
Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow



1
Graduate Institute of Biomedical Informatics, College of Medical Science and Technology, Taipei Medical University, Taipei 11031, Taiwan
2
Division of Pulmonary Medicine, Department of Internal Medicine, School of Medicine, College of Medicine, Taipei Medical University, Taipei 11031, Taiwan
3
Division of Pulmonary Medicine, Department of Internal Medicine, Taipei Medical University Hospital, Taipei 11031, Taiwan
4
IT and Bioinformatics Program, Division of Intramural, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA
*
Author to whom correspondence should be addressed.
Genes 2022, 13(4), 686; https://doi.org/10.3390/genes13040686
Submission received: 20 March 2022
/
Revised: 6 April 2022
/
Accepted: 12 April 2022
/
Published: 13 April 2022
(This article belongs to the Special Issue Bioinformatics Analysis for Diseases)
Abstract
:Several variants of the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) are emerging all over the world. Variant surveillance from genome sequencing has become crucial to determine if mutations in these variants are rendering the virus more infectious, potent, or resistant to existing vaccines and therapeutics. Meanwhile, analyzing many raw sequencing data repeatedly with currently available code-based bioinformatics tools is tremendously challenging to be implemented in this unprecedented pandemic time due to the fact of limited experts and computational resources. Therefore, in order to hasten variant surveillance efforts, we developed an installation-free cloud workflow for robust mutation profiling of SARS-CoV-2 variants from multiple Illumina sequencing data. Herein, 55 raw sequencing data representing four early SARS-CoV-2 variants of concern (Alpha, Beta, Gamma, and Delta) from an open-access database were used to test our workflow performance. As a result, our workflow could automatically identify mutated sites of the variants along with reliable annotation of the protein-coding genes at cost-effective and timely manner for all by harnessing parallel cloud computing in one execution under resource-limitation settings. In addition, our workflow can also generate a consensus genome sequence which can be shared with others in public data repositories to support global variant surveillance efforts.
1. Introduction
A novel coronavirus species caused a previously unidentified human pneumonia-like disease for the first time in Wuhan, China, on 12 December 2019 [1]. Later, the World Health Organization (WHO; Geneva, Switzerland) named this disease coronavirus disease 2019 (COVID-19) and the virus that caused it severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) on 11 February 2020 [2] and declared this COVID-19 a pandemic on 11 March 2020 [3] following the rapid increase in infected case numbers outside its place of origin within a short time. The virus has spread to almost all countries globally with a total of more than 270 millions cases and over 5 millions of deaths reported in for approximately 2 years since it was first detected in late 2019 [4]. Meanwhile, due to the nature of the virus itself, as the virus replicates and produces an error, replicates contain several mutations, and so the virus has evolved resulting in several variants which may be implicated in high rates of infection among human population [5]. Since May 2021, the WHO has used a new naming system for key SARS-CoV-2 variants with letters of the Greek alphabet to label variants of concern (VOCs) or variants of interest (VOIs) for easier identification by the public [6]. These correspond to their own official scientific names in the Phylogenetic Assignment of Named Global Outbreaks (PANGO) lineage nomenclature [7]. As of December 2021, among more than a thousand variants circulating around the world [8], there are five VOCs and eight VOIs with this new naming system [9] as shown in Table 1.
Meanwhile, in order to describe variants, it is critical to identify and track mutations from genome sequencing. The global health effort is shown by deposition of the sequences of SARS-CoV-2 samples from many countries in FASTA format in public data repositories, such as the Global Initiative on Sharing All Influenza Data (GISAID) EpiCoV™ [10] and the National Center for Biotechnology Information (NCBI; Bethesda, MD, USA) GenBank [11], where they have been made freely accessible since January 2020. Most of these sequences are coming from Illumina (San Diego, CA, USA) [12], which is considered the worldwide market leader for next-generation sequencing technology.
The challenge of processing SARS-CoV-2 raw sequencing data in the FASTQ format is that it is of a greater size compared to the genome sequence in FASTA format which also persists. Recently, a web-based COVID-19 analysis platform [13] was published that can process raw sequencing data in FASTQ format. However, this platform is computationally impractical for public use when dealing with large sample numbers during a pandemic event like COVID-19, since it can only deal with a single sample in each turn by default. A scalable workflow could be a solution for processing multiple samples at the same time by utilizing parallel computation [14]. Some available workflows for analyzing SARS-CoV-2 genomes are still in limited use for non-variant detection [15] or non-Illumina sequencing data [16]. Meanwhile, analyzing sequencing data of SARS-CoV-2 also remains challenging due to an enormous shortage of experts [17]. For example, many available bioinformatics tools for analyzing the SARS-CoV-2 genome [18] are complex due to the fact of their code-based utilization which need prior programming knowledge to install and implement. Therefore, implementing friendly workflows for analyzing abundant Illumina-based FASTQ raw sequencing data for variant detection purposes has becomes an urgent need during the pandemic.
In this study, we built a cloud-powered bioinformatics workflow that was optimized for mutation profiling of SARS-CoV-2 variants to accommodate limitations of both computational and expert resources. Our installation-free workflow on a specified public cloud platform can diminish the implementation barrier in resource-poor settings to allow reliable variant identification from SARS-CoV-2 Illumina raw sequencing data in a high-throughput manner by utilizing parallel computation. Mutation profiling that consists of mutation identification and annotation is the central focus of our cloud workflow, and it is complemented by the generation of consensus sequences of the SARS-CoV-2 genome which can subsequently be deposited in public data repositories to support global health efforts.
2. Materials and Methods
2.1. Data Selection
We selected sequencing data from four early VOCs (Alpha, Beta, Gamma, and Delta), which are considered highly transmissible variants in human populations [19]. These four VOC types also accounted for ~80% of the submitted SARS-CoV-2 sequences available on GISAID EpiCoV™ as of December 31, 2021, including Pango sub-lineages for each VOC, Q.1~Q.8 (aliases of B.1.1.7.1~B.1.1.7.8) sub-lineages of Alpha, B.1.351.1~B.1.351.5 sub-lineages of Beta, P.1.1~P.1.17 (aliases of B.1.1.28.1.1~B.1.1.28.1.17) sub-lineages with their own descendants of Gamma, and AY.1~AY.133 (aliases of B.1.617.2.1~B.1.617.2.133) sub-lineages with their own descendants of Delta.
