
Targeting the SARS-CoV-2 HR1 with Small Molecules as Inhibitors of the Fusion Process

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Pharmacophore-Based Virtual Screening
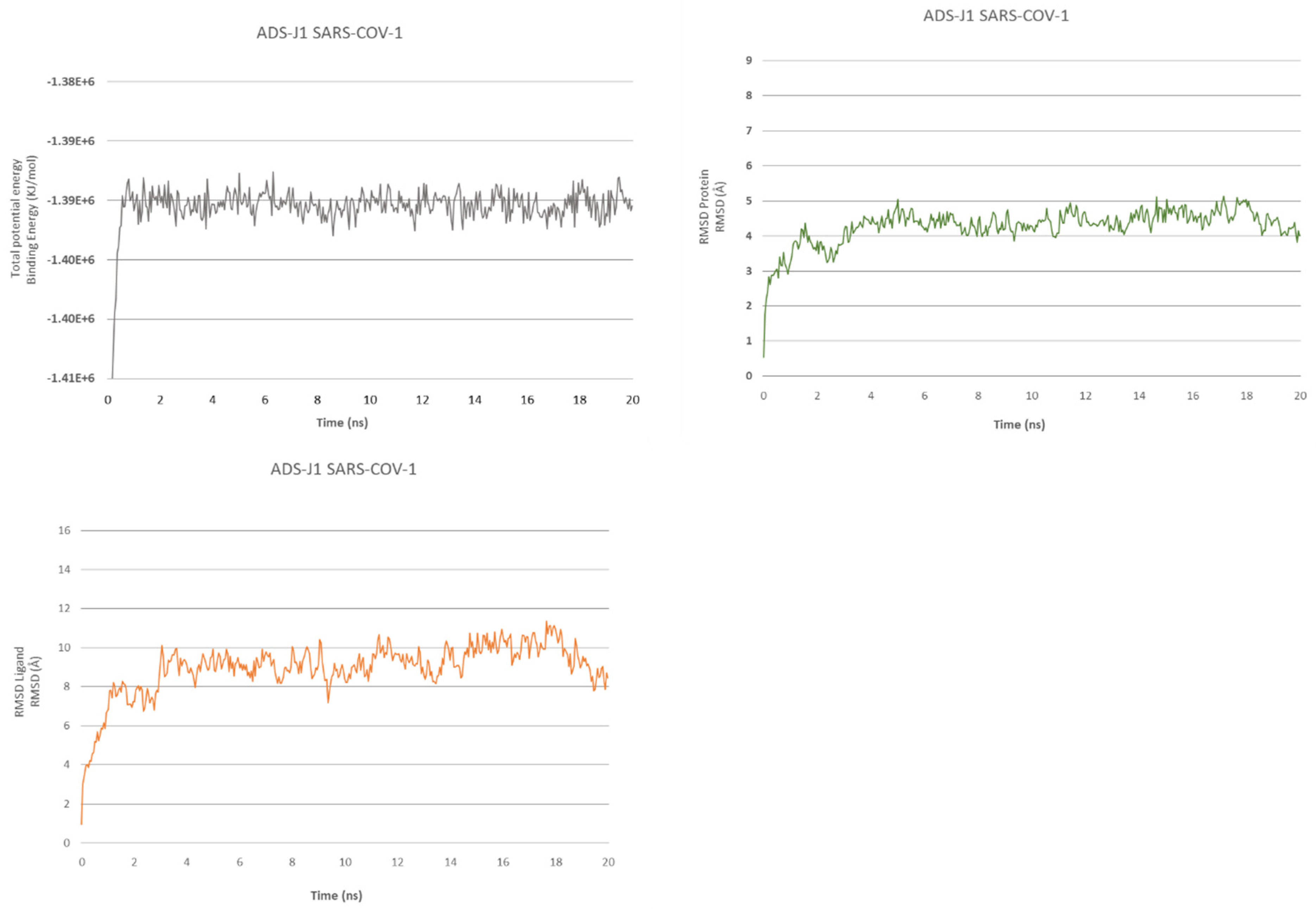
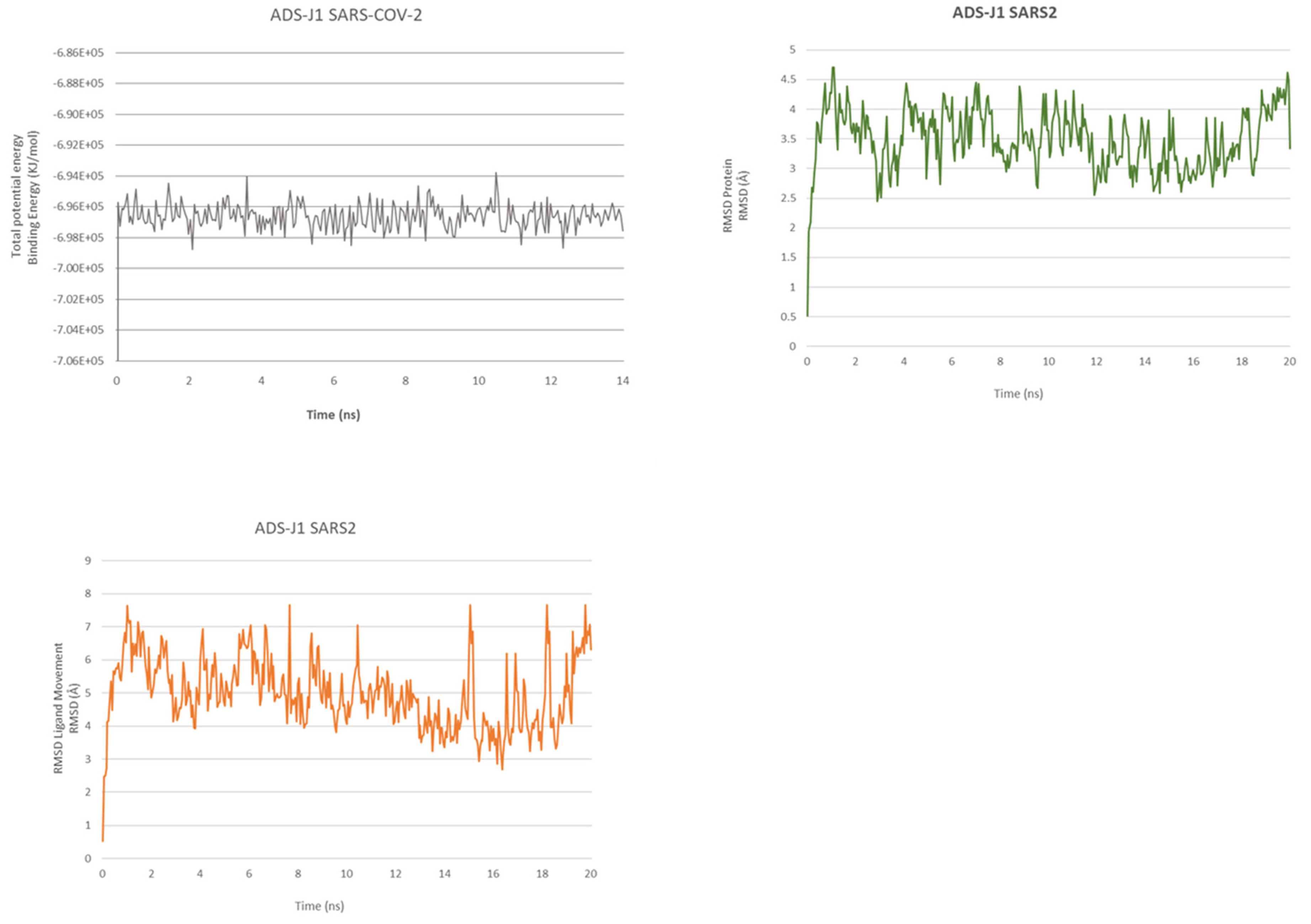
2.2. ADS-J1’s Docking and MD Simulation
2.3. Peptides Docking and Analysis
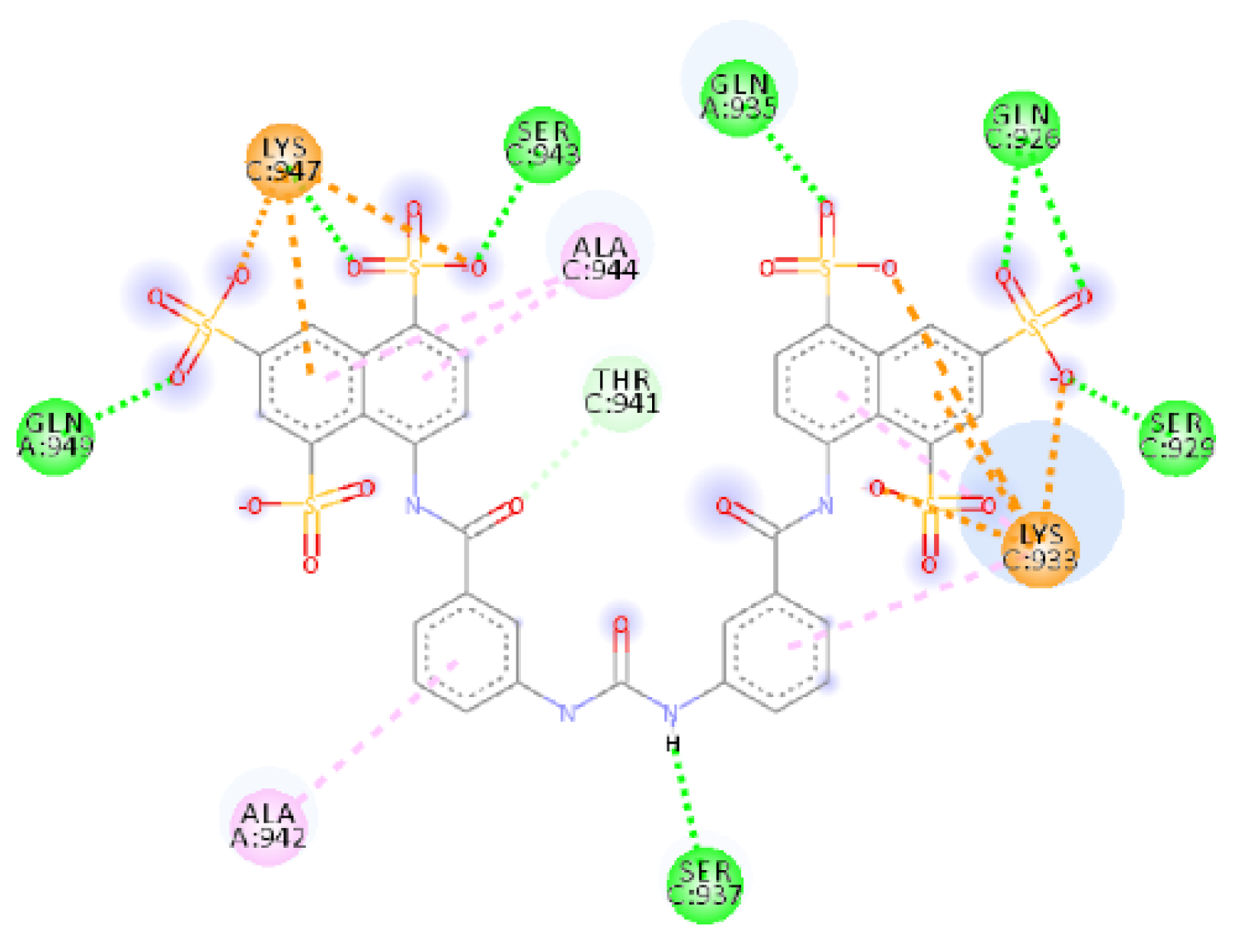
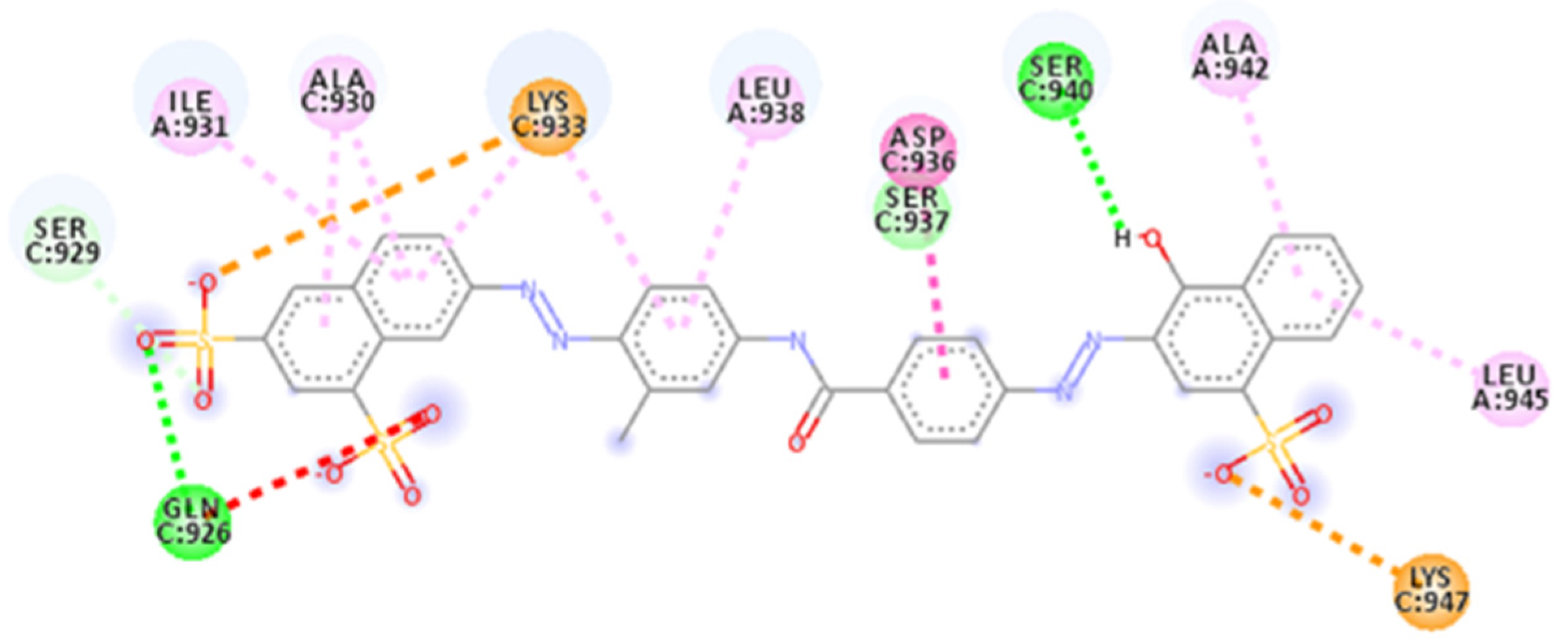
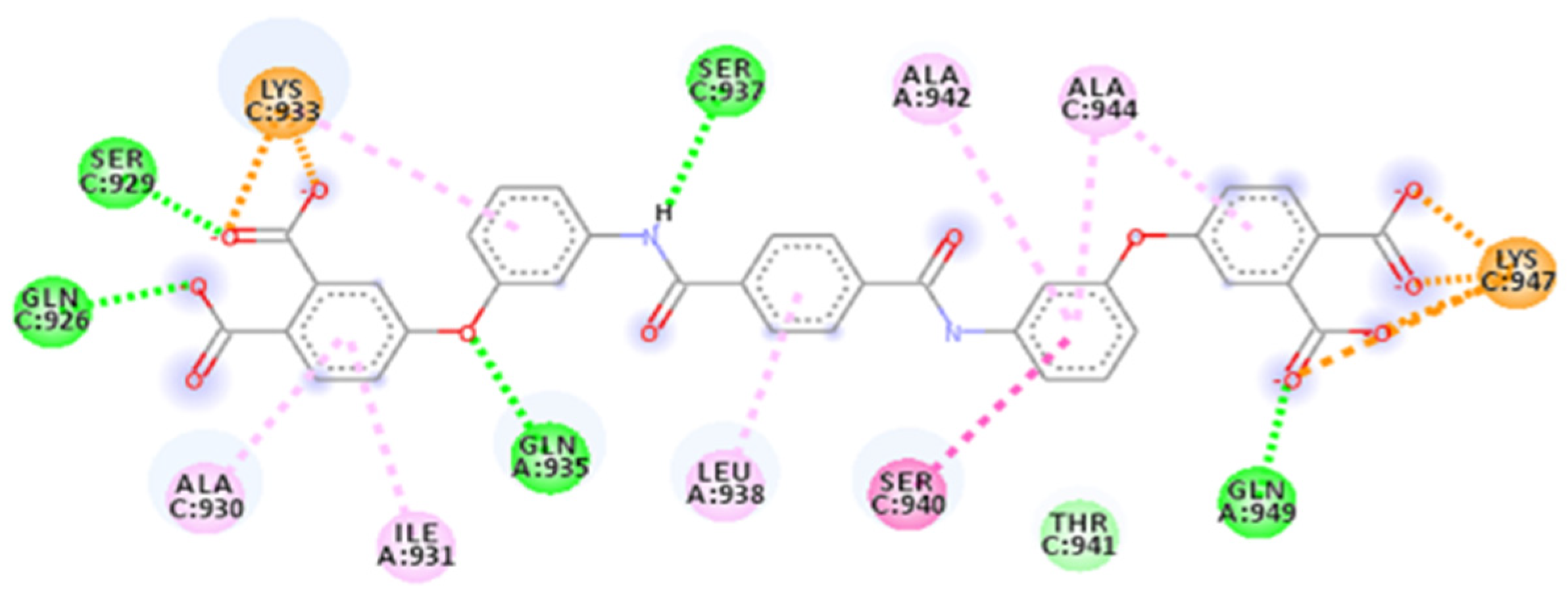
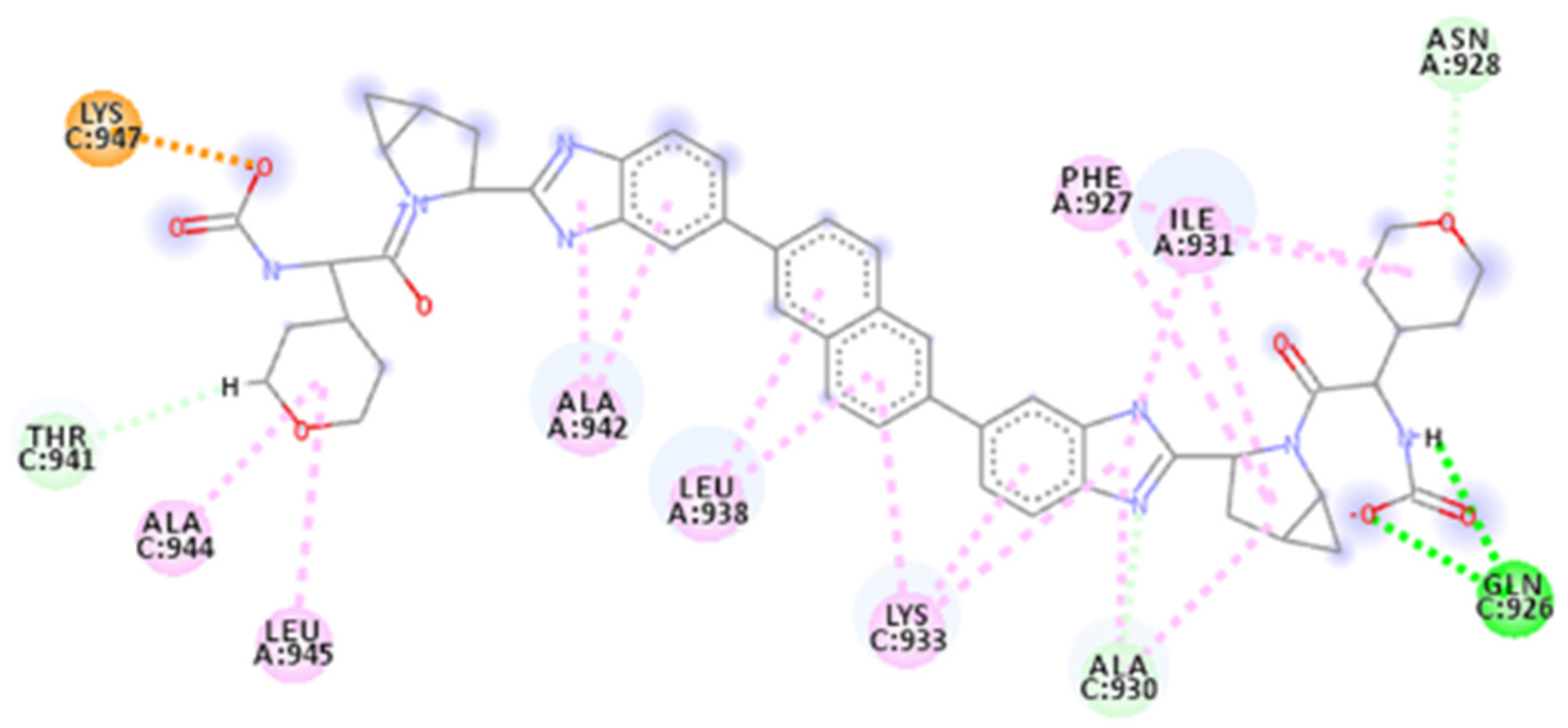
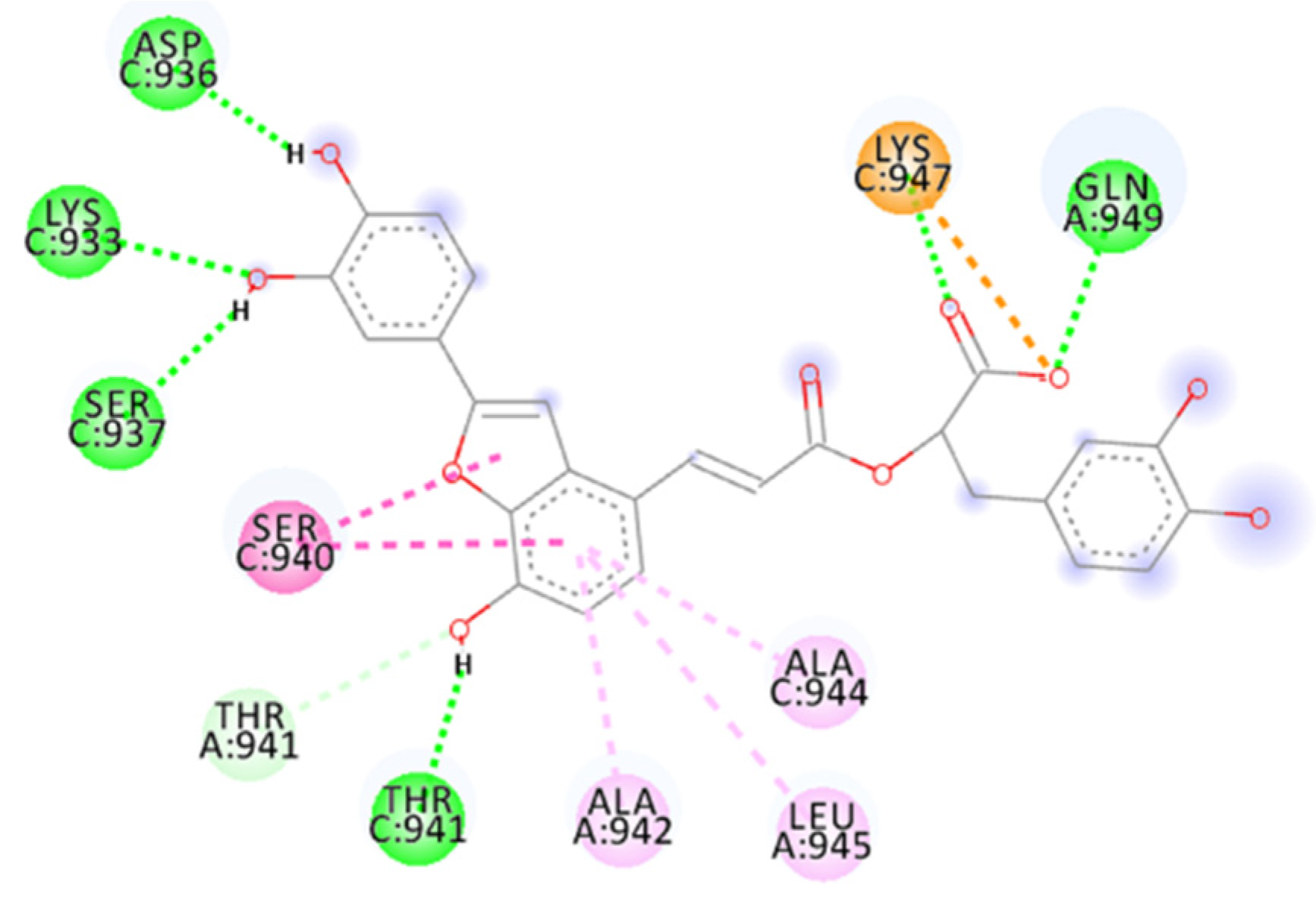
2.4. Focus on the Interactions of the Best-Scoring Compounds
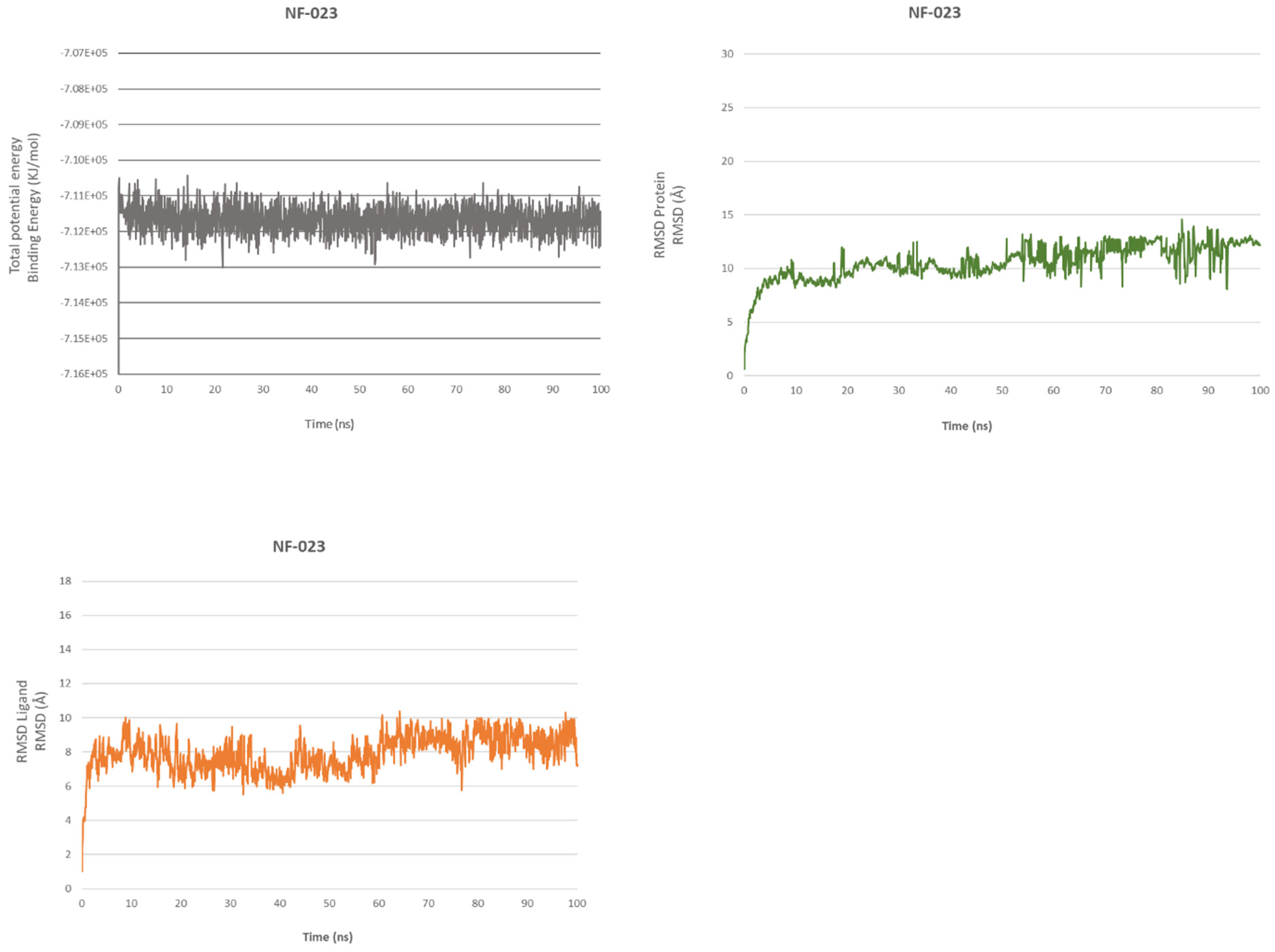
2.4.1. NF 023 Hydrate
2.4.2. ZINC00097961973
2.4.3. ZINC000150368097
2.4.4. ZINC000097996131
2.4.5. PubChem-66982178
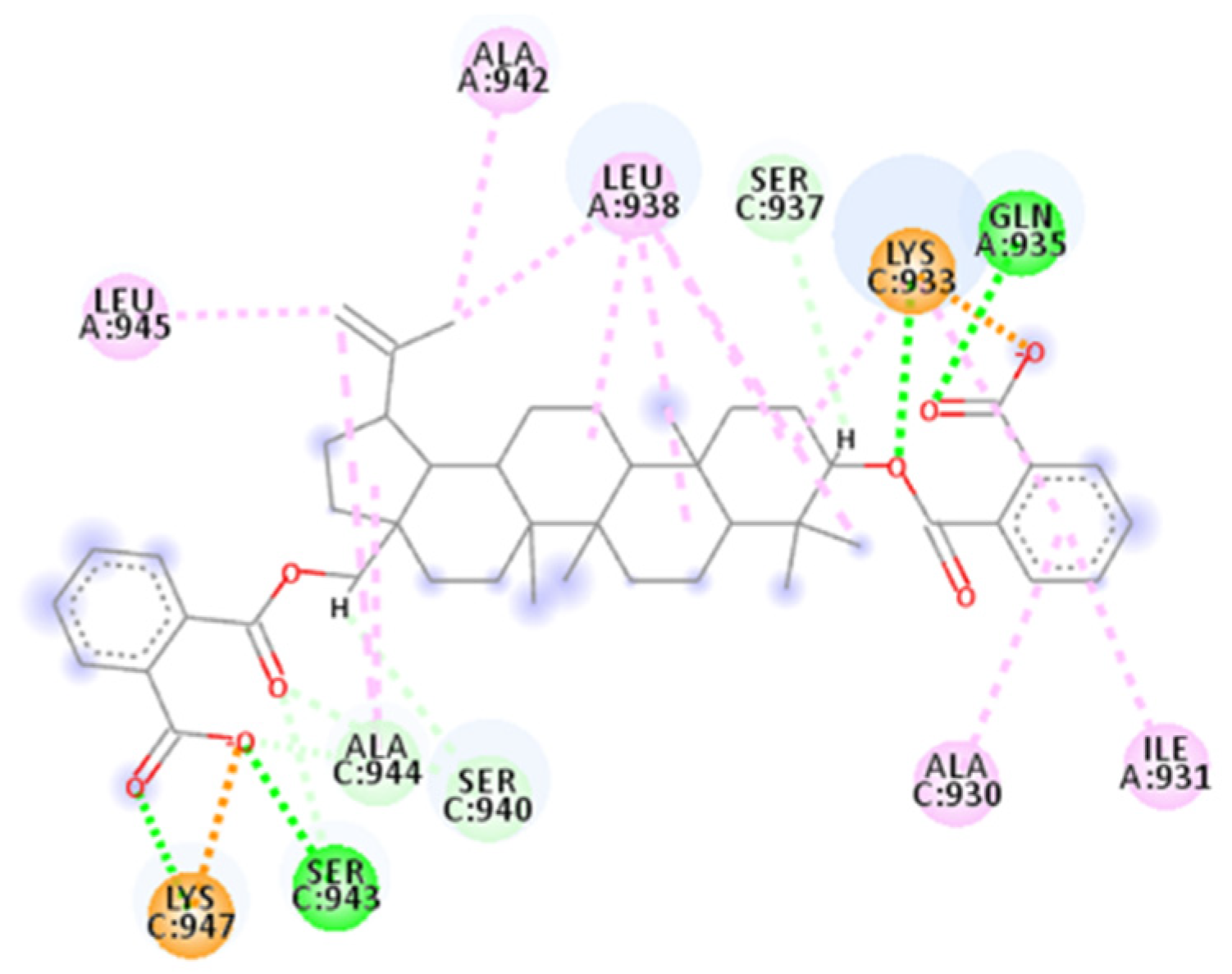
2.4.6. AP00094
2.4.7. AVP1227
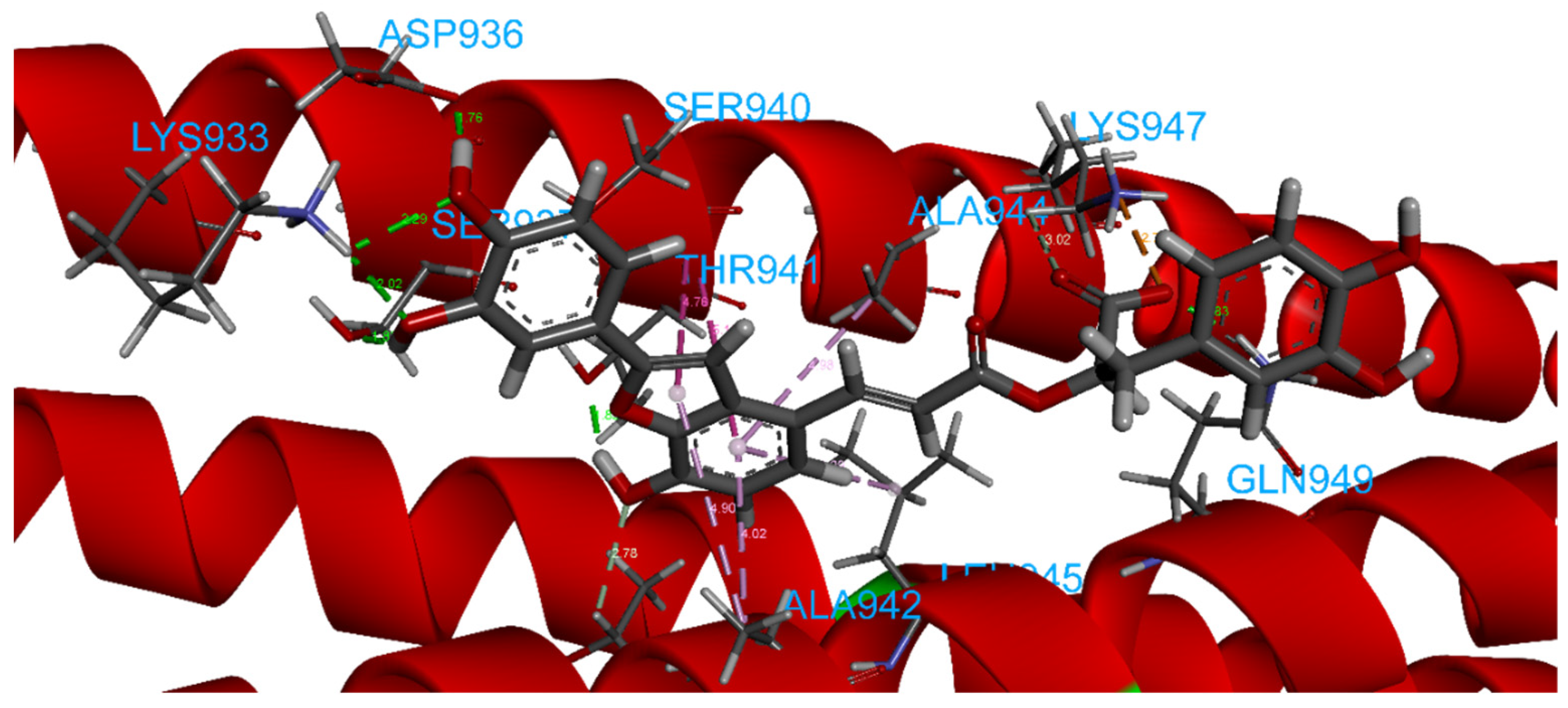
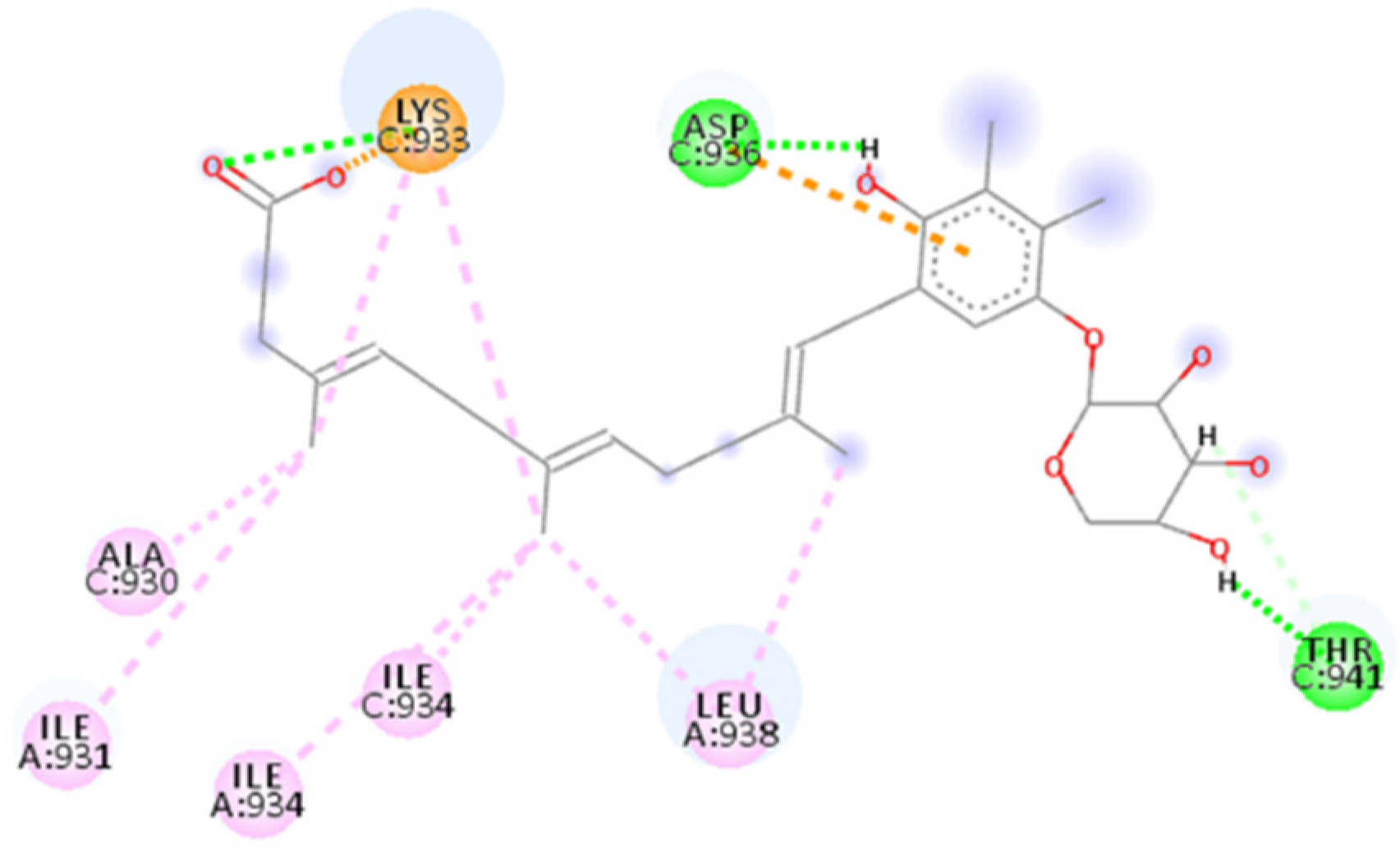
2.4.8. Salvianolic Acid C
2.4.9. ZINC000150346512
2.4.10. Marine_160925_88_2
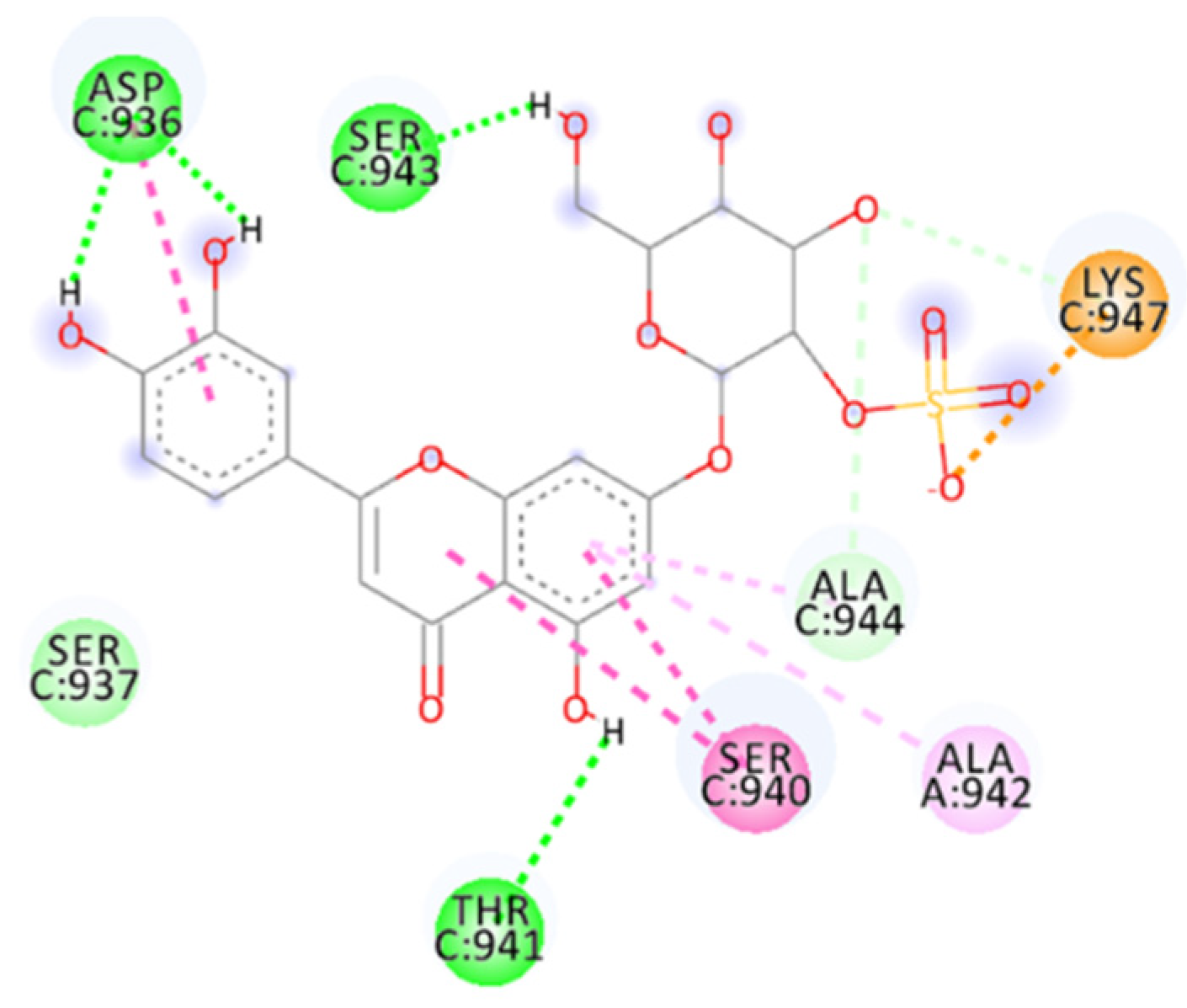
2.4.11. Thalassiolin A
2.4.12. SN00114935
3. Methods and Materials
3.1. PBVS and Selected Databases
3.2. Structure Preparation and Minimization
3.3. Peptides Docking through HDOCK Server
3.4. Molecular Docking
3.5. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lau, S.K.P.; Woo, P.C.Y.; Yip, C.C.Y.; Tse, H.; Tsoi, H.W.; Cheng, V.C.C.; Lee, P.; Tang, B.S.F.; Cheung, C.H.Y.; Lee, R.A.; et al. Coronavirus HKU1 and other coronavirus infections in Hong Kong. J. Clin. Microbiol. 2006, 44, 2063–2071. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Lau, S.K.P.; To, K.K.W.; Cheng, V.C.C.; Woo, P.C.Y.; Yuen, K.Y. Middle East Respiratory Syndrome Coronavirus: Another Zoonotic Betacoronavirus Causing SARS-Like Disease. Clin. Microbiol. Rev. 2015, 28, 465–522. [Google Scholar] [CrossRef]
- Cheng, V.C.C.; Lau, S.K.P.; Woo, P.C.Y.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007, 20, 660–694. [Google Scholar] [CrossRef]
- Chan, J.F.W.; To, K.K.W.; Tse, H.; Jin, D.Y.; Yuen, K.Y. Interspecies transmission and emergence of novel viruses: Lessons from bats and birds. Trends Microbiol. 2013, 21, 544–555. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 570, 270–273. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- McCarty, T.R.; Hathorn, K.E.; Redd, W.D.; Rodriguez, N.J.; Zhou, J.C.; Bazarbashi, A.N.; Njie, C.; Wong, D.; Trinh, Q.D.; Shen, L.; et al. How Do Presenting Symptoms and Outcomes Differ by Race/Ethnicity Among Hospitalized Patients With Coronavirus Disease 2019 Infection? Experience in Massachusetts. Clin. Infect. Dis. 2021, 73, E4131–E4138. [Google Scholar] [CrossRef]
