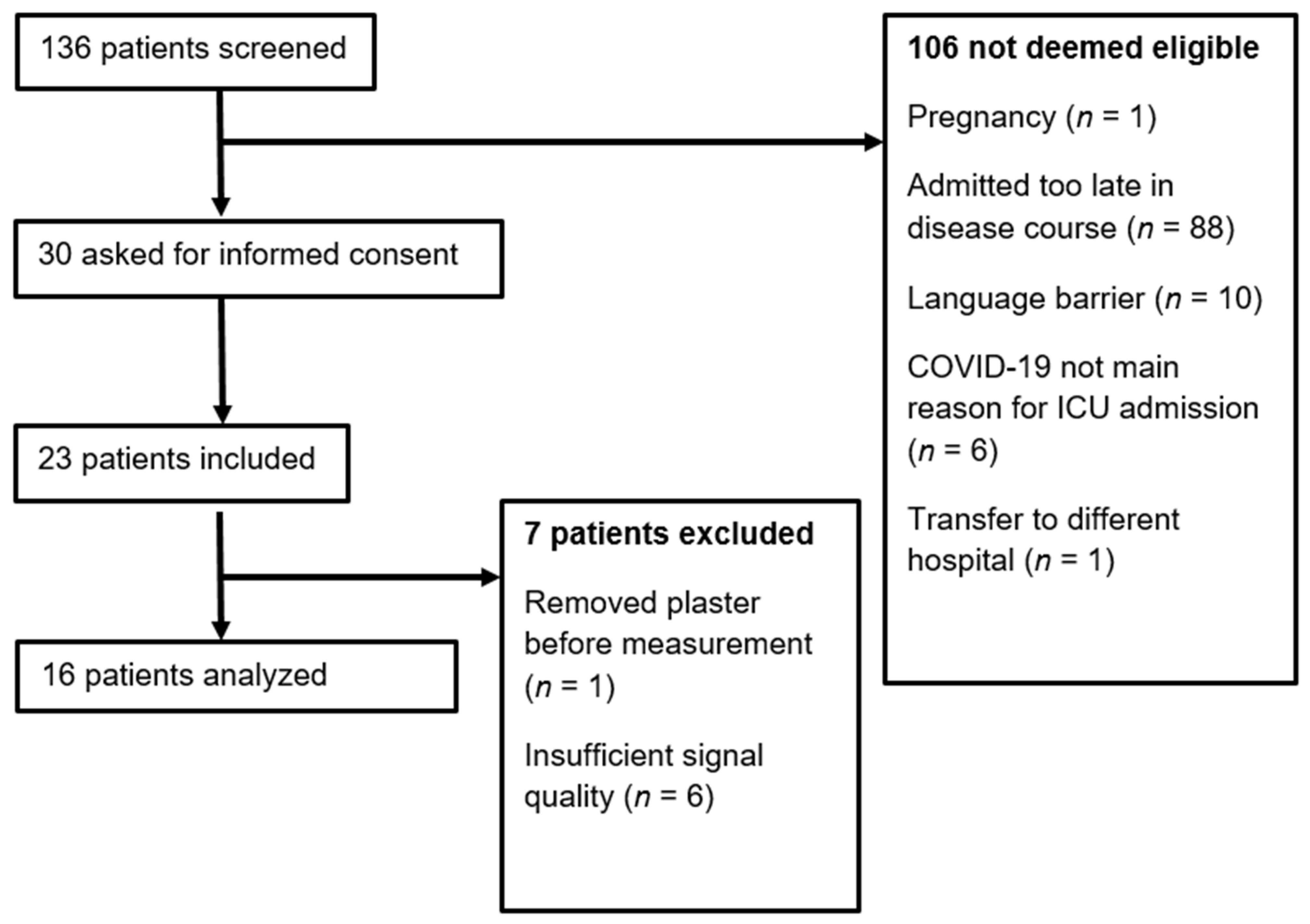
3.1. Descriptive Statistics
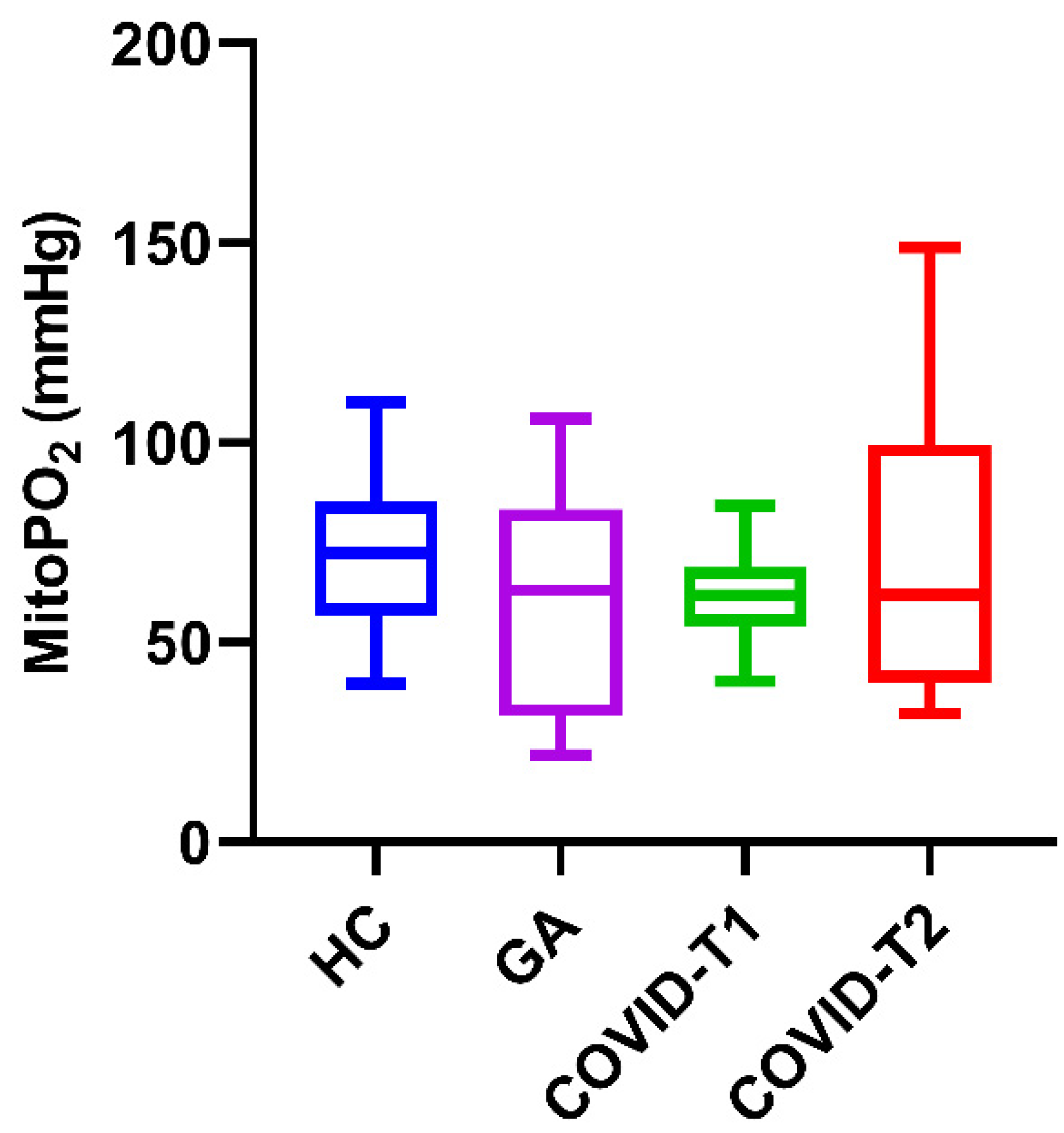
3.2. MitoPO2 Measurements
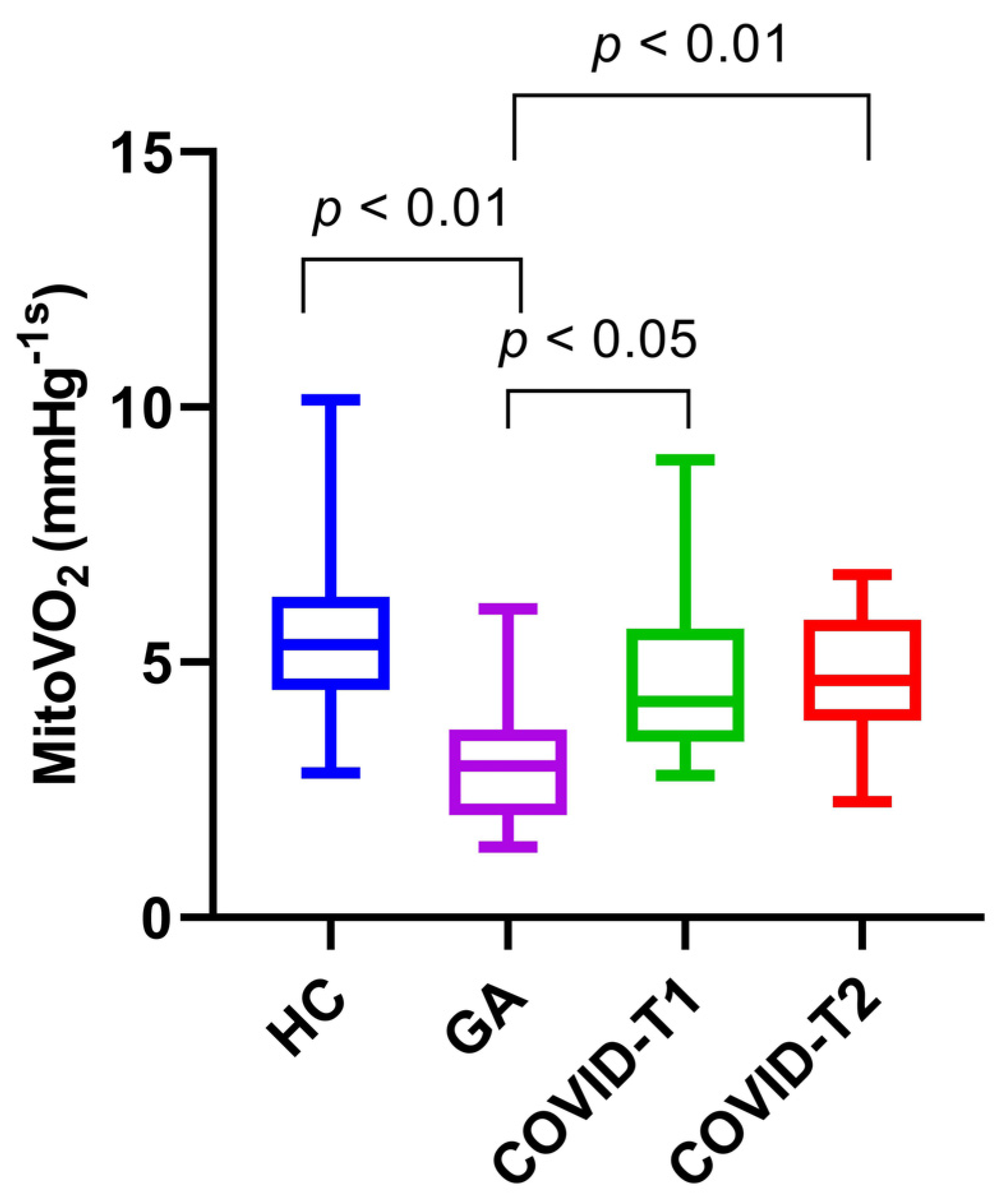
3.3. MitoVO2 Measurements
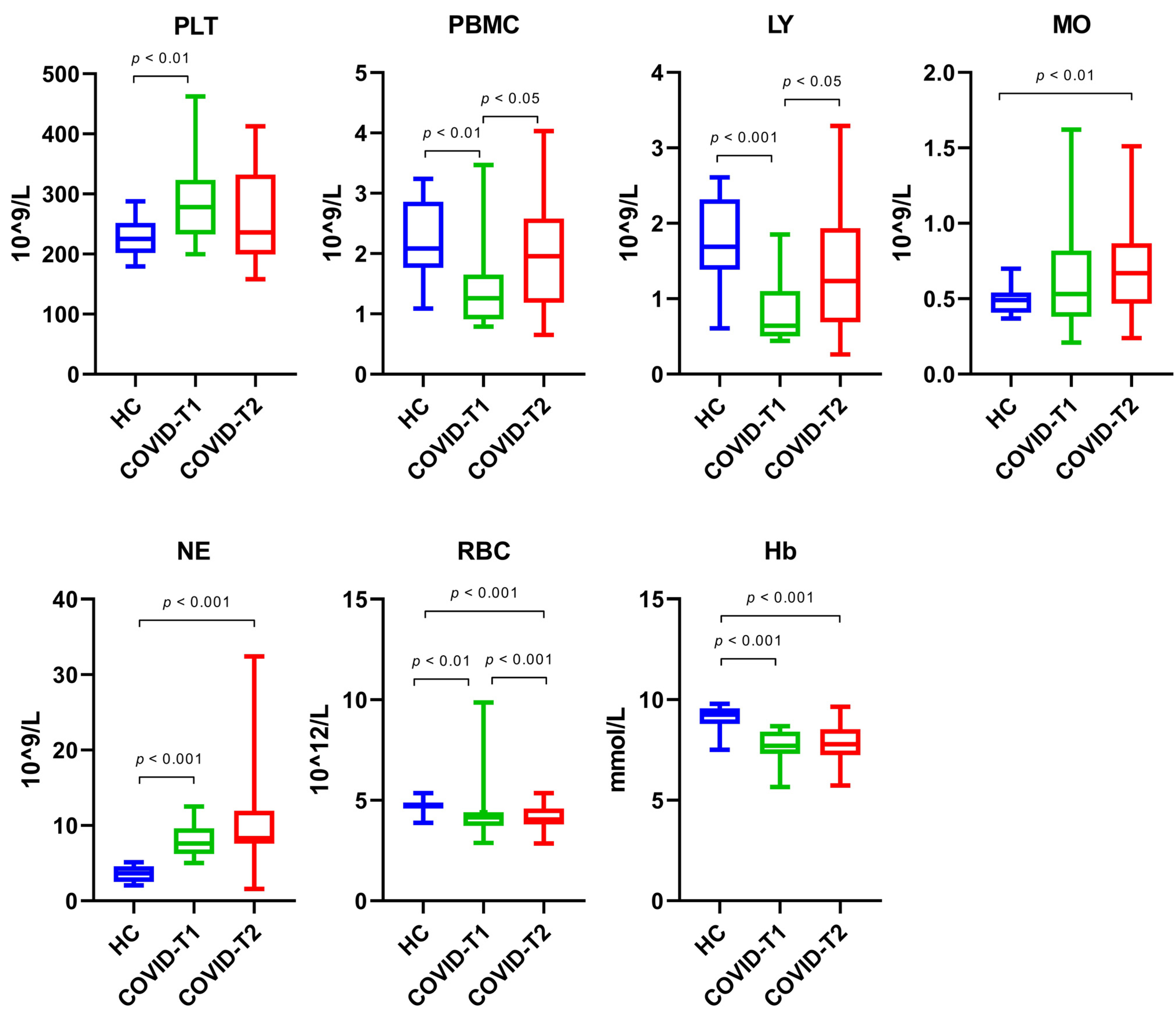
3.4. Whole Blood Cell Count
3.5. Isolated Blood Cell Count
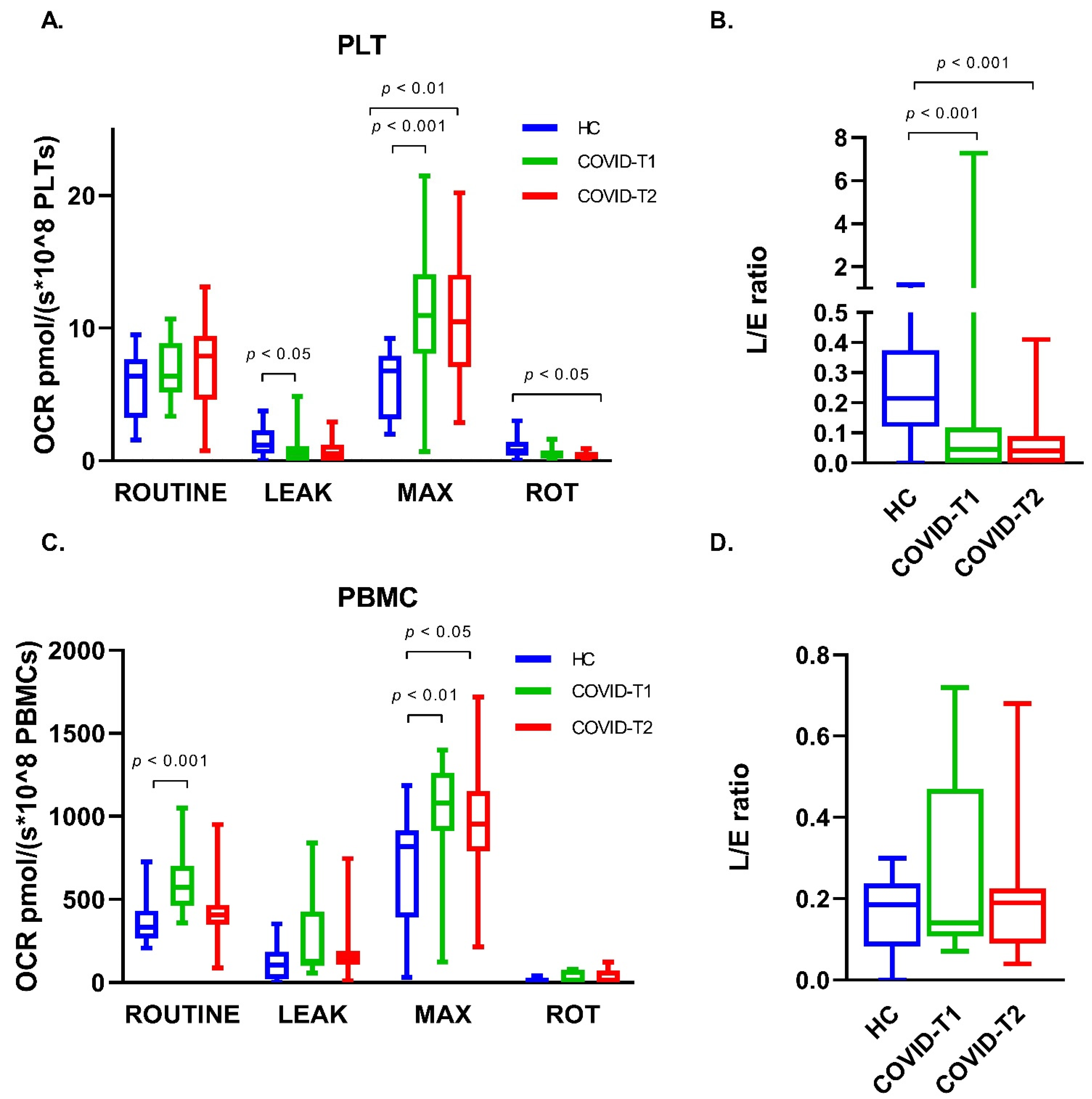
3.6. Platelet Oxygen Consumption
3.7. PBMC Oxygen Consumption
3.8. In Vivo and Ex Vivo Measurement Correlation Analysis
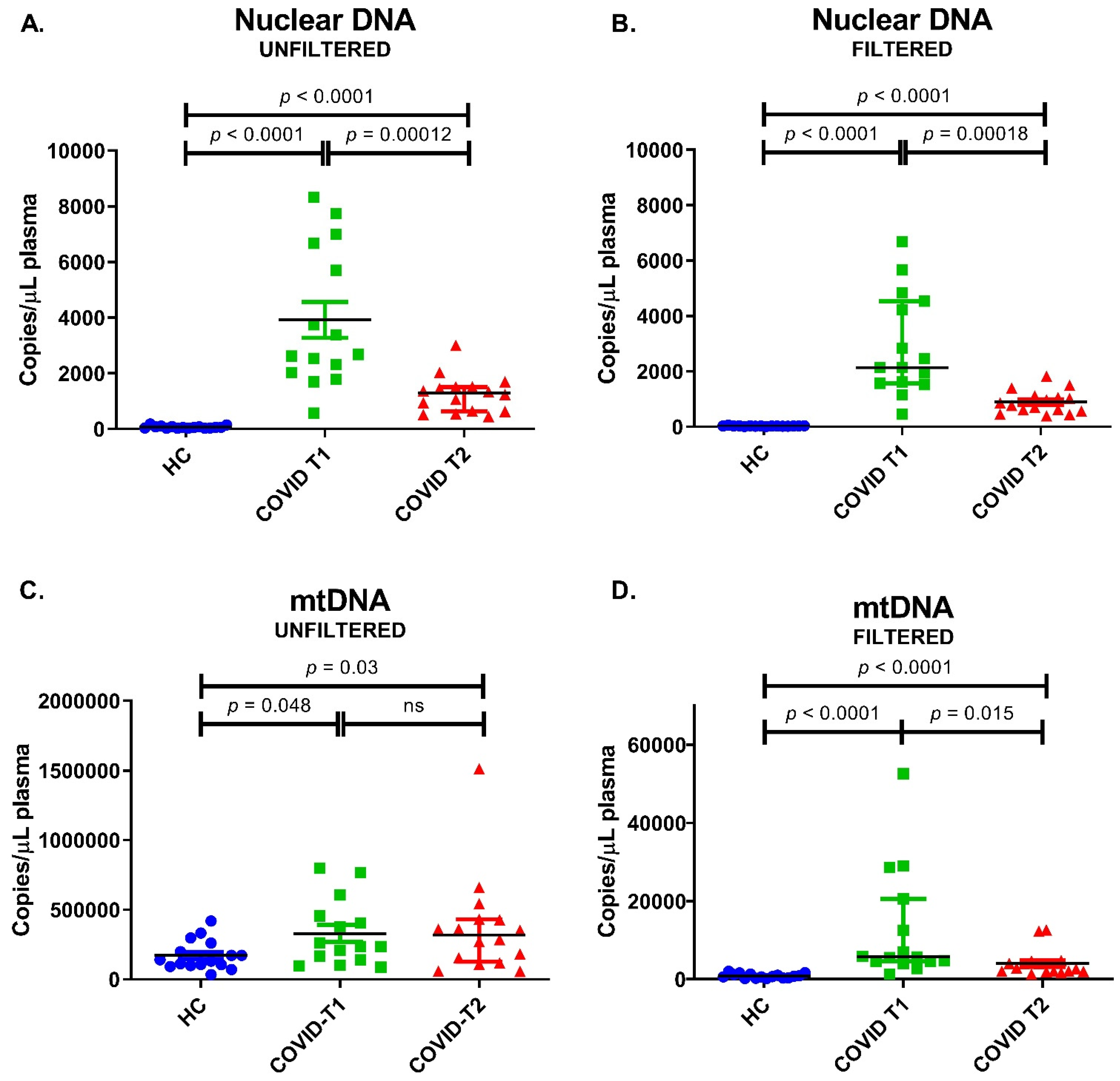
3.9. Nuclear DNA and mtDNA Amounts in Plasma
4. Discussion
4.1. MitoPO2 and mitoVO2
4.2. Mitochondrial DNA
4.3. PBMCs
4.4. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Coronavirus (COVID-19) Dashboard: WHO. 2021. Available online: https://covid19.who.int (accessed on 10 July 2022).
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Q.; Fan, Z.; Xie, R.; Wang, Z.; Lu, Y. Platelet mitochondrial dysfunction and the correlation with human diseases. Biochem. Soc. Trans. 2017, 45, 1213–1223. [Google Scholar] [CrossRef]
- Fink, M.P. Bench-to-bedside review: Cytopathic hypoxia. Crit. Care 2002, 6, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Mik, E.G.; Stap, J.; Sinaasappel, M.; Beek, J.F.; Aten, J.A.; van Leeuwen, T.; Ince, C. Mitochondrial PO2 measured by delayed fluorescence of endogenous protoporphyrin IX. Nat. Methods 2006, 3, 939–945. [Google Scholar] [CrossRef]
