
Computational Identification of Potential Anti-Inflammatory Natural Compounds Targeting the p38 Mitogen-Activated Protein Kinase (MAPK): Implications for COVID-19-Induced Cytokine Storm



Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Structure Retrieval and Processing
2.2. Screening Library of Compounds
2.3. Validation of Docking Protocol
2.4. Virtual Screening of Ligands
2.5. Pharmacological Profiling
2.6. Elucidation of the Protein-Ligand Interactions
2.7. Prediction of Biological Activities of Hit Compounds
2.8. Molecular Dynamics Simulation of Protein-Ligand Complexes
3. Results and Discussion
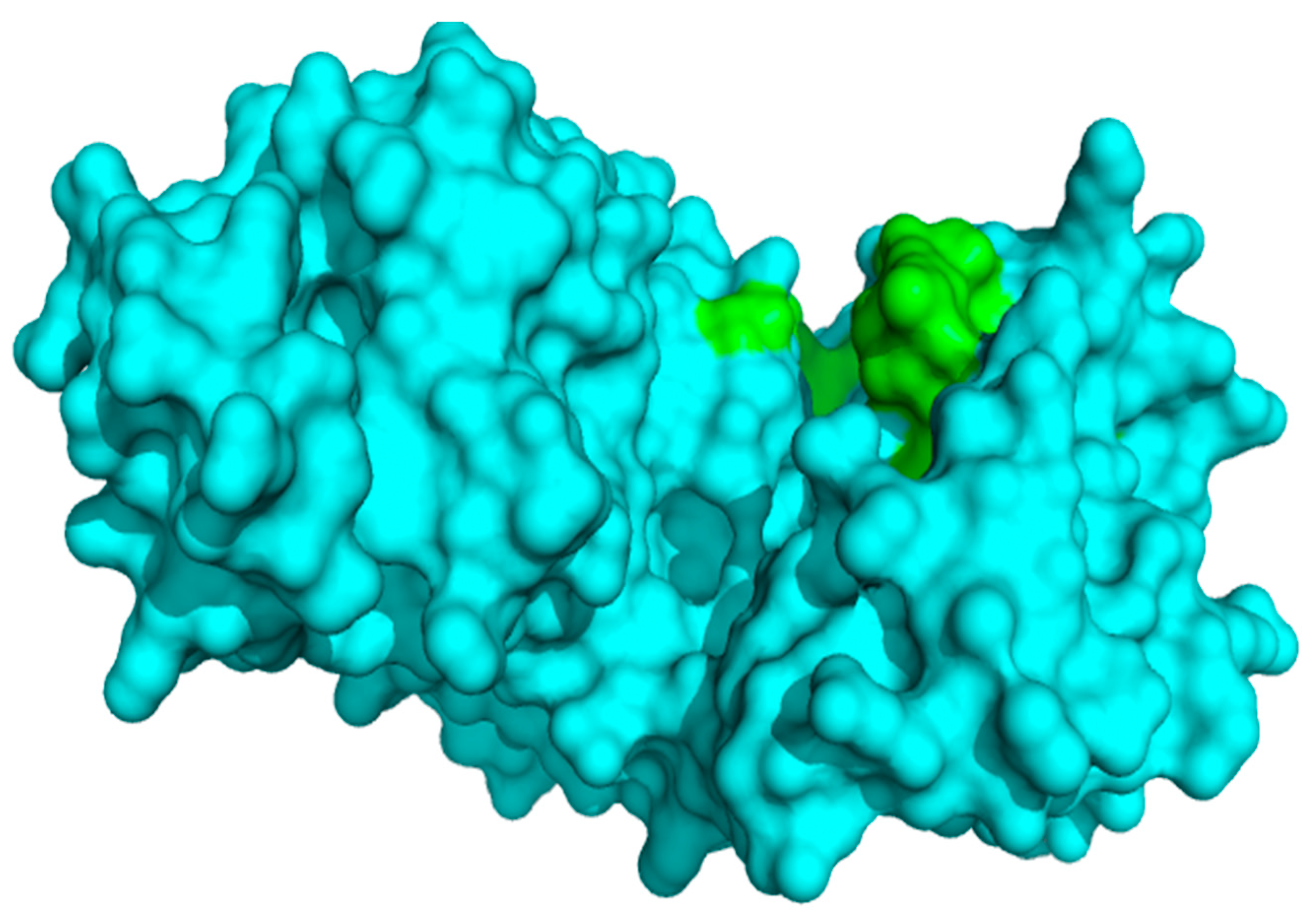
3.1. Protein Structure Retrieval and Analysis
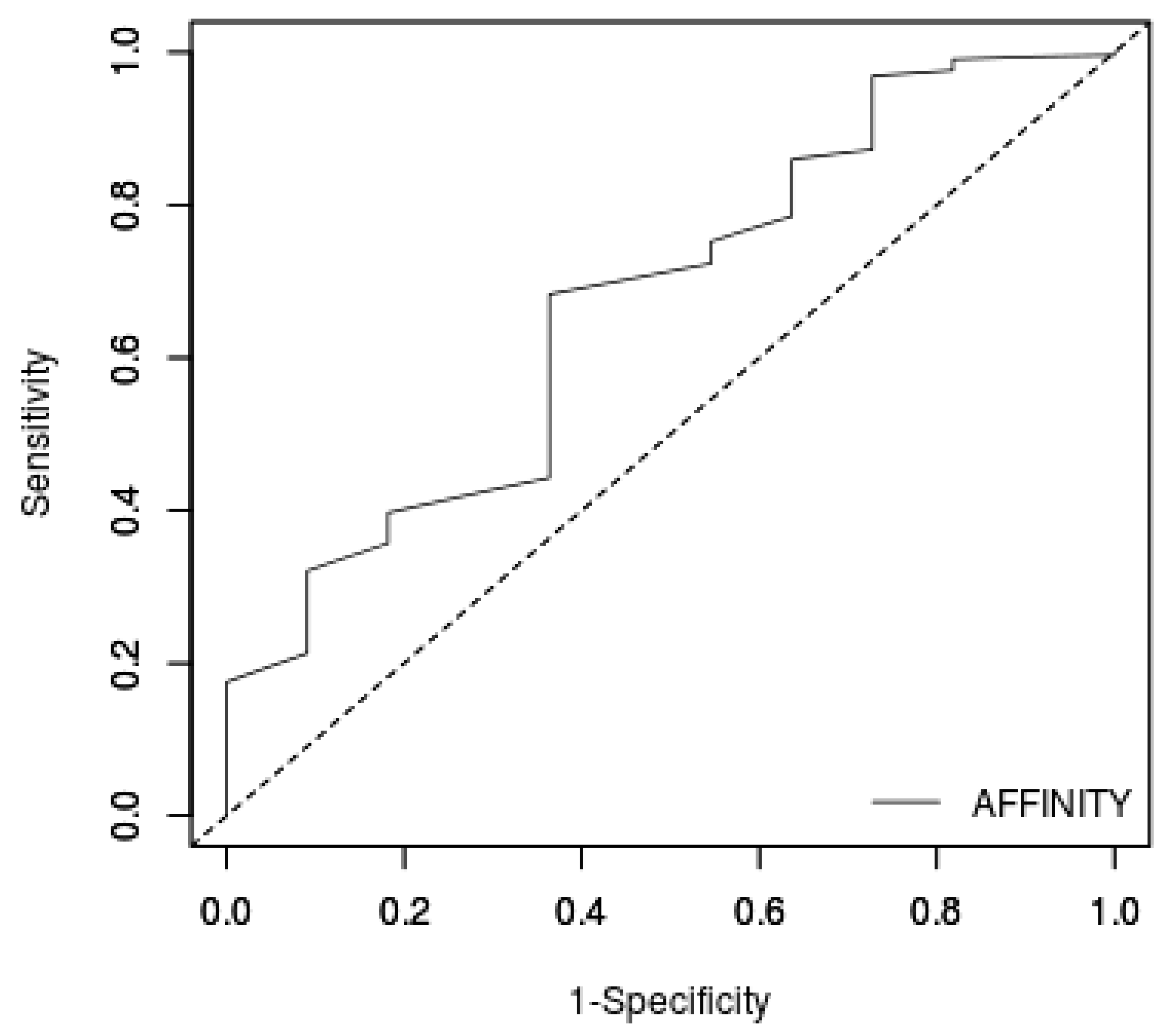
3.2. Docking Protocol Validation