- Martin, W.R.; Cheng, F.X. Repurposing of FDA-Approved Toremifene to Treat COVID-19 by Blocking the Spike Glycoprotein and NSP14 of SARS-CoV-2. J. Proteome Res. 2020, 19, 4670–4677. [Google Scholar] [CrossRef]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. Structural classification of neutralizing antibodies against the SARS-CoV-2 spike receptor-binding domain suggests vaccine and therapeutic strategies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Creech, C.B.; Walker, S.C.; Samuels, R.J. SARS-CoV-2 vaccines. JAMA 2021, 325, 1318–1320. [Google Scholar] [CrossRef]
- Baranov, P.V.; Henderson, C.M.; Anderson, C.B.; Gesteland, R.F.; Atkins, J.F.; Howard, M.T. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology 2005, 332, 498–510. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Ling, R.; Dai, Y.; Huang, B.; Huang, W.; Yu, J.; Lu, X.; Jiang, Y. In silico design of antiviral peptides targeting the spike protein of SARS-CoV-2. Peptides 2020, 130, 170328. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Xiong, H.; Farmar, J.; Debnath, A.K.; Tien, P. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef]
- Lu, L.; Liu, Q.; Zhu, Y.; Chan, K.-H.; Qin, L.; Li, Y.; Wang, Q.; Chan, J.F.-W.; Du, L.; Yu, F. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 3067. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Zhu, Y.; Yu, D.; Yan, H.; Chong, H.; He, Y. Design of potent membrane fusion inhibitors against SARS-CoV-2, an emerging coronavirus with high fusogenic activity. J. Virol. 2020, 94, e00635-20. [Google Scholar] [CrossRef]
- Xia, S.; Yan, L.; Xu, W.; Agrawal, A.S.; Algaissi, A.; Tseng, C.-T.K.; Wang, Q.; Du, L.; Tan, W.; Wilson, I.A. A pan-coronavirus fusion inhibitor targeting the HR1 domain of human coronavirus spike. Sci. Adv. 2019, 5, eaav4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, S.; Zheng, R.; Zhang, S.; Wang, S.; Chen, R.; Sun, K.; Zeng, H.; Zhou, J.; Wei, W. Global patterns of breast cancer incidence and mortality: A population-based cancer registry data analysis from 2000 to 2020. Cancer Commun. 2021, 41, 1183–1194. [Google Scholar] [CrossRef]