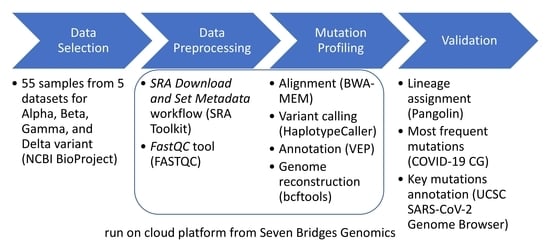
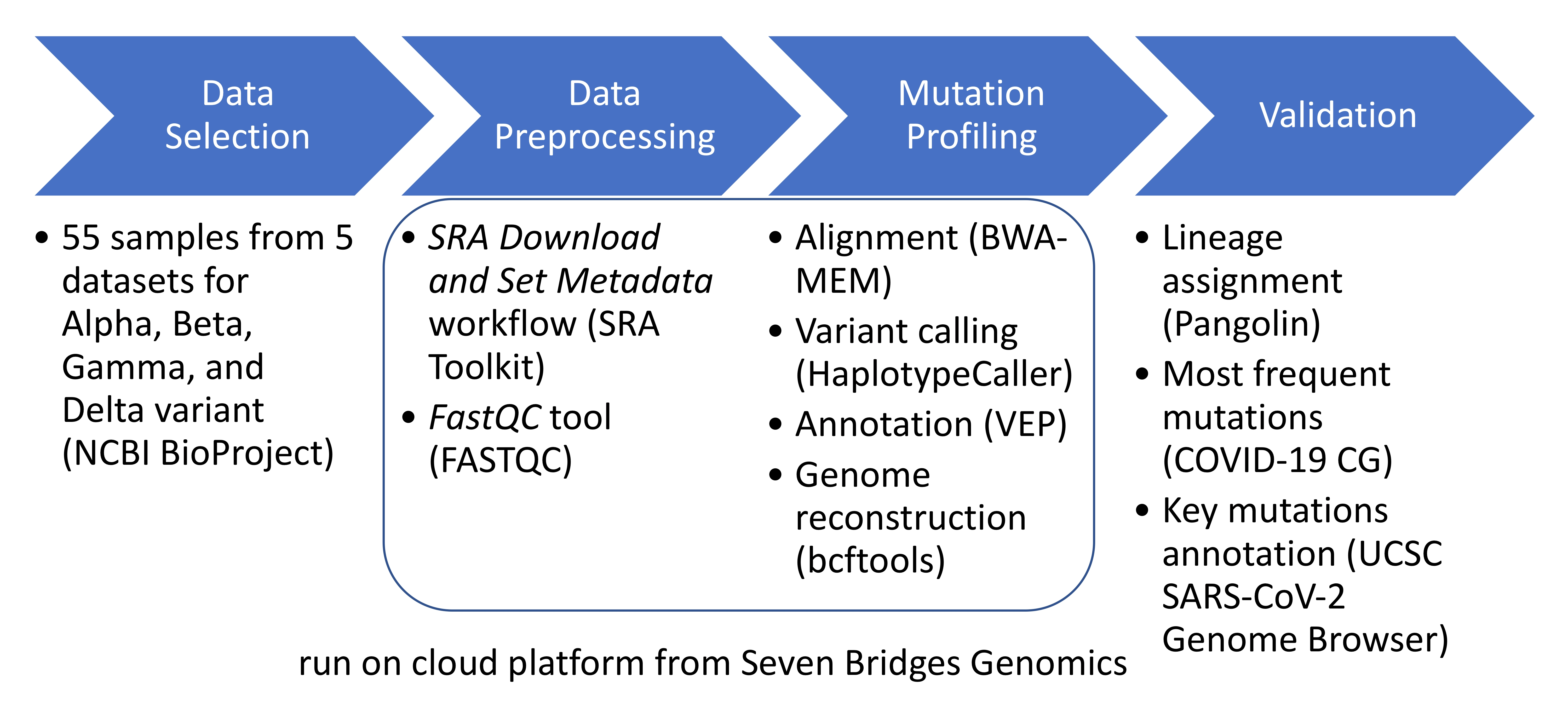
In this study, several data sets that collected raw sequencing data of SARS-CoV-2 from other wet-lab experiments were used to represent VOCs. The registered data sets from 2020~2021 were selected in the NCBI BioProject public repository [20] based on searches using “B.1.1.7” or “B.1.351” or “P.1” or “B.1.617.2” as keywords. The inclusion criteria were applied as follows: (1) data set from a virus organism group; (2) data sets from SARS-CoV-2 that infected humans; (3) data sets generated from Illumina sequencer types; (4) data sets with sequencing data files available in the Sequence Read Archive (SRA) database [21] at the NCBI; (5) data sets with clear numbers of VOCs; and (6) data sets with viral SARS-CoV-2 amplicon or whole-genome sequencing (WGS) data. Of 58 total BioProject data sets, 53 were ultimately excluded, leaving five data sets for further processing, which were PRJNA704235, PRJNA708134, PRJNA726840, PRJNA726871, and PRJNA733209. The flow chart diagram that shows the process of selecting these five data sets is shown in Figure 1, and a summary of each data set that accounts for a total of 55 samples subjected to more than 22 million raw sequencing reads is given in Table 2 (details of samples for each data set are available in a Supplementary Excel File, Data sets tab).
2.2. Data Preprocessing
We used the Cancer Genomics Cloud (CGC, https://cgc.sbgenomics.com, accessed on 13 November 2021) platform [22] from Seven Bridges Genomics (SBG; Boston, MA, USA) to further process our five selected data sets. CGC facilitates the workflow development from scratch through the Rabix Composer [23] using Common Workflow Language (CWL) [24], which is emerging as a workflow definition standard to describe analytical pipelines of bioinformatics tools for portable, scalable, and reproducible analyses. Meanwhile, the CGC also has hundreds of predefined tools or workflows which can be used for bioinformatics analytical purposes inside the platform that are publicly accessible through its web-based interface.
In the beginning, we used a predefined workflow called SRA Download and Set Metadata that implements SRA fasterq-dump (v.2.10.8) from the SRA Toolkit [25] to facilitate the automatic transfer of FASTQ raw sequencing files for all selected VOC data sets from the SRA database into the CGC platform in one execution turn. We provided the SRA metadata files in TXT format which contained the SRA accession numbers (the full list is available in a Supplementary Excel File, SRA Metadata tab) representing the FASTQ raw sequencing file for each sample of our selected VOC data sets as the input. All output read files from this workflow were then used as input for another predefined tool in the CGC called FastQC (v. 0.11.4) which implements the stand-alone FASTQC program [26] to evaluate the sequencing quality of each file. Only files that had an overall good per-base sequence quality assessment according to this tool were used for further mutation profiling purposes by our cloud workflow.
2.3. Mutation Profiling
2.3.1. Cloud Workflow Design
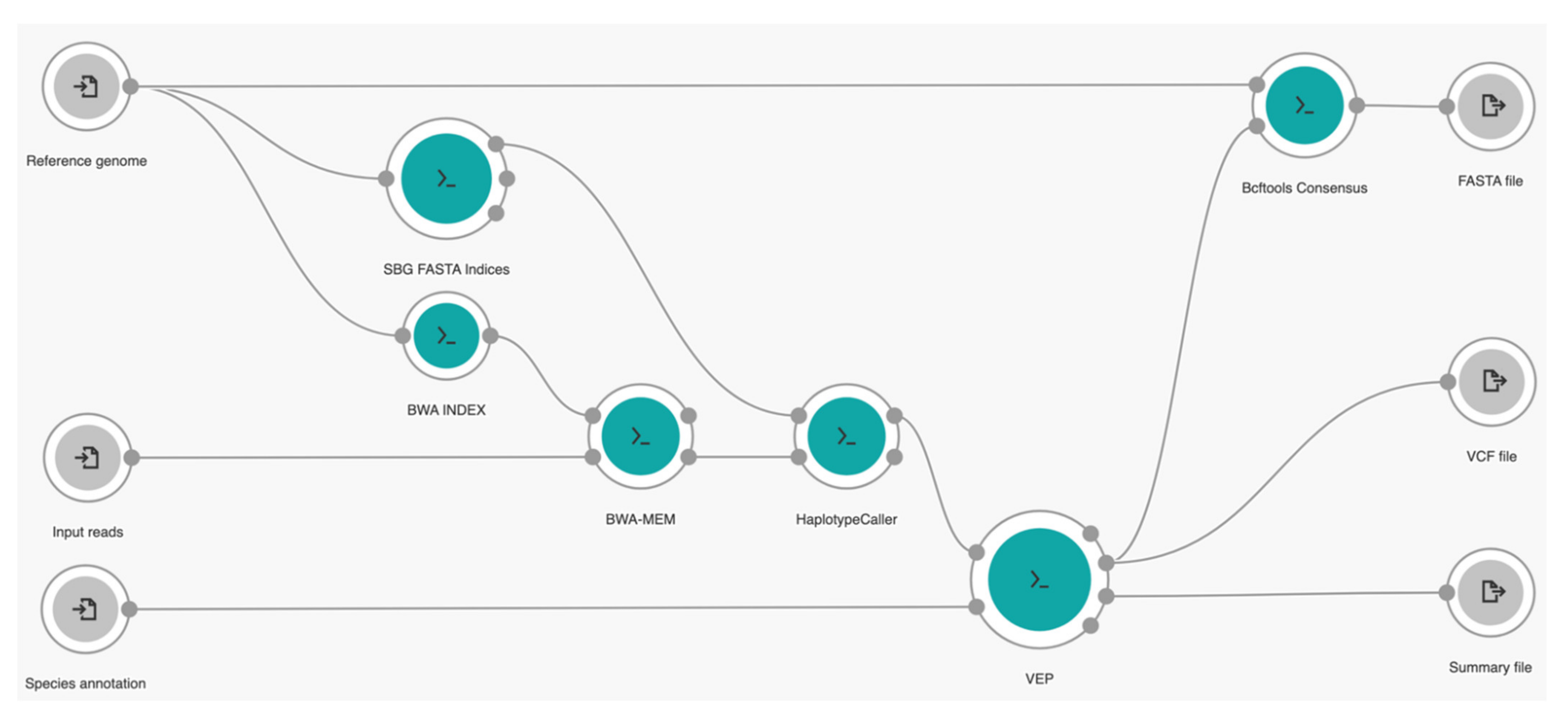
Our own tailored workflow is a series of several well-established bioinformatics tools, starting from alignment, variant calling, and variant annotation, to genome reconstruction built in the CGC platform. We used Burrows-Wheeler Alignment Maximal Exact Matches (BWA-MEM) for alignment [27], HaplotypeCaller from the Genome Analysis ToolKit (GATK) for variant calling [28], the Ensembl Variant Effect Predictor (VEP) for annotation [29], and BCFtools for consensus genome reconstruction [30]. Our workflow can run multiple times in parallel to process each sample per instance from Amazon Web Services (AWS; Amazon, Seattle, WA, USA) or the Google Cloud Platform (GCP; Google, Mountain View, CA, USA) by enabling the batch-mode option based on specified metadata criteria of input files on the platform. A layout of our cloud workflow is given in Figure 2.