- Harms, F.A.; de Boon, W.M.I.; Balestra, G.M.; Bodmer, S.I.A.; Johannes, T.; Stolker, R.J.; Mik, E.G. Oxygen-dependent delayed fluorescence measured in skin after topical application of 5-aminolevulinic acid. J. Biophotonics 2011, 4, 731–739. [Google Scholar] [CrossRef]
- Mik, E.G.; Johannes, T.; Zuurbier, C.J.; Heinen, A.; Houben-Weerts, J.H.; Balestra, G.M.; Stap, J.; Beek, J.F.; Ince, C. In Vivo Mitochondrial Oxygen Tension Measured by a Delayed Fluorescence Lifetime Technique. Biophys. J. 2008, 95, 3977–3990. [Google Scholar] [CrossRef] [Green Version]
- Harms, F.A.; Voorbeijtel, W.J.; Bodmer, S.I.; Raat, N.J.; Mik, E.G. Cutaneous respirometry by dynamic measurement of mitochondrial oxygen tension for monitoring mitochondrial function in vivo. Mitochondrion 2013, 13, 507–514. [Google Scholar] [CrossRef]
- Ubbink, R.; Bettink, M.A.W.; van Weteringen, W.; Mik, E.G. Mitochondrial oxygen monitoring with COMET: Verification of calibration in man and comparison with vascular occlusion tests in healthy volunteers. Int. J. Clin. Monit. Comput. 2020, 35, 1357–1366. [Google Scholar] [CrossRef]
- Bettink, M.A.W.; Harms, F.A.; Dollee, N.; Specht, P.A.; Raat, N.J.; Schoonderwoerd, G.; Mik, E.G. Non-invasive versus ex vivo measurement of mitochondrial function in an endotoxemia model in rat: Toward monitoring of mitochondrial therapy. Mitochondrion 2020, 50, 149–157. [Google Scholar] [CrossRef]