- LaBonte, J.; Lebbos, J.; Kirkpatrick, P. Enfuvirtide. Nat. Rev. Drug Discov. 2003, 2, 345–346. [Google Scholar] [CrossRef]
- Chu, L.H.M.; Chan, S.H.; Tsai, S.N.; Wang, Y.; Cheng, C.H.K.; Wong, K.B.; Waye, M.M.Y.; Ngai, S.M. Fusion core structure of the severe acute respiratory syndrome coronavirus (SARS-CoV): In search of potent SARS-CoV entry inhibitors. J. Cell. Biochem. 2008, 104, 2335–2347. [Google Scholar] [CrossRef]
- Singh, R.; Bhardwaj, V.K.; Sharma, J.; Purohit, R.; Kumar, S. In-silico evaluation of bioactive compounds from tea as potential SARS-CoV-2 nonstructural protein 16 inhibitors. J. Tradit. Complement. Med. 2022, 12, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bhardwaj, V.K.; Sharma, J.; Kumar, D.; Purohit, R. Identification of potential plant bioactive as SARS-CoV-2 Spike protein and human ACE2 fusion inhibitors. Comput. Biol. Med. 2021, 136, 104631. [Google Scholar] [CrossRef]
- Chauhan, M.; Bhardwaj, V.K.; Kumar, A.; Kumar, V.; Kumar, P.; Enayathullah, M.G.; Thomas, J.; George, J.; Kumar, B.K.; Purohit, R.; et al. Theaflavin 3-gallate inhibits the main protease (M(pro)) of SARS-CoV-2 and reduces its count in vitro. Sci. Rep. 2022, 12, 13146. [Google Scholar] [CrossRef]
- Kashyap, P.; Bhardwaj, V.K.; Chauhan, M.; Chauhan, V.; Kumar, A.; Purohit, R.; Kumar, A.; Kumar, S. A ricin-based peptide BRIP from Hordeum vulgare inhibits M(pro) of SARS-CoV-2. Sci. Rep. 2022, 12, 12802. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Rajendran, V.; Purohit, R.; Kumar, S. Identification of bioactive molecules from tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 3449–3458. [Google Scholar] [CrossRef]
- Singh, R.; Bhardwaj, V.K.; Purohit, R. Potential of turmeric-derived compounds against RNA-dependent RNA polymerase of SARS-CoV-2: An in-silico approach. Comput. Biol. Med. 2021, 139, 104965. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Rajendran, V.; Purohit, R.; Kumar, S. Bioactive Molecules of Tea as Potential Inhibitors for RNA-Dependent RNA Polymerase of SARS-CoV-2. Front. Med. 2021, 8, 684020. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bhardwaj, V.K.; Das, P.; Purohit, R. A computational approach for rational discovery of inhibitors for non-structural protein 1 of SARS-CoV-2. Comput. Biol. Med. 2021, 135, 104555. [Google Scholar] [CrossRef]