In general, our workflow can be explained as follows. In the beginning, our workflow needs three different input files provided by the user: the raw sequencing read files as FASTQ, a reference genome as FASTA, and species cache information as zipped files. For this study of SARS-CoV-2 mutation profiling purposes, we used previously preprocessed files of selected VOC data sets as input reads, the original genome sequence of viral SARS-CoV-2 ribonucleic acid (RNA) with a length of 29,903 nucleotides [31] available in NCBI GenBank (NC_045512.2) as our reference genome, and annotation of a SARS-CoV-2 species cache file provided by Ensembl Genomes [32] for easier annotation purposes (available at http://ftp.ebi.ac.uk/ensemblgenomes/pub/viruses/variation/vep/sars_cov_2_vep_101_ASM985889v3.tar.gz, accessed on 13 November 2021). Then, the BWA INDEX tool was used to transform the reference genome in the full-text index in Minute space (FM-index) prior to the alignment process. At the same time, the reference genome was also processed by SBG FASTA Indices to create the FASTA dictionary and index which are later required for running GATK tools. The raw reads were aligned to the reference genome in the FM-index by BWA-MEM tool (v.0.7.17) and embedded with Biobambam2 sortmadup (v.2.0.87) [33] internally to detect and remove duplicate reads by selecting “RemoveDuplicates” for the duplicate detection option in the workflow settings. Next, the HaplotypeCaller tool (v. 4.0.2.0) was used to perform variant calling of aligned reads to generate the information of nucleic acid change positions for mutation identification purposes in the variant call format (VCF) output file [34], a standard format for recording genome polymorphism data, such as single nucleotide variations (SNVs), insertions/deletions (indels), and structural variants. Therefore, the VEP tool (v.101.0) was further used for detailed annotations of this VCF file by selecting “True” for the output sequence ontology variant class and to pick one line or block of consequence data per variant, including the transcript-specific column option, and “sars_cov_2” for the species option to generate the output of detailed annotation of VCF with its corresponding summary file. Finally, the Bcftools Consensus tool (v.1.9) was used to generate the genome sequence of raw sequencing data in FASTA format based on this VCF file and the reference genome.
2.3.2. Validation
All genome sequences reconstructed by our workflow were then passed to the web-based Pango lineage assigner tool (Pangolin, https://pangolin.cog-uk.io; v.3.1.19, with Pango lineages v. accessed on 20 January 2022) [35] in a multi-FASTA way to confirm the related Pango lineage mentioned in each BioProject data set. Meanwhile, we selected the most frequent mutations, a measure determined by dividing the number of corresponding mutations of the specified variant group by the total sample available on the same variant group that was present in ≥90% of the samples in our data sets within each VOC group. The same criteria were also applied to select the most frequent mutation for each VOC in the COVID-19 CoV Genetics (CG; https://covidcg.org/?tab=report accessed on 31 December 2021) web-based tool (v. 2.4.5) [36] to validate the mutation identification results from our workflow that occurred in larger genomics population samples by directly comparing the nucleotide features from our detailed annotation of the VCF output into this COVID-19 CG web-based tool of related Pango lineages. For this validation purpose, we used COVID-19 CG data as per 31 December 2021 that considered 965,917 Alpha, 27,098 Beta, 53,556 Gamma, and 151,863 Delta samples enabled by data from GISAID EpiCoV™. Meanwhile, another web-based tool called, the University of California Santa Cruz (UCSC) SARS-CoV-2 Genome Browser [37] was used to validate annotations of key mutations found in the most frequent mutations (available at https://genome.ucsc.edu/cgi-bin/hgTracks?db=wuhCor1 accessed on 31 December 2021) based on its 2021 updated database [38].
3. Results
3.1. Cloud Workflow Performance
In our experiment, 98% of reads (22,406,134 of 22,888,595) across 110 paired-ended read files of 55 samples passed our data preprocessing step that were used as input for our defined cloud workflow in mutation profiling steps. The 98% of reads came from data sets that had no additional file when downloaded as paired ended files containing forward and reverse reads.
All data preprocessing and mutation profiling steps were performed in the CGC platform under the availability of diverse computed-optimized AWS instances. For twenty minutes, US$ 2.22, with default instance configuration settings, was spent during this study as shown in more detail in Table 3. The data preprocessing step took the most time, while our workflow that focused on mutation profiling purposes took less time due to its parallel computation capability, which works very efficiently in a timely manner to simultaneously process multiple samples in one execution turn.
3.2. Mutation Profiling
Our workflow successfully identified 96% of samples (53 of 55) after the lineage of constructed genome sequence output for each sample was confirmed through the web-based Pangolin tool which belonged to lineages or sub-lineages of the respective lineage mentioned in corresponding data set metadata information with two samples withdrawn: SRR13907331 and SRR13907335 from PRJNA708134 Alpha group which belonged to B.1.1.528 and B.1.1.263 lineage respectively (the constructed FASTA genome file for all 55 samples is available as a zipped file in the Supplementary Materials). Some of the samples were also confirmed to belong to the sub-lineage of the respective lineage: two samples of the Q.4 sub-lineage in the Alpha group (SRR13907332 and SRR13907333 from PRJNA708134) and one sample of the P.1.14 sub-lineage in the Gamma group (SRR14736561 from PRJNA733209). From these 96% confirmed samples, 328 mutations with SNVs dominated at 273, and the remaining 55 indels were found by taking the output of detailed annotation of the VCF file for each sample from our workflow (the summary list of mutations from these 96% confirmed samples is available in Supplementary Materials: Table S1: MutProfil “Mutation tab”, with the mutation details in the matrix format available in SNVs and Indels tab in the same file).