3.3. Pre-Filtering of Library and Molecular Docking Studies
3.4. Pharmacological Profiling of Hit Compounds
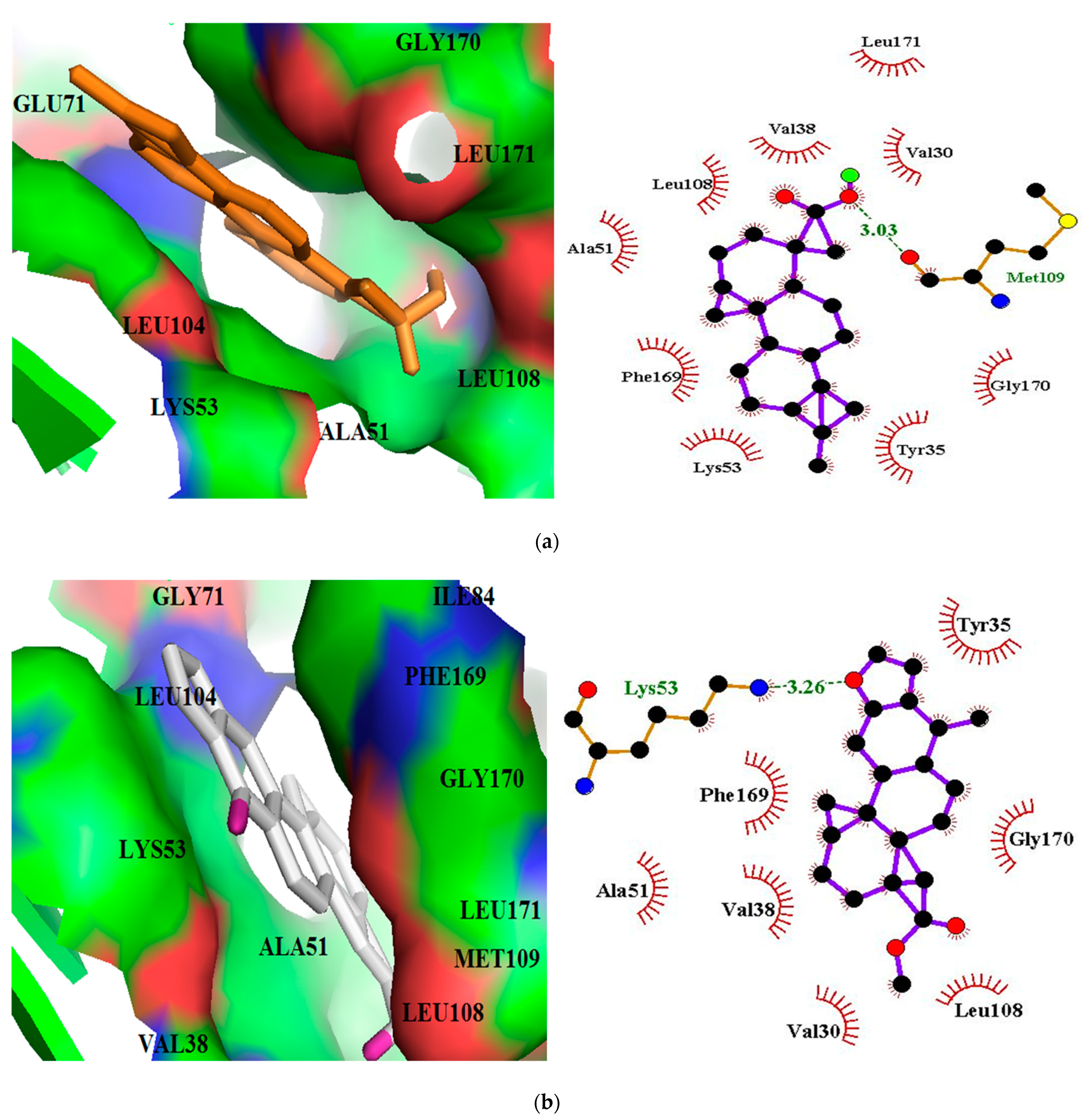
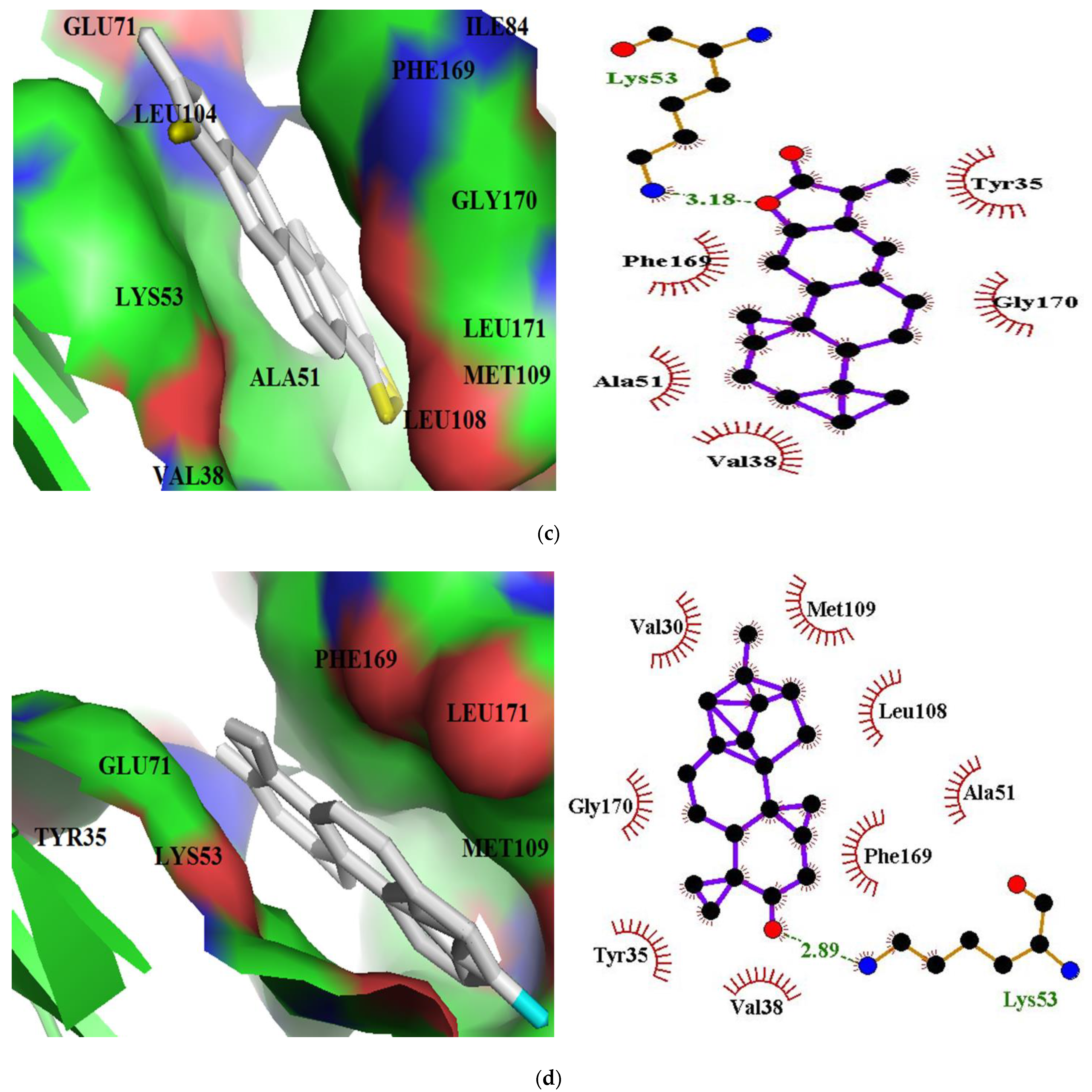
3.5. Visualization and 2-D Representation of Protein-Ligand Interactions