- Bhardwaj, V.K.; Singh, R.; Das, P.; Purohit, R. Evaluation of acridinedione analogs as potential SARS-CoV-2 main protease inhibitors and their comparison with repurposed anti-viral drugs. Comput. Biol. Med. 2021, 128, 104117. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; Kumar Bhardwaj, V.; Singh, R.; Rajendran, V.; Purohit, R.; Kumar, S. An in-silico evaluation of different bioactive molecules of tea for their inhibition potency against non structural protein-15 of SARS-CoV-2. Food Chem. 2021, 346, 128933. [Google Scholar] [CrossRef]
- Singh, R.; Bhardwaj, V.K.; Das, P.; Bhattacherjee, D.; Zyryanov, G.V.; Purohit, R. Benchmarking the ability of novel compounds to inhibit SARS-CoV-2 main protease using steered molecular dynamics simulations. Comput. Biol. Med. 2022, 146, 105572. [Google Scholar] [CrossRef]
- Floresta, G.; Zagni, C.; Gentile, D.; Patamia, V.; Rescifina, A. Artificial Intelligence Technologies for COVID-19 De Novo Drug Design. Int. J. Mol. Sci. 2022, 23, 3261. [Google Scholar] [CrossRef]
- Cardullo, N.; Catinella, G.; Floresta, G.; Muccilli, V.; Rosselli, S.; Rescifina, A.; Bruno, M.; Tringali, C. Synthesis of Rosmarinic Acid Amides as Antioxidative and Hypoglycemic Agents. J. Nat. Prod. 2019, 82, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Amata, E.; Gentile, D.; Romeo, G.; Marrazzo, A.; Pittalà, V.; Salerno, L.; Rescifina, A. Fourfold Filtered Statistical/Computational Approach for the Identification of Imidazole Compounds as HO-1 Inhibitors from Natural Products. Mar. Drugs 2019, 17, 113. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Pistarà, V.; Amata, E.; Dichiara, M.; Damigella, A.; Marrazzo, A.; Prezzavento, O.; Punzo, F.; Rescifina, A. Molecular modeling studies of pseudouridine isoxazolidinyl nucleoside analogues as potential inhibitors of the pseudouridine 5ʹ-monophosphate glycosidase. Chem. Biol. Drug Des. 2018, 91, 519–525. [Google Scholar] [CrossRef]
- Floresta, G.; Patamia, V.; Gentile, D.; Molteni, F.; Santamato, A.; Rescifina, A.; Vecchio, M. Repurposing of FDA-Approved Drugs for Treating Iatrogenic Botulism: A Paired 3D-QSAR/Docking Approach. ChemMedChem 2020, 15, 256–262. [Google Scholar] [CrossRef]