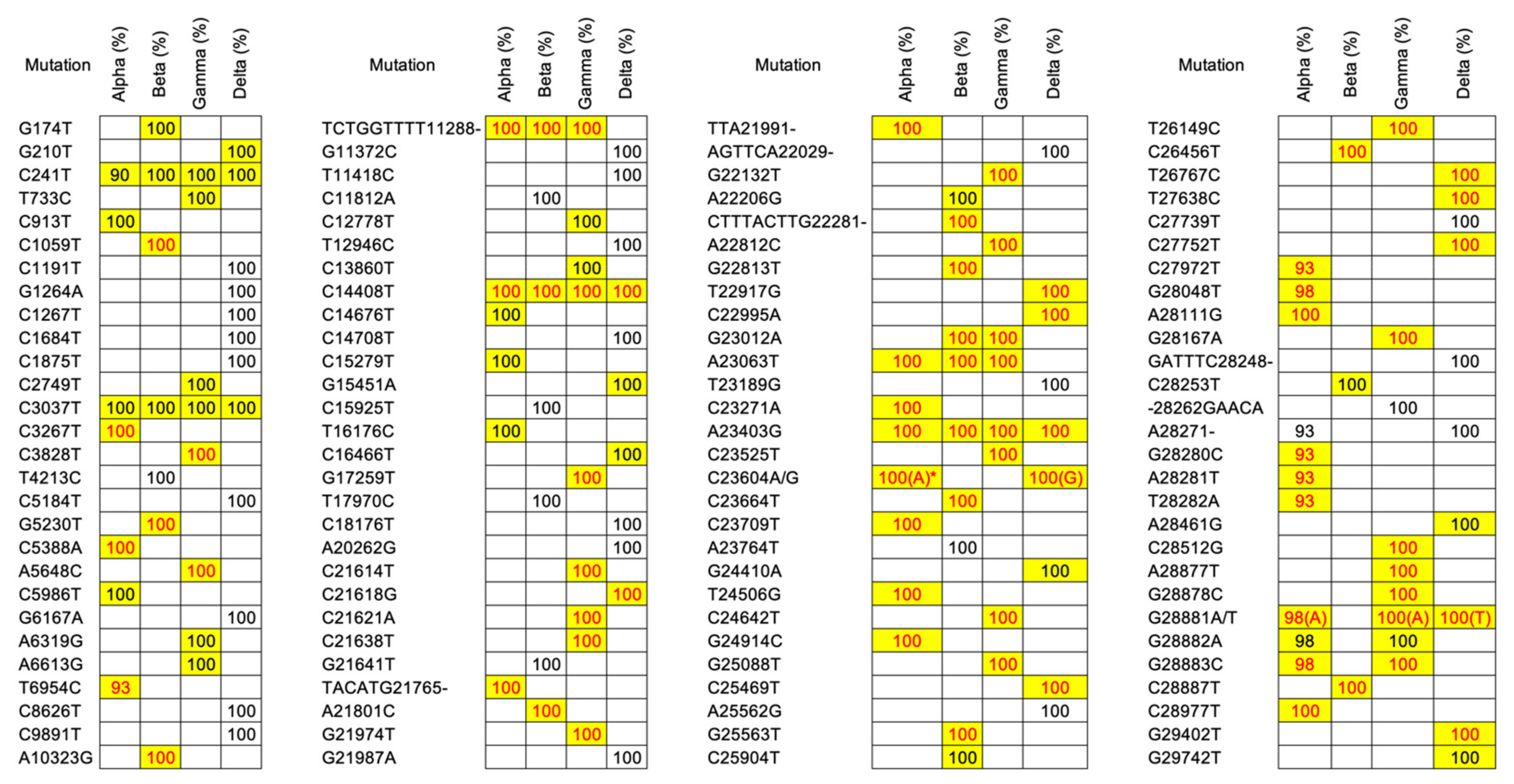
We further proceeded to validate these mutations in order to observe the occurrence of common mutations in each VOC group that could generally well represent or characterize the corresponding VOCs. There were 112 most frequent mutations consisting of 104 SNVs and 8 indels (one insertion and seven deletions) across the four VOCs; 32 for Alpha, 27 for Beta, 34 for Gamma, and 42 for Delta (a special explanation for Delta must be given since it included only a single sample which was insufficient to derive the most frequent mutations; therefore, we assumed all mutations in this sample were the most frequent mutations). Our observations of these most frequent mutations also matched very well with the COVID-19 CG. Specifically, all of the most frequent mutations listed in the COVID-19 CG for each VOC (31 for Alpha, 21 for Beta, 33 for Gamma, and 20 for Delta) belonged to the list of our most frequent mutations. Meanwhile, all key mutations mentioned in the UCSC SARS-CoV-2 Genome Browser for each VOC (23 for Alpha, 15 for Beta, 24 for Gamma, and 12 for Delta) were also found in these validated most frequent mutations (see Figure 3 for a list of the most frequent and key mutations across the four VOCs). All of these key mutations occurred in the protein-coding gene region that has moderate or high mutation consequences based on detailed annotation information of the codon change from our cloud workflow output which can provide quick insights into how the mutation impacts the encoded protein (see Table 4, Table 5, Table 6 and Table 7 for more annotation details of key mutations of Alpha, Beta, Gamma, and Delta, respectively). The annotation of mutations that occurred in the open reading frame 1ab (ORF1ab) protein-coding gene from our results strongly contrasted with results in the UCSC SARS-CoV-2 Genome Browser since the annotation file did not cover all 16 non-structural proteins (nsps; nsp1~16) encoded from ORF1ab [39] due to the complexity of the annotation process in this gene region [40]. Meanwhile, results for the remaining protein-coding genes revealed a consensus with many mutations mainly found in spike (S) protein-coding genes in all VOC groups.
4. Discussion
We present herein a workflow that integrates several publicly available bioinformatics tools, ranging from BWA-MEM, HaplotypeCaller, and VEP to BCFtools, that can be run in a publicly available cloud platform, CGC, to process raw Illumina sequencing data in FASTQ format stored in a public SRA database from systematic open-access data sets selected in the NCBI BioProject based on some public available data for SARS-CoV-2, including the public SARS-CoV-2 reference genome in the NCBI GenBank and SARS-CoV-2 annotation data from Ensembl Genomes, for mutation profiling of SARS-CoV-2 variants. Our workflow results were highly concordant in terms of genome consensus sequence identity (to the sub-lineage level), and mutation identification and annotation when cross-validated with other dedicated SARS-CoV-2 resources, ranging from the web-based Pangolin tool to COVID-19 CG and UCSC SARS-CoV-2 Genome Browser. In addition, this study also suggested that when different data sets that come from different experiments are taken into consideration, we can still conclude the similar most frequent mutations even with limited sample sizes. Taking our experiment as an example, samples in the Alpha and Gamma groups that came from more than one data set within the sub-lineage found for each group, Q.4 for Alpha and P.1.14 for Gamma, still reflected the most frequent mutations found in larger sample sizes available from other resources. This phenomenon can be explained by the concordance of genomes deposited in the larger sample size in public repository data which were also generated from many experiments around the world resulting in high diversity levels similar to our distinct data sets used in the Alpha and Gamma groups. Meanwhile, our workflow also ensures that there is an unbiased bioinformatics analysis across data, since we used raw sequencing data as our input and applied uniform analytical tools to process these data from the beginning. This approach is critical when dealing with data that come from multiple data sets from different experiments as seen in this study due to different experiments that may have been conducted by different sequencing data analyses based on each lab’s protocol settings. Finally, our workflow that integrated many publicly available resources ensures its reproducibility for use by a wide variety of users from diverse backgrounds in their workspace by extending its capability to process their own sequencing data sets in real-world settings beyond the open-access data sets shown in this study by dragging and dropping their own sequencing files along with all necessary data using the CGC web uploader to make it run.