- Harms, F.A.; Bodmer, S.I.A.; Raat, N.J.; Mik, E.G. Non-invasive monitoring of mitochondrial oxygenation and respiration in critical illness using a novel technique. Crit. Care 2015, 19, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibellini, L.; De Biasi, S.; Paolini, A.; Borella, R.; Boraldi, F.; Mattioli, M.; Lo Tartaro, D.; Fidanza, L.; Caro-Maldonado, A.; Meschiari, M.; et al. Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with COVID-19 pneumonia. EMBO Mol. Med. 2020, 12, e13001. [Google Scholar] [CrossRef]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol. Physiol. 2021, 320, C57–C65. [Google Scholar] [CrossRef]
- Nakahira, K.; Kyung, S.-Y.; Rogers, A.J.; Gazourian, L.; Youn, S.; Massaro, A.F.; Quintana, C.; Osorio, J.C.; Wang, Z.; Zhao, Y.; et al. Circulating Mitochondrial DNA in Patients in the ICU as a Marker of Mortality: Derivation and Validation. PLoS Med. 2013, 10, e1001577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagirath, V.C.; Dwivedi, D.J.; Liaw, P.C. Comparison of the Proinflammatory and Procoagulant Properties of Nuclear, Mitochondrial, and Bacterial DNA. Shock 2015, 44, 265–271. [Google Scholar] [CrossRef]
- Krychtiuk, K.A.; Ruhittel, S.; Hohensinner, P.J.; Koller, L.; Kaun, C.; Lenz, M.; Bauer, B.; Wutzlhofer, L.; Draxler, D.F.; Maurer, G.; et al. Mitochondrial DNA and Toll-Like Receptor-9 Are Associated with Mortality in Critically Ill Patients. Crit. Care Med. 2015, 43, 2633–2641. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Aguayo, J.J.; Garza-Veloz, I.; Badillo-Almaráz, J.I.; Bernal-Silva, S.; Martínez-Vázquez, M.C.; Juárez-Alcalá, V.; Vargas-Rodríguez, J.R.; Gaeta-Velasco, M.L.; González-Fuentes, C.; Ávila-Carrasco, L.; et al. Mitochondria and Mitochondrial DNA: Key Elements in the Pathogenesis and Exacerbation of the Inflammatory State Caused by COVID-19. Medicina 2021, 57, 928. [Google Scholar] [CrossRef]
- SSingh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef]
- Vincent, J.L.; Moreno, R.; Takala, J.; Willatts, S.; De Mendonça, A.; Bruining, H.; Reinhart, C.K.; Suter, P.; Thijs, L.G. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. Intensive Care Med. 1996, 22, 707–710. [Google Scholar] [CrossRef]
- The ARDS Definition of Task Force; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute Respiratory Distress Syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]
- Knaus, W.A.; Draper, E.A.; Wagner, D.P.; Zimmerman, J.E. APACHE II: A severity of disease classification system. Crit. Care Med. 1985, 13, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Ubbink, R.; Bettink, M.A.W.; Janse, R.; Harms, F.A.; Johannes, T.; Münker, F.M.; Mik, E.G. A monitor for Cellular Oxygen METabolism (COMET): Monitoring tissue oxygenation at the mitochondrial level. Int. J. Clin. Monit. Comput. 2016, 31, 1143–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodmer, S.I.A.; Balestra, G.M.; Harms, F.A.; Johannes, T.; Raat, N.J.H.; Stolker, R.J.; Mik, E.G. Microvascular and mitochondrial PO2 simultaneously measured by oxygen-dependent delayed luminescence. J. Biophotonics 2011, 5, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golub, A.S.; Popel, A.S.; Zheng, L.; Pittman, R.N. Analysis of phosphorescence in heterogeneous systems using distributions of quencher concentration. Biophys. J. 1997, 73, 452–465. [Google Scholar] [CrossRef] [Green Version]
- Harms, F.A.; Stolker, R.J.; Mik, E.G. Cutaneous Respirometry as Novel Technique to Monitor Mitochondrial Function: A Feasibility Study in Healthy Volunteers. PLoS ONE 2016, 11, e0159544. [Google Scholar]
- Puskarich, M.A.; Kline, J.A.; Watts, J.A.; Shirey, K.; Hosler, J.; Jones, A.E. Early alterations in platelet mitochondrial function are asso-ciated with survival and organ failure in patients with septic shock. J. Crit. Care 2016, 31, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Sjövall, F.; Morota, S.; Hansson, M.J.; Friberg, H.; Gnaiger, E.; Elmér, E. Temporal increase of platelet mitochondrial respiration is negatively associated with clinical outcome in patients with sepsis. Crit. Care 2010, 14, R214. [Google Scholar] [CrossRef] [Green Version]
- Petrus, A.; Ratiu, C.; Noveanu, L.; Lighezan, R.; Rosca, M.; Muntean, D.M.; Duicu, O. Assessment of Mitochondrial Respiration in Human Platelets. Rev. Chim. 2017, 68, 768–771. [Google Scholar] [CrossRef]
- Sumbalova, Z.G.-S.L.; Calabria, E.; Volani, C.; Gnaiger, E. O2k-Protocols: Isolation of peripheral blood mononuclear cells and platelets from human blood for HRR. Mitochondr. Physiol. Netw. 2018, 17, 1–16. [Google Scholar]