- Floresta, G.; Gentile, D.; Perrini, G.; Patamia, V.; Rescifina, A. Computational Tools in the Discovery of FABP4 Ligands: A Statistical and Molecular Modeling Approach. Mar. Drugs 2019, 17, 624. [Google Scholar] [CrossRef] [Green Version]
- Floresta, G.; Rescifina, A.; Abbate, V. Structure-Based Approach for the Prediction of Mu-opioid Binding Affinity of Unclassified Designer Fentanyl-Like Molecules. Int. J. Mol. Sci. 2019, 20, 2311. [Google Scholar] [CrossRef]
- Gentile, D.; Floresta, G.; Patamia, V.; Chiaramonte, R.; Mauro, G.L.; Rescifina, A.; Vecchio, M. An Integrated Pharmacophore/Docking/3D-QSAR Approach to Screening a Large Library of Products in Search of Future Botulinum Neurotoxin A Inhibitors. Int. J. Mol. Sci. 2020, 21, 9470. [Google Scholar] [CrossRef]
- Xia, S.; Lan, Q.; Zhu, Y.; Wang, C.; Xu, W.; Li, Y.; Wang, L.; Jiao, F.; Zhou, J.; Hua, C.; et al. Structural and functional basis for pan-CoV fusion inhibitors against SARS-CoV-2 and its variants with preclinical evaluation. Signal Transduct. Target. Ther. 2021, 6, 288. [Google Scholar] [CrossRef]
- Simmaco, M.; Mignogna, G.; Canofeni, S.; Miele, R.; Mangoni, M.L.; Barra, D. Temporins, antimicrobial peptides from the European red frog Rana temporaria. Eur. J. Biochem. 1996, 242, 788–792. [Google Scholar] [CrossRef]
- Mangoni, M. Temporins, anti-infective peptides with expanding properties. Cell. Mol. Life Sci. CMLS 2006, 63, 1060–1069. [Google Scholar] [CrossRef]
- Swithenbank, L.; Cox, P.; Harris, L.G.; Dudley, E.; Sinclair, K.; Lewis, P.; Cappiello, F.; Morgan, C. Temporin A and Bombinin H2 Antimicrobial Peptides Exhibit Selective Cytotoxicity to Lung Cancer Cells. Scientifica 2020, 2020, 3526286. [Google Scholar] [CrossRef]
- Liu, X.; Huang, Y.; Cheng, M.; Pan, L.; Si, Y.; Li, G.; Niu, Y.; Zhao, L.; Zhao, J.; Li, X. Screening and rational design of hepatitis C virus entry inhibitory peptides derived from GB virus A NS5A. J. Virol. 2013, 87, 1649–1657. [Google Scholar] [CrossRef]
- Yang, C.; Pan, X.; Xu, X.; Cheng, C.; Huang, Y.; Li, L.; Jiang, S.; Xu, W.; Xiao, G.; Liu, S. Salvianolic acid C potently inhibits SARS-CoV-2 infection by blocking the formation of six-helix bundle core of spike protein. Signal Transduct. Target. Ther. 2020, 5, 220. [Google Scholar] [CrossRef]