Understanding SARS-CoV-2 mutations is also very helpful to elucidate genomic variability that allows the virus to evade host immunity and acquire drug resistance. For example, some vaccines to prevent the COVID-19 infection were developed to target proteins of SARS-CoV-2 with the S protein as the main protein target due to its role in initiating viral entry into host cells [41]. Some mutations found in this S protein made the current vaccines less effective since they can impact antibody neutralization which makes variants within these mutations exhibit resistance to antibody-mediated immunity induced by the vaccines [42]. Meanwhile, the antiviral drug, remdesivir, is also mainly used to inhibit the SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) protein (nsp12) that plays a role in viral polymerase which is essential for replicating viral RNA [43]. Any mutation to this RdRp makes the administration of remdesivir become ineffective since it affects the binding affinity of the drug [44]. Therefore, our workflow that utilizes genomics technologies is valuable for understanding the molecular underpinning of SARS-CoV-2 through profiling its mutation capability. Furthermore, the direct annotation from sequencing data within our cloud workflow can provide quick insights into where mutations exist in protein-coding genes to derive additional insights into SARS-CoV-2. Take the most frequent mutations from each VOC group in our experiment for example, there were four mutations found which overlapped in four VOCs, which were C241T, C3037T, C14408T, and A23403G. These mutations showed strong allelic associations since they were the dominant types found in the SARS-CoV-2 genome in a previous study [45]. Two non-synonymous mutations of this quartet, C14408T and A23403G, that respectively correspond to the RdRp protein P4715L and S protein D614G were found to be key mutations in this study and may impact current treatment options, while D614G was associated with an increase of human-to-human transmission efficiency [46]. They also showed significant positive correlations with fatality rates by Toyoshima et al. [47], which may explain why the disease severity increased when infected by these four VOCs [48], while the other two did not belong to the key mutation list in our experiment due to their location in the 5’ untranslated region (C241T) and having low coding consequential impacts of a synonymous mutation in the nsp3 (C3037T), which is known as the largest encoded multi-domain protein in CoV genera [49]. In addition, the raw sequencing data used as input for our cloud workflow open the possibility of precisely identifying existing or future SARS-CoV-two variants, which cannot be conducted with the current existing diagnostics test assay of reverse-transcription polymerase chain reaction (RT-PCR). Unlike sequencing, RT-PCR does not facilitate detecting changes in the SARS-CoV-2 viral genome which is key to detecting mutations in the viral variants [50].
Our workflow can be a solution to a lack of resources for analyzing the SARS-CoV-2 genome. Our workflow has an affordable cost which will be beneficial for implementation especially in those countries with limited computational and expert resources. Given the fact that many VOCs and VOIs that appeared in developing countries as determined by the United Nations [51], such as VOC Beta in South Africa, VOC Gamma and VOI Zeta in Brazil, VOC Delta and VOI Kappa in India, VOI Theta in the Philippines, VOI Lambda in Peru, and VOI Mu in Colombia, may have been circulating around the world including in those least developed countries with resource-poor settings and limited access to computational and expert resources, hence our workflow with fewer requirements is accessible online by everyone through their own local machine with internet access which can be a solution to tackle resource-limitation issues. Previously, without the workflow systems, each code-based bioinformatics tool that constructed the workflow must be installed manually in the local computer, required prior programming knowledge to perform the analysis, and works independently to analyze one sample per run [52], which all are time-consuming and not feasible especially in the time of pandemic. Even though, with workflow systems, an understanding of the syntax was still required to set up the environment first before running the entire workflow, for example, COVseq [53] and V-pipe [54] that required to install the workflow engine of Snakemake [55], resulting in an additional time that may delay mutation profiling process. Therefore, our CWL-based workflow takes advantage of particular cloud environment by packed together several pre-defined bioinformatics tools into an installation-free workflow by just copying and pasting the code in JSON format on the CGC platform. This will ensure that users with no prior programming knowledge can also directly benefit from our workflow by not necessarily deploying the analytical tools one by one anymore, since the analytical process is automatically done end-to-end along the workflow. In addition, since our workflow works through the cloud that is capable of parallel computation, the same workflow can be run in many instances to ensure robustness, while users do not necessarily have to manually perform the same analysis for each sample over and over again. Finally, our workflow can also provide great support to the global health effort of tracking the occurrence of variants around the world. Our cloud workflow is able to generate sequences of SARS-CoV-2 in FASTA format from raw sequencing data in a short time which can be deposited in public data repositories such as GISAID EpiCoV™ or NCBI GenBank. Later, these sequences can be used for any further downstream analysis, if needed, like phylogenetics analysis to track transmission, flag key mutations, or estimate reproduction numbers [56].
Our present workflow has several limitations. Although our workflow uses standardized workflow definitions in CWL, since our workflow utilizes the CGC platform-agnostic code, it might not work optimally outside the CGC platform. Therefore, prior registration in the CGC platform is needed in order to use our cloud workflow. Meanwhile, parallel computations on the CGC platform can only perform up to 80 samples in one execution turn by default. If a user wishes to use a larger number, a request should be made to the CGC team in advance. Furthermore, our workflow only works with Illumina sequencing data as the input due to genomics technological differences that exist behind each sequencing platform, while Illumina works in short read sequencing by a synthesis approach [57].
5. Conclusions
There are a number of SARS-CoV-2 variants circulating around the world which have made the end of the novel COVID-19 pandemic unpredictable. It is important to know particular variants based on the identified mutations of the genes, since such variants may have different virulence activities. Cloud-powered diagnostic tools have accelerated the identification of viral mutations through sequencing which has become critical to fighting COVID-19. Our workflow that collects a set of well-established bioinformatics tools for mutation profiling purposes, including viral mutation identification and annotation, can be an option to meet this demand. Our workflow designed for Illumina sequencing works in parallel, in a rapid and cost-effective manner suitable for resource-poor settings and may be best suited for application worldwide for detecting current and emerging variants so that the spread of specific variants can be limited as early as possible. In addition, our workflow is able to generate a genome sequence of SARS-CoV-2 to support global genomics surveillance by accession to public data repositories. In the future, our cloud workflow capability can be extended to predict upcoming variants with a prominent machine learning approach due to the abundant SARS-CoV-2 sequences available in public data repositories. Furthermore, our workflow can also be easily operated to detect variants of other pathogens species beyond SARS-CoV-2 as long as sequencing data, reference genome, and the related species annotation data are available.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/genes13040686/s1, Detailed information on the data sets used in this paper and their identification performance are available in the Supplementary Materials: Table S1: MutProfil. The generated genomic sequences of all samples used in this study are also available as a zipped file: MutProfil.zip.
Author Contributions
Conceptualization, H.G.-M.L. and Y.-C.G.L.; data curation, H.G.-M.L. and S.-H.H.; formal analysis, H.G.-M.L. and Y.-C.G.L.; funding acquisition, S.-H.H., Y.C.F. and Y.-C.G.L.; investigation, S.-H.H.; Methodology, H.G.-M.L.; Project administration, Y.-C.G.L.; Resources, Y.C.F. and Y.-C.G.L.; Software, H.G.-M.L.; Supervision, Y.C.F. and Y.-C.G.L.; Validation, S.-H.H. and Y.-C.G.L.; Visualization, H.G.-M.L.; Writing—original draft, H.G.-M.L.; Writing—review and editing, S.-H.H., Y.C.F. and Y.-C.G.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the Ministry of Science and Technology (MOST) (grant MOST 109-2221-E-038-016) of the Taiwanese government to Y.-C.G.L., the Taipei Medical University Hospital (W0303, 109TMUH-SP-02) to S.-H.H. and the Fund from the Division of Intramural Research Program, National Institute of Neurological Disorders and Stroke, National Institutes of Health, USA to Y.C.F. The Seven Bridges Cancer Research Data Commons Cloud Resource has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health (Contract No. HHSN261201400008C and ID/IQ Agreement No. 17×146 under Contract No. HHSN261201500003I and 75N91019D00024).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The workflow source code is freely available for reproducible purposes on the Cancer Genomics Cloud platform at https://github.com/hendrick0403/MutProfil (accessed on 20 January 2022) since 2 April 2022. Meanwhile, five VOC data sets used in this study can be found in the NCBI BioProject public repository (https://www.ncbi.nlm.nih.gov/bioproject, accessed on 3 November 2021) with accession codes of PRJNA704235, PRJNA708134, PRJNA726840, PRJNA726871, and PRJNA733209.