- Di Caro, V.; Walko, T.D., 3rd; Bola, R.A.; Hong, J.D.; Pang, D.; Hsue, V.; Au, A.K.; Halstead, E.S.; Carcillo, J.A.; Clark, R.S. Plasma Mitochondrial DNA—A Novel DAMP in Pediatric Sepsis. Shock 2016, 45, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control. An Introduction to OXPHOS Analysis, 5th ed.; Bioenergetic Communications: Innsbruck, Austria, 2020. [Google Scholar]
- Smolková, K.; Bellance, N.; Scandurra, F.; Génot, E.; Gnaiger, E.; Plecitá-Hlavatá, L.; Ježek, P.; Rossignol, R. Mitochondrial bioenergetic adaptations of breast cancer cells to aglycemia and hypoxia. J. Bioenerg. Biomembr. 2010, 42, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Gnaiger, E.; Steinlechner-Maran, R.; Méndez, G.; Eberl, T.; Margreiter, R. Control of mitochondrial and cellular respiration by oxygen. J. Bioenerg. Biomembr. 1995, 27, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar]
- Neu, C.; Baumbach, P.; Plooij, A.K.; Skitek, K.; Götze, J.; Von Loeffelholz, C.; Schmidt-Winter, C.; Coldewey, S.M. Non-invasive Assessment of Mitochondrial Oxygen Metabolism in the Critically Ill Patient Using the Protoporphyrin IX-Triplet State Lifetime Technique—A Feasibility Study. Front. Immunol. 2020, 11, 757. [Google Scholar] [CrossRef]
- Düsseldorf HHU. G*Power: Statistical Power Analyses for Windows and Mac: Heinrich Heine Universität Düsseldorf. Available online: https://www.psychologie.hhu.de/arbeitsgruppen/allgemeine-psychologie-und-arbeitspsychologie/gpower.html (accessed on 23 July 2020).