- Rowley, D.C.; Hansen, M.S.; Rhodes, D.; Sotriffer, C.A.; Ni, H.; McCammon, J.A.; Bushman, F.D.; Fenical, W. Thalassiolins A–C: New marine-derived inhibitors of HIV cDNA integrase. Bioorg. Med. Chem. 2002, 10, 3619–3625. [Google Scholar] [CrossRef]
- Regalado, E.L.; Rodríguez, M.; Menéndez, R.; Concepción, Á.A.; Nogueiras, C.; Laguna, A.; Rodríguez, A.A.; Williams, D.E.; Lorenzo-Luaces, P.; Valdés, O. Repair of UVB-damaged skin by the antioxidant sulphated flavone glycoside thalassiolin B isolated from the marine plant Thalassia testudinum Banks ex König. Mar. Biotechnol. 2009, 11, 74–80. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Modeling 2004, 10, 155–164. [Google Scholar] [CrossRef]
- Yan, Y.; Tao, H.; He, J.; Huang, S.-Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. YASARA View—molecular graphics for all devices—from smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA—A self-parameterizing force field. Proteins Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef]
- Xu, Y.; Lou, Z.; Liu, Y.; Pang, H.; Tien, P.; Gao, G.F.; Rao, Z. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J. Biol. Chem. 2004, 279, 49414–49419. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Krieger, E.; Dunbrack, R.L.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pK a prediction with hydrogen bonding network optimization. In Computational Drug Discovery and Design; Springer: Berlin/Heidelberg, Germany, 2012; pp. 405–421. [Google Scholar]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Maier, J.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Galimberti, M.; Barbera, V.; Guerra, S.; Bernardi, A. Facile functionalization of sp2 carbon allotropes with a biobased Janus molecule. Rubber Chem. Technol. 2017, 90, 285–307. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Molecules | Conformers | Cutoffs | Results |
|---|---|---|---|---|
| FDA | 1856 | 21,850 | - | 1884 |
| MNP | 14,064 | 164,952 | −6.00 | 19 |
| MolPort | 14,064 | 164,952 | −6.00 | 145 |
| ZINC | 13,190,317 | 123,399,574 | −6.00 | 149 |
| ChemSpace2 | 50,181,678 | 250,205,463 | - | 50 |
| PubChem | 93,067,404 | 450,708,705 | −7.00 | 98 |
| CHEMBL25 | 1,752,844 | 23,136,925 | - | 128 |
| SNP | 274,363 | 2,928,422 | −6.00 | 30 |
| N. | Database | Name/Class | Id | HDOCK Score | Source |