Acknowledgments
The authors wish to thank the Seven Bridges Genomics bioinformatics team for providing the predefined tools and workflow used in this study and their technical support during our defined workflow execution. We also gratefully acknowledge the authors, originating and submitting laboratories of the SARS-CoV-2 data sets from the NCBI BioProject, SARS-CoV-2 annotation file from Ensembl Genomes, SARS-CoV-2 sequences from the NCBI GenBank for our reference genome, and GISAID EpiCoV™ as the source to cross-validate our results. This manuscript was edited by Wallace Academic Editing.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naming the Coronavirus Disease (COVID-19) and the Virus that Causes It. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance/naming-the-coronavirus-disease-(covid-2019)-and-the-virus-that-causes-it (accessed on 31 December 2021).
- Mahase, E. COVID-19: WHO declares pandemic because of “alarming levels” of spread, severity, and inaction. BMJ 2020, 368, m1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Lauring, A.S.; Hodcroft, E.B. Genetic Variants of SARS-CoV-2-What Do They Mean? JAMA 2021, 325, 529–531. [Google Scholar] [CrossRef]
- WHO Announces Simple, Easy-to-Say Labels for SARS-CoV-2 Variants of Interest and Concern. Available online: https://www.who.int/news/item/31-05-2021-who-announces-simple-easy-to-say-labels-for-sars-cov-2-variants-of-interest-and-concern (accessed on 31 December 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- O’Toole, A.; Hill, V.; Pybus, O.G.; Watts, A.; Bogoch, I.I.; Khan, K.; Messina, J.P.; COVID, T.; UK, G. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2 with grinch. Wellcome Open Res. 2021, 6, 121. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 31 December 2021).
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data-from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2022, 50, D161–D164. [Google Scholar] [CrossRef]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef]
- Gong, Z.; Zhu, J.W.; Li, C.P.; Jiang, S.; Ma, L.N.; Tang, B.X.; Zou, D.; Chen, M.L.; Sun, Y.B.; Song, S.H.; et al. An online coronavirus analysis platform from the National Genomics Data Center. Zool. Res. 2020, 41, 705–708. [Google Scholar] [CrossRef]
- Strozzi, F.; Janssen, R.; Wurmus, R.; Crusoe, M.R.; Githinji, G.; di Tommaso, P.; Belhachemi, D.; Moller, S.; Smant, G.; de Ligt, J.; et al. Scalable workflows and reproducible data analysis for genomics. In Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1910, pp. 723–745. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.G.; Hsiao, S.H.; Lee, Y.G. Orchestrating an Optimized Next-Generation Sequencing-Based Cloud Workflow for Robust Viral Identification during Pandemics. Biology 2021, 10, 1023. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Krautwurst, S.; Spott, R.; Lohde, M.; Jundzill, M.; Marquet, M.; Holzer, M. Porecov-An Easy to Use, Fast, and Robust Workflow for SARS-CoV-2 Genome Reconstruction via Nanopore Sequencing. Front. Genet. 2021, 12, 711437. [Google Scholar] [CrossRef] [PubMed]
- Perkel, J.M. Workflow systems turn raw data into scientific knowledge. Nature 2019, 573, 149–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hufsky, F.; Lamkiewicz, K.; Almeida, A.; Aouacheria, A.; Arighi, C.; Bateman, A.; Baumbach, J.; Beerenwinkel, N.; Brandt, C.; Cacciabue, M.; et al. Computational strategies to combat COVID-19: Useful tools to accelerate SARS-CoV-2 and coronavirus research. Brief. Bioinform. 2021, 22, 642–663. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.; Archer, B.; Laurenson-Schafer, H.; Jinnai, Y.; Konings, F.; Batra, N.; Pavlin, B.; Vandemaele, K.; van Kerkhove, M.D.; Jombart, T.; et al. Increased transmissibility and global spread of SARS-CoV-2 variants of concern as at June 2021. Eurosurveillance 2021, 26, 2100509. [Google Scholar] [CrossRef]
- Barrett, T.; Clark, K.; Gevorgyan, R.; Gorelenkov, V.; Gribov, E.; Karsch-Mizrachi, I.; Kimelman, M.; Pruitt, K.D.; Resenchuk, S.; Tatusova, T.; et al. BioProject and BioSample databases at NCBI: Facilitating capture and organization of metadata. Nucleic Acids Res. 2012, 40, D57–D63. [Google Scholar] [CrossRef]
- Katz, K.; Shutov, O.; Lapoint, R.; Kimelman, M.; Brister, J.R.; O’Sullivan, C. The Sequence Read Archive: A decade more of explosive growth. Nucleic Acids Res. 2022, 50, D387–D390. [Google Scholar] [CrossRef]
- Lau, J.W.; Lehnert, E.; Sethi, A.; Malhotra, R.; Kaushik, G.; Onder, Z.; Groves-Kirkby, N.; Mihajlovic, A.; DiGiovanna, J.; Srdic, M.; et al. The Cancer Genomics Cloud: Collaborative, Reproducible, and Democratized-A New Paradigm in Large-Scale Computational Research. Cancer Res. 2017, 77, e3–e6. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, G.; Ivkovic, S.; Simonovic, J.; Tijanic, N.; Davis-Dusenbery, B.; Kural, D. Rabix: An open-source workflow executor supporting recomputability and interoperability of workflow descriptions. In Proceedings of the Pacific Symposium on Biocomputing, Kohala Coast, HI, USA, 3–7 January 2017; Volume 22, pp. 154–165. [Google Scholar] [CrossRef] [Green Version]
- Amstutz, P.; Crusoe, M.R.; Tijanić, N.; Chapman, B.; Chilton, J.; Heuer, M.; Kartashov, A.; Leehr, D.; Ménager, H.; Nedeljkovich, M.; et al. Common Workflow Language, v1.0. Figshare 2016. [Google Scholar] [CrossRef]
- Team, S.T.D. The NCBI SRA Toolkit. Available online: https://github.com/ncbi/sra-tools (accessed on 31 December 2021).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 31 December 2021).
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yates, A.D.; Allen, J.; Amode, R.M.; Azov, A.G.; Barba, M.; Becerra, A.; Bhai, J.; Campbell, L.I.; Carbajo Martinez, M.; Chakiachvili, M.; et al. Ensembl Genomes 2022: An expanding genome resource for non-vertebrates. Nucleic Acids Res. 2022, 50, D996–D1003. [Google Scholar] [CrossRef]
- Tischler, G.; Leonard, S. biobambam: Tools for read pair collation based algorithms on BAM files. Source Code Biol. Med. 2014, 9, 13. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- O’Toole, A.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Chen, A.T.; Altschuler, K.; Zhan, S.H.; Chan, Y.A.; Deverman, B.E. COVID-19 CG enables SARS-CoV-2 mutation and lineage tracking by locations and dates of interest. eLife 2021, 10, e63409. [Google Scholar] [CrossRef]
- Fernandes, J.D.; Hinrichs, A.S.; Clawson, H.; Gonzalez, J.N.; Lee, B.T.; Nassar, L.R.; Raney, B.J.; Rosenbloom, K.R.; Nerli, S.; Rao, A.A.; et al. The UCSC SARS-CoV-2 Genome Browser. Nat. Genet. 2020, 52, 991–998. [Google Scholar] [CrossRef]
- Navarro Gonzalez, J.; Zweig, A.S.; Speir, M.L.; Schmelter, D.; Rosenbloom, K.R.; Raney, B.J.; Powell, C.C.; Nassar, L.R.; Maulding, N.D.; Lee, C.M.; et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021, 49, D1046–D1057. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.; Al-Mulla, F.; Wei, D.Q.; Abubaker, J. Remdesivir MD Simulations Suggest a More Favourable Binding to SARS-CoV-2 RNA Dependent RNA Polymerase Mutant P323L Than Wild-Type. Biomolecules 2021, 11, 919. [Google Scholar] [CrossRef]
- Yang, H.C.; Chen, C.H.; Wang, J.H.; Liao, H.C.; Yang, C.T.; Chen, C.W.; Lin, Y.C.; Kao, C.H.; Lu, M.J.; Liao, J.C. Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations. Proc. Natl. Acad. Sci. USA 2020, 117, 30679–30686. [Google Scholar] [CrossRef]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef]
- Lin, L.; Liu, Y.; Tang, X.; He, D. The Disease Severity and Clinical Outcomes of the SARS-CoV-2 Variants of Concern. Front. Public Health 2021, 9, 775224. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Ascoli, C.A. Could mutations of SARS-CoV-2 suppress diagnostic detection? Nat. Biotechnol. 2021, 39, 274–275. [Google Scholar] [CrossRef] [PubMed]
- World Economic Situation and Prospects. Available online: https://www.un.org/development/desa/dpad/wp-content/uploads/sites/45/WESP2020_Annex.pdf (accessed on 31 December 2021).