- Cohen, P.J. Effect of Anesthetics on Mitochondrial Function. Anesthesiology 1973, 39, 153–164. [Google Scholar] [CrossRef]
- Harris, R.A.; Munroe, J.; Farmer, B.; Kim, K.C.; Jenkins, P. Action of halothane upon mitochondrial respiration. Arch. Biochem. Biophys. 1971, 142, 435–444. [Google Scholar] [CrossRef]
- Miró, O.; Barrientos, A.; Alonso, J.R.; Casademont, J.; Jarreta, D.; Urbano-Márquez, A.; Cardellach, F. Effects of general anaesthetic procedures on mitochondrial function of human skeletal muscle. Eur. J. Clin. Pharmacol. 1999, 55, 35–41. [Google Scholar] [CrossRef]
- Niezgoda, J.; Morgan, P.G. Anesthetic considerations in patients with mitochondrial defects. Pediatr. Anesth. 2013, 23, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Niederer, L.E.; Miller, H.; Haines, K.L.; Molinger, J.; Whittle, J.; MacLeod, D.B.; McClave, S.A.; Wischmeyer, P.E. Prolonged progressive hypermetabolism during COVID-19 hospitalization undetected by common predictive energy equations. Clin. Nutr. ESPEN 2021, 45, 341–350. [Google Scholar] [CrossRef]
- Rosenthal, M.D.; Moore, F.A. Persistent Inflammation, Immunosuppression, and Catabolism: Evolution of Multiple Organ Dys-function. Surg. Infect. 2016, 17, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.P.; Li, M.; Lu, X.L.; Zhu, Y.M.; Ou-Yang, W.-X.; Xiao, Z.H.; Qiu, J.; Li, S.J. Use of plasma mitochondrial DNA levels for determining disease severity and prognosis in pediatric sepsis: A case control study. BMC Pediatr. 2018, 18, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, S.T.; Franken, L.; Adamzik, M.; Schumak, B.; Scherag, A.; Engler, A.; Schönborn, N.; Walden, J.; Koch, S.; Baba, H.A.; et al. Mitochondrial DNA. Anesthesiology 2016, 124, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Aguayo, J.J.; Garza-Veloz, I.; Vargas-Rodríguez, J.R.; Martinez-Vazquez, M.C.; Avila-Carrasco, L.; Bernal-Silva, S.; González-Fuentes, C.; Comas-García, A.; Alvarado-Hernández, D.E.; Centeno-Ramirez, A.S.H.; et al. Peripheral Blood Mitochondrial DNA Levels Were Modulated by SARS-CoV-2 Infection Severity and Its Lessening Was Associated with Mortality Among Hospitalized Patients With COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 754708. [Google Scholar] [CrossRef]
- Scozzi, D.; Cano, M.; Ma, L.; Zhou, D.; Zhu, J.H.; O’Halloran, J.A.; Goss, C.W.; Rauseo, A.M.; Liu, Z.; Sahu, S.K.; et al. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 2021, 6, e143299. [Google Scholar] [CrossRef] [PubMed]
- Bohn, M.K.; Hall, A.; Sepiashvili, L.; Jung, B.; Steele, S.; Adeli, K. Pathophysiology of COVID-19: Mechanisms Underlying Disease Severity and Progression. Physiology 2020, 35, 288–301. [Google Scholar] [CrossRef]
- Chiu, R.W.K.; Chan, L.Y.S.; Lam, N.Y.L.; Tsui, N.B.Y.; Ng, E.K.-O.; Rainer, T.; Lo, Y.M.D. Quantitative Analysis of Circulating Mitochondrial DNA in Plasma. Clin. Chem. 2003, 49, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Garrabou, G.; Morén, C.; López, S.; Tobías, E.; Cardellach, F.; Miró, Ò.; Casademont, J. The Effects of Sepsis on Mitochondria. J. Infect. Dis. 2011, 205, 392–400. [Google Scholar] [CrossRef] [Green Version]
- Belikova, I.; Lukaszewicz, A.C.; Faivre, V.; Damoisel, C.; Singer, M.; Payen, D. Oxygen consumption of human peripheral blood mononuclear cells in severe human sepsis. Crit. Care Med. 2007, 35, 2702–2708. [Google Scholar]
- Sjövall, F.; Morota, S.; Persson, J.; Hansson, M.J.; Elmér, E. Patients with sepsis exhibit increased mitochondrial respiratory capacity in peripheral blood immune cells. Crit. Care 2013, 17, R152. [Google Scholar] [CrossRef] [Green Version]
- Jeger, V.; Djafarzadeh, S.; Jakob, S.M.; Takala, J. Mitochondrial function in sepsis. Eur. J. Clin. Investig. 2013, 43, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.; Yano, M.; Rosenberg, A.S.; Puig, M. Lessons Learned to Date on COVID-19 Hyperinflammatory Syndrome: Consid-erations for Interventions to Mitigate SARS-CoV-2 Viral Infection and Detrimental Hyperinflammation. Front. Immunol. 2020, 11, 1131. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COVID-19 Patients | Healthy Control | General Anesthesia | p-Value | |