|---|---|---|---|---|---|
| HR1–HR2 SARS-CoV-2 | −245.71 | — | |||
| HR1 SARS seq. EK1 seq. | −197.92 | — | |||
| HR1 SARS2 seq. EK1 seq. | −203.98 | — | |||
| 1 | AVPdb | P3 | AVP1841 | −229.37 | SARS-CoV-1 spike protein |
| 2 | AVPdb | I10L/V13L | AVP1510 | −225.29 | HCV non-structural protein 5A |
| 3 | AVPdb | FP4 | AVP1754 | −225.03 | Mimetic for the SOCS protein |
| 4 | AVPdb | gH625 | AVP1250 | −224.33 | HSV-1 H glycoprotein (gH) |
| 5 | AVPdb | BLfcin 17–31 | AVP1853 | −223.25 | Bovine lactoferrin |
| 6 | AVPdb | — | AVP1039 | −222.93 | HCV envelope glycoprotein (E1, E2) |
| 7 | APD3 | Cecropin A | AP00139 | −219.63 | Giant silk moth |
| 8 | AVPdb | — | AVP1033 | −213.17 | HCV envelope glycoprotein (E1, E2) |
| 9 | AVPdb | — | AVP1034 | −212.76 | HCV envelope glycoprotein (E1, E2) |
| 10 | AVPdb | Gly137–Arg151 | AVP0430 | −211.43 | HSV glycoprotein (gC) |
| 11 | AVPdb | C5A | AVP1504 | −210.86 | HCV non-structural protein 5A |
| 12 | AVPdb | SEQ ID NO:52 | AVP1463 | −210.81 | HCV envelope glycoprotein (E1, E2) |
| 13 | AVPdb | CL58.1 | AVP1174 | −208.69 | Human claudin-1 (CLDN1) |
| 14 | AVPdb | GBVA4 | AVP1226 | −208.62 | GBVA non-structural protein 5A |
| 15 | AVPdb | FP3 | AVP1753 | −207.82 | Mimetic for the SOCS protein |
| 16 | AVPdb | SEQ ID NO:54 | AVP1465 | −206.96 | HCV envelope glycoprotein (E1, E2) |
| 17 | AVPdb | I6L/I10L | AVP1509 | −206.74 | HCV non-structural protein 5A |
| 18 | AVPdb | c01 | AVP0968 | −206.67 | Phage display |
| 19 | AVPdb | — | AVP0778 | −204.47 | HSV-1 B glycoprotein (gB) |
| 20 | AVPdb | — | AVP0708 | −203.73 | HSV-1 B glycoprotein (gB) |
| 21 | APD3 | Human neutrophil peptide-3 | AP00178 | −203.42 | Monocytes; saliva; Homo sapiens |
| 22 | AVPdb | — | AVP0793 | −202.75 | HSV-1 B glycoprotein (gB) |
| 23 | APD3 | human neutrophil peptide-1 | AP00176 | −202.20 | Monocytes; saliva; Homo sapiens |
| 24 | AVPdb | — | AVP0709 | −201.84 | HSV-1 B glycoprotein (gB) |
| 25 | AVPdb | SEQ ID NO:53 | AVP1464 | −199.84 | HCV envelope glycoprotein (E1, E2) |
| 26 | AVPdb | CL58+2 | AVP1184 | −199.27 | Human claudin-1 (CLDN1) |
| 27 | AVPdb | EPK209 | AVP1129 | −198.29 | FeLV transmembrane protein (TM) |
| 28 | AVPdb | RTD3 | AVP1910 | −197.60 | Rhesus theta-defensin |
| 29 | AVPdb | I10V | AVP1513 | −197.00 | HCV non-structural protein 5A |