- Wratten, L.; Wilm, A.; Goke, J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat. Methods 2021, 18, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, M.; Zhang, N.; Harbers, L.; Milia, M.G.; Brossa, S.; Huong Nguyen, T.T.; Cerutti, F.; Berrino, E.; Sapino, A.; Bienko, M.; et al. COVseq is a cost-effective workflow for mass-scale SARS-CoV-2 genomic surveillance. Nat. Commun. 2021, 12, 3903. [Google Scholar] [CrossRef] [PubMed]
- Posada-Cespedes, S.; Seifert, D.; Topolsky, I.; Jablonski, K.P.; Metzner, K.J.; Beerenwinkel, N. V-pipe: A computational pipeline for assessing viral genetic diversity from high-throughput data. Bioinformatics 2021, 37, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Koster, J.; Rahmann, S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics 2012, 28, 2520–2522. [Google Scholar] [CrossRef] [Green Version]
- Hodcroft, E.B.; de Maio, N.; Lanfear, R.; MacCannell, D.R.; Minh, B.Q.; Schmidt, H.A.; Stamatakis, A.; Goldman, N.; Dessimoz, C. Want to track pandemic variants faster? Fix the bioinformatics bottleneck. Nature 2021, 591, 30–33. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
Figure 1.
Flowchart diagram for selecting the data sets.

Figure 2.
Graphical representation of our cloud workflow.

Figure 3.
The most frequent mutations found in our variant of concern (VOC) samples. Mutation percentages highlighted in yellow denote the most common mutations listed in the COVID-19 CG for corresponding VOCs, while red mutation percentages denote key mutations listed by the UCSC SARS-CoV-2 Genome Browser. * Most mutations found in the sample, unless for its sub-lineage (Q.4) with C23604G.
Figure 3.
The most frequent mutations found in our variant of concern (VOC) samples. Mutation percentages highlighted in yellow denote the most common mutations listed in the COVID-19 CG for corresponding VOCs, while red mutation percentages denote key mutations listed by the UCSC SARS-CoV-2 Genome Browser. * Most mutations found in the sample, unless for its sub-lineage (Q.4) with C23604G.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
SARS-CoV-2 variants of concern (VOCs) and variants of interest (VOIs).
| Variant. | Lineage * | Alias | Place of Origin | Time of Origin | Type | Designation |
|---|---|---|---|---|---|---|
| α (Alpha) | B.1.1.7 | - | United Kingdom | September 2020 | VOC | 18 December 2020 |
| β (Beta) | B.1.351 | - | South Africa | May 2020 | VOC | 18 December 2020 |
| γ (Gamma) | P.1 | B.1.1.28.1 | Brazil | November 2020 | VOC | 11 January 2021 |
| δ (Delta) | B.1.617.2 | - | India | October 2020 | VOC | 11 May 2021 |
| ε (Epsilon) | B.1.427/B.1.429 | - | United States | March 2020 | VOI | 5 March 2021 |
| ζ (Zeta) | P.2 | B.1.1.28.2 | Brazil | April 2020 | VOI | 17 March 2021 |
| η (Eta) | B.1.525 | - | multiple countries | December 2020 | VOI | 17 March 2021 |
| θ (Theta) | P.3 | B.1.1.28.3 | Philippines | January 2021 | VOI | 24 March 2021 |
| ι (Iota) | B.1.526 | - | United States | November 2020 | VOI | 24 March 2021 |
| κ (Kappa) | B.1.617.1 | - | India | October 2020 | VOI | 4 April 2021 |
| λ (Lambda) | C.37 | B.1.1.1.37 | Peru | December 2020 | VOI | 14 June 2021 |
| μ (Mu) | B.1.621 | - | Colombia | January 2021 | VOI | 30 August 2021 |
| ο (Omicron) | B.1.1.529 | - | multiple countries | November 2021 | VOC | 26 November 2021 |
SARS-CoV-2, severe acute respiratory syndrome coronavirus 2. * Includes all descendent lineages.
Table 2.
Summary of selected data sets.
| Data Set | No. of Samples | No. of Reads | Sequencer Type | Sequencing Strategy | |||
|---|---|---|---|---|---|---|---|
| Alpha | Beta | Gamma | Delta | ||||
| PRJNA704235 | - | - | 3 | - | 1,483,054 | MiniSeq | WGS |
| PRJNA708134 | 39 | - | - | - | 16,060,411 | NovaSeq | amplicon |
| PRJNA726840 | 3 | 8 | - | - | 2,801,089 | iSeq | WGS |
| PRJNA726871 | - | - | - | 1 | 128,048 | MiSeq | amplicon |
| PRJNA733209 | - | - | 1 | - | 2,415,993 | MiniSeq | WGS |
| Total | 42 | 8 | 4 | 1 | 22,888,595 | - | - |
WGS, whole-genome sequencing.
Table 3.
Summary of the cloud workflow performance.
| Step | Name | Instance 1 | Time 2 | Cost 3 |
|---|---|---|---|---|
| Data preprocessing | SRA Download and Set Metadata workflow | c4.8xlarge a | 7 | 0.09 |
| FastQC tool | c4.2xlarge b | 6 | 0.04 | |
| Mutation Profiling | Our cloud workflow * | c4.2xlarge | 7 | 2.09 |
| Total | 20 | 2.22 | ||
1 From Amazon Web Services; 2 in minutes; 3 in US$. a Thirty-six virtual central processing units (vCPUs), 60 gigabytes (GB) of memory, and 1024 GB attached storage; b 8v CPUs, 15 GB of memory, and 1024 GB attached storage. * In parallel with one instance per sample that cost on average US $0.04 per instance.
Table 4.