|---|---|---|---|---|
| Gender (M/F) | 12/4 | 12/4 | 7/2 | p = 0.913 |
| Age | 59 (52–65) | 57 (53–63) | 66 (58–70) | p = 0.397 |
| Body Mass Index | 31.45 (27.54–34.47) | 24.91 (22.93–27.34) | 26.49 (24.22–28.49) | p = 0.002 |
| Comorbidity (n,%) | ||||
| • Hypertension | 8 (50%) | 0 | 1 (11%) | p = 0.002 |
| • Coronary artery disease | 1 (6.25%) | 0 | 5 (55%) | p < 0.001 |
| • Diabetes Mellitus | 3 (18.6%) | 0 | 1 (11%) | p = 0.200 |
| • Obesity | 10 (62.5%) | 1 (6.25%) | 2 (22%) | p = 0.004 |
| • Morbid Obesity | 2 (12.5%) | 0 | 0 | p = 0.193 |
| • Astma or COPD | 2 (12.5%) | 1 (6.25%) | 1 (11%) | p = 0.827 |
| • Kidney disease | 1 (6.25%) | 0 | 0 | p = 0.449 |
| Time between intubation and first measurement | ||||
| • 1 days | 5 (31%) | n.a. | n.a. | n.a. |
| • 2 days | 6 (38%) | n.a. | n.a. | n.a. |
| • 3 days | 4 (25%) | n.a. | n.a. | n.a. |
| SOFA score on ICU admission | 4.50 (3.00–8.00) | n.a. | n.a. | n.a. |
| Apache II score | ||||
| • COVID-T1 | 22.00 (18.25–23.75) | n.a. | n.a. | n.a. |
| • COVID-T2 | 9.50 (7.25–19.75) | n.a | n.a | n.a. |
| ARDS score (n,%) | ||||
| • Mild | 3 (19%) | n.a. | n.a. | n.a. |
| • Moderate | 10 (62.5%) | n.a. | n.a. | n.a. |
| • Severe | 2 (12.5%) | n.a. | n.a. | n.a. |
| Prone position (n,%) | 11 (69%) | n.a. | n.a. | n.a |
| CT-scan phenotype | ||||
| • Ground glass opacities, without consolidations and pulmonary embolisms | 13 (81.25%) | n.a. | n.a. | n.a. |
| • Pulmonary embolism in combination with ground glass opacities | 2 (12.5%) | n.a. | n.a. | n.a. |
| • Consolidations accompanied by pulmonary fibrosis and ground glass opacities consistent with ARDS | 1 (6.25%) | n.a. | n.a. | n.a. |
| Whole blood cell count | ||||
| • Platelets | 278 (233–324) | 225 (202–252) | n.a. | p = 0.003 |
| • Peripheral blood mononuclear cells | 1.26 (0.91–1.65) | 2.09 (1.76–2.86) | n.a. | p = 0.002 |
| • Lymphocytes | 0.64 (0.50–1.10) | 1.69 (1.39–2.32) | n.a. | p < 0.001 |
| • Monocytes | 0.53 (0.38–0.82) | 0.49 (0.41–0.54) | n.a. | p = 0.514 |
| • Neutrophils | 7.62 (6.20–9.60) | 3.66 (2.52–4.59) | p < 0.001 | |
| • Red blood cell | 4.15 (3.73–4.41) | 4.74 (4.60–4.90) | n.a. | p = 0.003 |
| • Hemoglobin | 7.71 (7.31–8.41) | 9.25 (8.79–9.58) | n.a. | p < 0.001 |
| Unfiltered Plasma | Filtered Plasma | |||
|---|---|---|---|---|
| mtDNA-Containing Particles + Free Circulating mtDNA | Free Circulating mtDNA | |||
| Nuclear DNA (β-globin) | HC (n = 16) | 55 (33–87) | 28 (22–36) | copy/µL plasma |
| COVID-T1 (n = 15) | 2680 (2030–6670) | 2140 (1572–4540) | ||
| COVID-T2 (n = 16) | 1290 (637–1510) | 818 (576–1101) | ||
| mtDNA (mtND1) | HC (n = 16) | 135,000 (101,350–245,500) | 729 (353–1282) | |
| COVID-T1 (n = 15) | 236,000 (141,000–455,000) | 5720 (4600–20,600) | ||
| COVID-T2 (n = 16) | 319,500 (127,000–430,000) | 2750 (1980–4490) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Streng, L.W.J.M.; de Wijs, C.J.; Raat, N.J.H.; Specht, P.A.C.; Sneiders, D.; van der Kaaij, M.; Endeman, H.; Mik, E.G.; Harms, F.A. In Vivo and Ex Vivo Mitochondrial Function in COVID-19 Patients on the Intensive Care Unit. Biomedicines 2022, 10, 1746. https://doi.org/10.3390/biomedicines10071746
Streng LWJM, de Wijs CJ, Raat NJH, Specht PAC, Sneiders D, van der Kaaij M, Endeman H, Mik EG, Harms FA. In Vivo and Ex Vivo Mitochondrial Function in COVID-19 Patients on the Intensive Care Unit. Biomedicines. 2022; 10(7):1746. https://doi.org/10.3390/biomedicines10071746
Chicago/Turabian StyleStreng, Lucia W. J. M., Calvin J. de Wijs, Nicolaas J. H. Raat, Patricia A. C. Specht, Dimitri Sneiders, Mariëlle van der Kaaij, Henrik Endeman, Egbert G. Mik, and Floor A. Harms. 2022. "In Vivo and Ex Vivo Mitochondrial Function in COVID-19 Patients on the Intensive Care Unit" Biomedicines 10, no. 7: 1746. https://doi.org/10.3390/biomedicines10071746