| 30 | AVPdb | — | AVP1042 | −196.98 | HCV envelope glycoprotein (E1, E2) |
| 31 | APD3 | Lactoferricin B | AP00026 | −196.90 | Bos taurus |
| 32 | APD3 | Temporin A | AP00094 | −196.37 | European common frog |
| 33 | AVPdb | EPK210 | AVP1130 | −196.26 | FeLV transmembrane protein (TM) |
| 34 | AVPdb | — | AVP0740 | −195.56 | HSV-1 B glycoprotein (gB) |
| 35 | AVPdb | SARSWW-IV | AVP0549 | −193.86 | SARS-CoV-1 spike protein |
| 36 | AVPdb | SEQ ID NO:62 | AVP1473 | −193.55 | HCV envelope glycoprotein (E1, E2) |
| 37 | AVPdb | C18-p1b | AVP0966 | −193.09 | Phage display |
| 38 | AVPdb | CL-9 | AVP1191 | −193.03 | Human claudin-9 (CLDN9) |
| 39 | AVPdb | E1 | AVP0869 | −192.85 | FGF-4 signal sequence |
| 40 | AVPdb | c03 | AVP0969 | −192.15 | Phage display |
| 41 | AVPdb | 1OAN1 | AVP1058 | −191.92 | Synthetic |
| 42 | AVPdb | GBAV5 | AVP1227 | −191.20 | GBVA non-structural protein 5A |
| 43 | AVPdb | — | AVP0710 | −190.85 | HSV-1 B glycoprotein (gB) |
| 44 | AVPdb | I10A | AVP1514 | −190.75 | HCV non-structural protein 5A |
| 45 | AVPdb | GBVA8 | AVP1230 | −190.62 | GBVA non-structural protein 5A |
| 46 | AVPdb | — | AVP0741 | −190.33 | HSV-1 B glycoprotein (gB) |
| 47 | APD3 | Melittin | AP00146 | −189.77 | Honeybee venom |
| 48 | AVPdb | B6 | AVP1087 | −189.22 | FGF-4 signal sequence |
| 49 | AVPdb | — | AVP0684 | −188.46 | HSV-1 B glycoprotein (gB) |
| 50 | AVPdb | CL58-2 | AVP1183 | −188.25 | Human claudin-1 (CLDN1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, D.; Coco, A.; Patamia, V.; Zagni, C.; Floresta, G.; Rescifina, A. Targeting the SARS-CoV-2 HR1 with Small Molecules as Inhibitors of the Fusion Process. Int. J. Mol. Sci. 2022, 23, 10067. https://doi.org/10.3390/ijms231710067
Gentile D, Coco A, Patamia V, Zagni C, Floresta G, Rescifina A. Targeting the SARS-CoV-2 HR1 with Small Molecules as Inhibitors of the Fusion Process. International Journal of Molecular Sciences. 2022; 23(17):10067. https://doi.org/10.3390/ijms231710067
Chicago/Turabian StyleGentile, Davide, Alessandro Coco, Vincenzo Patamia, Chiara Zagni, Giuseppe Floresta, and Antonio Rescifina. 2022. "Targeting the SARS-CoV-2 HR1 with Small Molecules as Inhibitors of the Fusion Process" International Journal of Molecular Sciences 23, no. 17: 10067. https://doi.org/10.3390/ijms231710067