Key mutations in the Alpha VOC.
| Mutation | Type | Level | Protein Coding Gene Annotation | Codon Change | Consequence for the AA Sequence | Corresponding Protein Annotation of UCSC SARS-CoV-2 |
|---|---|---|---|---|---|---|
| C3267T | SNV | moderate | ORF1ab | aCt3002aTt | T1001I | nsp3 |
| C5388A | SNV | moderate | ORF1ab | gCt5123gAt | A1708D | nsp3 |
| T6954C | SNV | moderate | ORF1ab | aTa6689aCa | I2230T | nsp3 |
| TCTGGTTTT11288- | deletion | moderate | ORF1ab | TCTGGTTTT 11023:11031- | SGF3675:3677- | nsp6 |
| C14408T | SNV | moderate | ORF1ab | cCt14144cTt | P4715L | nsp12 |
| TACATG21765- | deletion | moderate | S | aTACATGtc 203:208atc | IHV68:70I | S |
| TTA21991- | deletion | moderate | S | gtTTAt 429-431gtt | VY143:144V | S |
| A23063T | SNV | moderate | S | Aat1501Tat | N501Y | S |
| C23271A | SNV | moderate | S | gCt1709gAt | A570D | S |
| A23403G | SNV | moderate | S | gAt1841gGt | D614G | S |
| C23604A | SNV | moderate | S | cCt2042cAt | P681H | S |
| C23709T | SNV | moderate | S | aCa2147aTa | T716I | S |
| T24506G | SNV | moderate | S | Tca2944Gca | S982A | S |
| G24914C | SNV | moderate | S | Gac3352Cac | D1118H | S |
| C27972T | SNV | high | ORF8 | Caa79Taa | Q27stop | ORF8 |
| G28048T | SNV | moderate | ORF8 | aGa155aTa | R52I | ORF8 |
| A28111G | SNV | moderate | ORF8 | tAc218tGc | Y73C | ORF8 |
| G28280C | SNV | moderate | N | Gat7Cat | D3H | N |
| A28281T | SNV | moderate | N | gAt8gTt | D3V | N |
| T28282A | SNV | moderate | N | gaT9gaA | D3E | N |
| G28881A | SNV | moderate | N | aGg608aAg | R203K | N |
| G28883C | SNV | moderate | N | Gga610Cga | G204R | N |
| C28977T | SNV | moderate | N | tCt704tTt | S235F | N |
AA, amino acid; SNV, single nucleotide variation; ORF, open reading frame; nsp, non-structural protein; S, spike; N, nucleocapsid.
Table 5.
Key mutations in the Beta VOC.
| Mutation | Type | Level | Protein Coding Gene Annotation | Codon Change | Consequence for the AA Sequence | Corresponding Protein Annotation of UCSC SARS-CoV-2 |
|---|---|---|---|---|---|---|
| C1059T | SNV | moderate | ORF1ab | aCc794aTc | T265I | nsp2 |
| G5230T | SNV | moderate | ORF1ab | aaG4965aaT | K1655N | nsp3 |
| A10323G | SNV | moderate | ORF1ab | aAg10058aGg | K3353R | nsp5 |
| TCTGGTTTT11288- | deletion | moderate | ORF1ab | TCTGGTTTT 11023:11031- | SGF3675:3677- | nsp6 |
| C14408T | SNV | moderate | ORF1ab | cCt14144cTt | P4715L | nsp12 |
| A21801C | SNV | moderate | S | gAt239gCt | D80A | S |
| CTTTACTTG22281- | deletion | moderate | S | aCTTTACTTGct719-727act | TLLA240:243T | S |
| G22813T | SNV | moderate | S | aaG1251aaT | K417N | S |
| G23012A | SNV | moderate | S | Gaa1450Aaa | E484K | S |
| A23063T | SNV | moderate | S | Aat1501Tat | N501Y | S |
| A23403G | SNV | moderate | S | gAt1841gGt | D614G | S |
| C23664T | SNV | moderate | S | gCa2102gTa | A701V | S |
| G25563T | SNV | moderate | ORF3a | caG171caT | Q57H | ORF3a |
| C26456T | SNV | moderate | E | cCt212cTt | P71L | E |
| C28887T | SNV | moderate | N | aCt614aTt | T205I | N |
E, envelope.
Table 6.
Key mutations in the Gamma VOC.
| Mutation | Type | Level | Protein Coding Gene Annotation | Codon Change | Consequence for the AA Sequence | Corresponding Protein Annotation of UCSC SARS-CoV-2 |
|---|---|---|---|---|---|---|
| C3828T | SNV | moderate | ORF1ab | tCa3563tTa | S1188L | nsp3 |
| A5648C | SNV | moderate | ORF1ab | Aaa5383Caa | K1795Q | nsp3 |
| TCTGGTTTT11288- | deletion | moderate | ORF1ab | TCTGGTTTT 11023:11031- | SGF3675:3677- | nsp6 |
| C14408T | SNV | moderate | ORF1ab | cCt14144cTt | P4715L | nsp12 |
| G17259T | SNV | moderate | ORF1ab | gaG16995gaT | E5665D | nsp13 |
| C21614T | SNV | moderate | S | Ctt52Ttt | L18F | S |
| C21621A | SNV | moderate | S | aCc59aAc | T20N | S |
| C21638T | SNV | moderate | S | Cct76Tct | P26S | S |
| G21974T | SNV | moderate | S | Gat412Tat | D138Y | S |
| G22132T | SNV | moderate | S | agG570agT | R190S | S |
| A22812C | SNV | moderate | S | aAg1250aCg | K417T | S |
| G23012A | SNV | moderate | S | Gaa1450Aaa | E484K | S |
| A23063T | SNV | moderate | S | Aat1501Tat | N501Y | S |
| A23403G | SNV | moderate | S | gAt1841gGt | D614G | S |
| C23525T | SNV | moderate | S | Cat1963Tat | H655Y | S |
| C24642T | SNV | moderate | S | aCt3080aTt | T1027I | S |
| G25088T | SNV | moderate | S | Gtt3526Ttt | V1176F | S |
| T26149C | SNV | moderate | ORF3a | Tcc757Ccc | S253P | ORF3a |
| G28167A | SNV | moderate | ORF8 | Gaa274Aaa | E92K | ORF8 |
| C28512G | SNV | moderate | N | cCa239cGa | P80R | N |
| A28877T | SNV | moderate | N | Agt604Tgt | S202C | N |
| G28878C | SNV | moderate | N | aGt605aCt | S202T | N |
| G28881A | SNV | moderate | N | aGg608aAg | R203K | N |
| G28883C | SNV | moderate | N | Gga610Cga | G204R | N |
Table 7.
Key mutations in the Delta VOC.
| Mutation. | Type | Level | Protein Coding Gene Annotation | Codon Change | Consequence for the AA Sequence | Corresponding Protein Annotation of UCSC SARS-CoV-2 |
|---|---|---|---|---|---|---|
| C14408T | SNV | moderate | ORF1ab | cCt14144cTt | P4715L | nsp12 |
| C21618G | SNV | moderate | S | aCa56aGa | T19R | S |
| T22917G | SNV | moderate | S | cTg1355cGg | L452R | S |
| C22995A | SNV | moderate | S | aCa1433aAa | T478K | S |
| A23403G | SNV | moderate | S | gAt1841gGt | D614G | S |
| C23604G | SNV | moderate | S | cCt2042cGt | P681R | S |
| C25469T | SNV | moderate | ORF3a | tCa77tTa | S26L | ORF3a |
| T26767C | SNV | moderate | M | aTc245aCc | I82T | M |
| T27638C | SNV | moderate | ORF7a | gTt245gCt | V82A | ORF7a |
| C27752T | SNV | moderate | ORF7a | aCa359aTa | T120I | ORF7a |
| G28881T | SNV | moderate | N | aGg608aTg | R203M | N |
| G29402T | SNV | moderate | N | Gat1129Tat | D377Y | N |
M, membrane.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lim, H.G.-M.; Hsiao, S.-H.; Fann, Y.C.; Lee, Y.-C.G. Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow. Genes 2022, 13, 686. https://doi.org/10.3390/genes13040686
AMA Style
Lim HG-M, Hsiao S-H, Fann YC, Lee Y-CG. Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow. Genes. 2022; 13(4):686. https://doi.org/10.3390/genes13040686
Chicago/Turabian StyleLim, Hendrick Gao-Min, Shih-Hsin Hsiao, Yang C. Fann, and Yuan-Chii Gladys Lee. 2022. "Robust Mutation Profiling of SARS-CoV-2 Variants from Multiple Raw Illumina Sequencing Data with Cloud Workflow" Genes 13, no. 4: 686. https://doi.org/10.3390/genes13040686
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.