3.6. Biological Activity Prediction
3.7. The Rationale for the Selection of Compounds
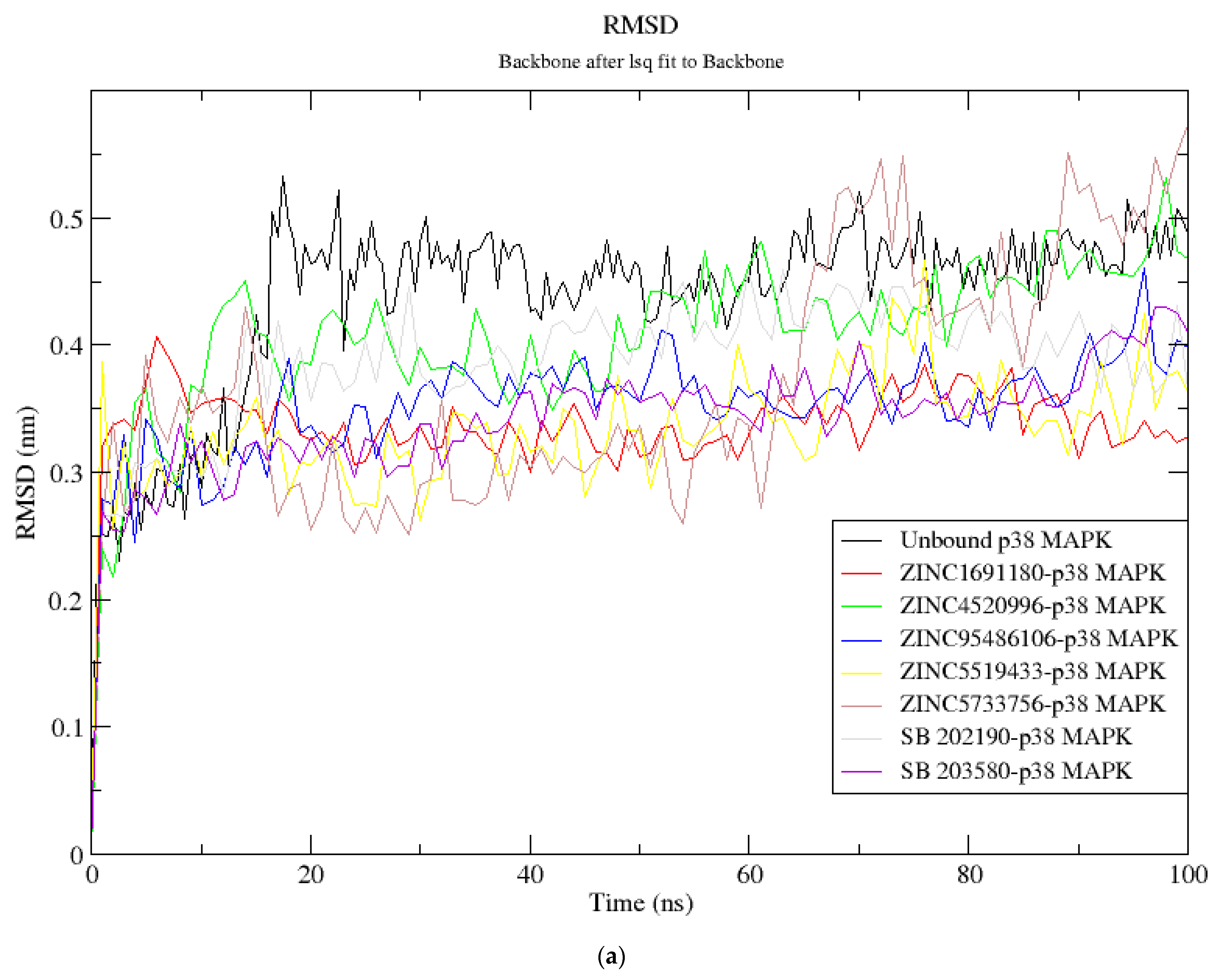
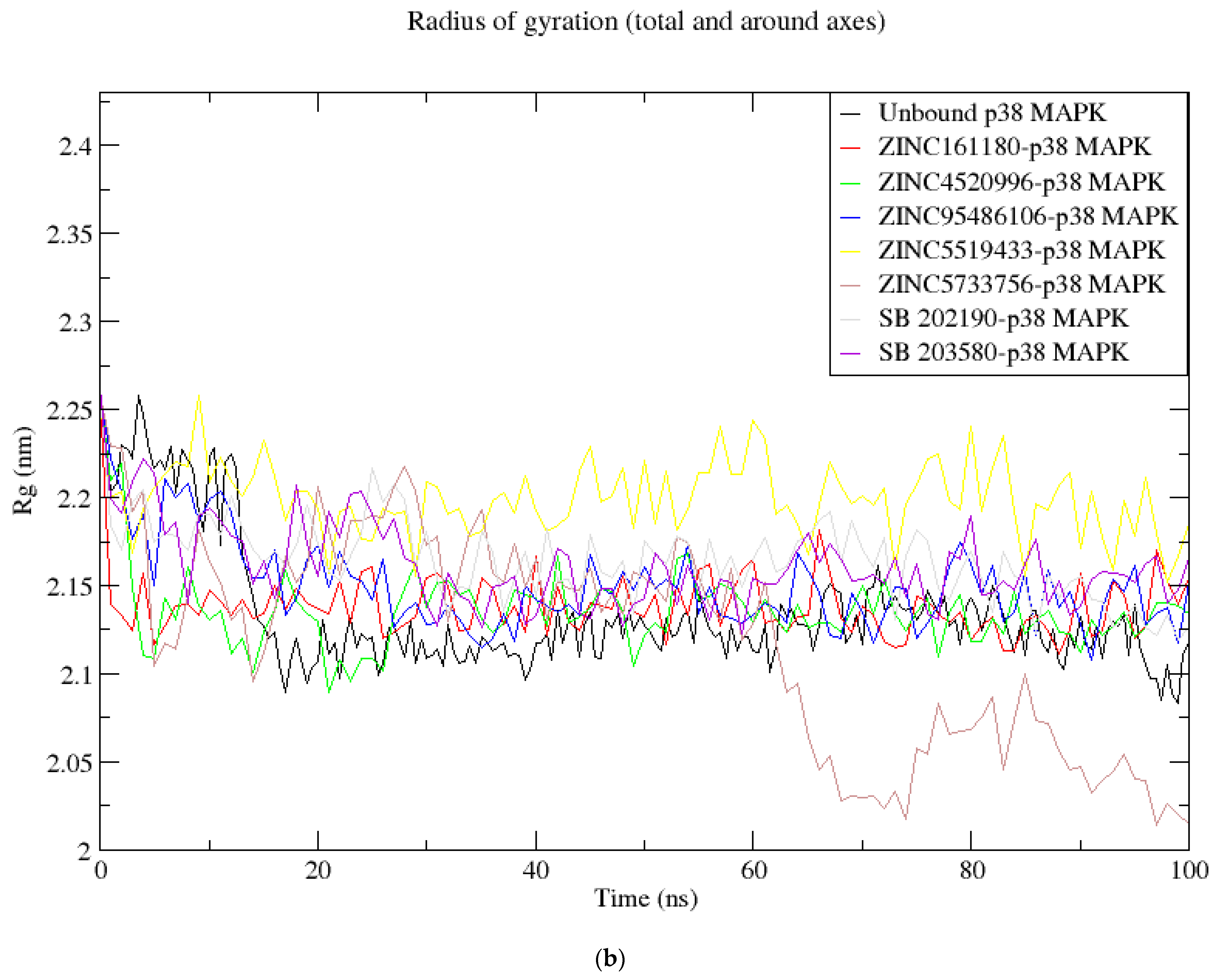
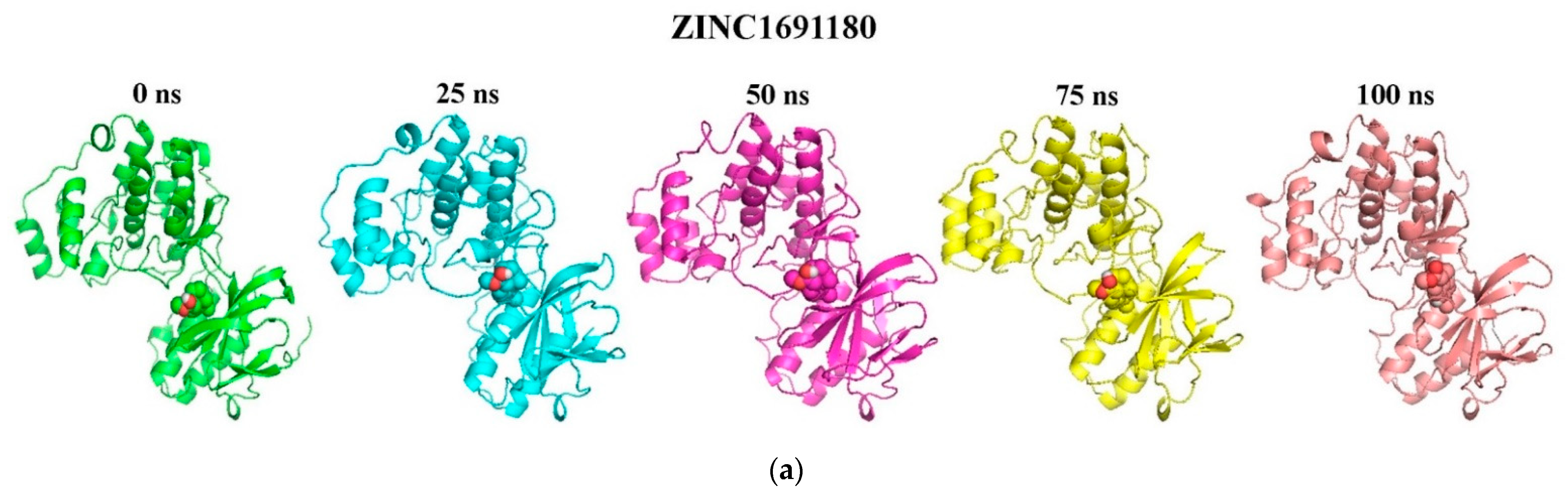
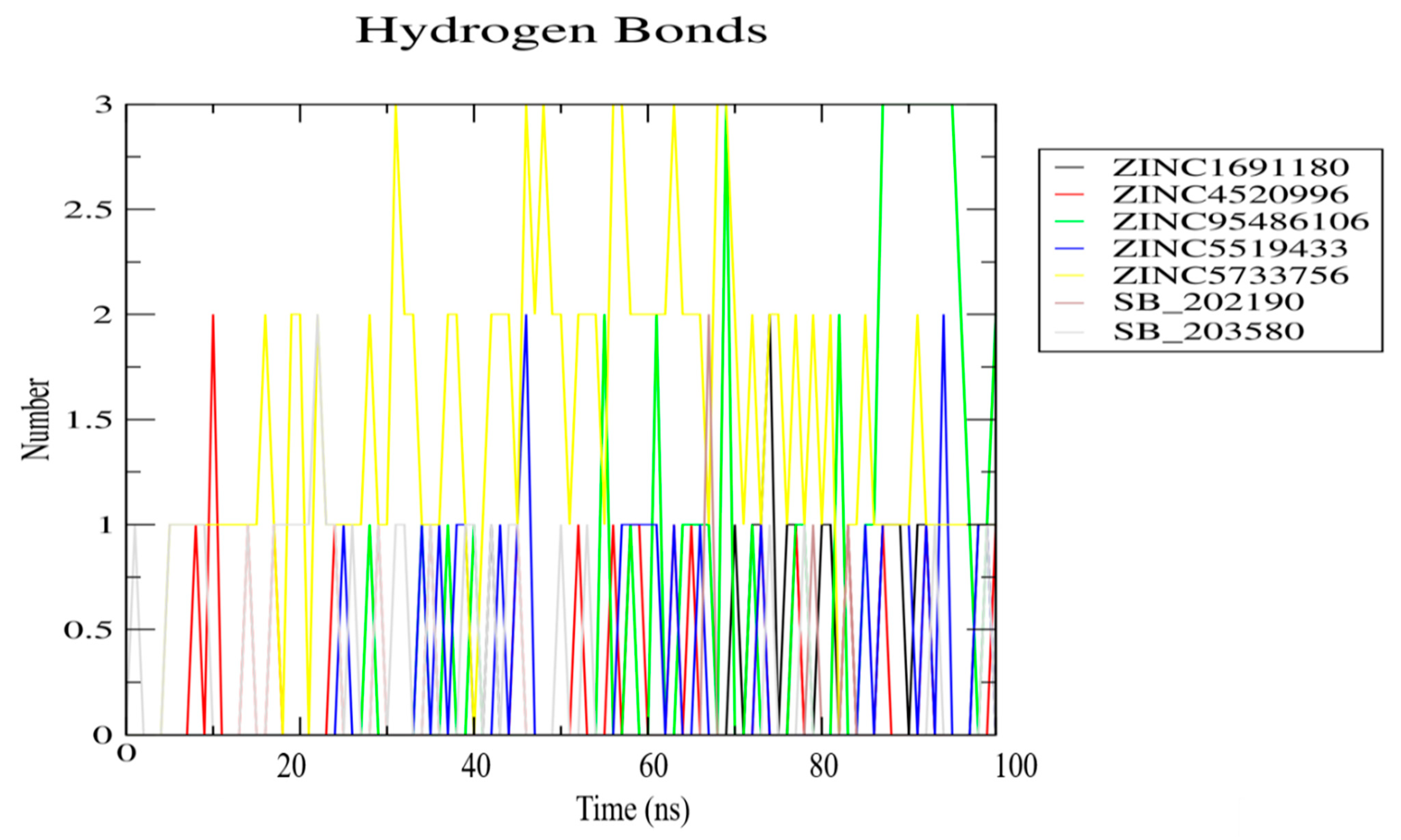
3.8. Molecular Dynamics Simulation of Selected Compounds
3.9. Evaluation of Selected Compounds via MM-PBSA Calculations
4. Summary and Potential Implication of the Study on COVID-19-Induced Cytokine Storm
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, D.; Heymann, D. Q&A: The novel coronavirus outbreak causing COVID-19. BMC Med. 2020, 18, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabi, A.F.; Al Zoubi, S.M.; Kasasbeh, A.G.; Salameh, M.D.; Al-Nasser, D.A. SARS-CoV-2 and Coronavirus Disease 2019: What We Know So Far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus genomics and bioinformatics analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, N.G.; Klepac, P.; Liu, Y.; Prem, K.; Jit, M.; Pearson, C.A.B.; Quilty, B.J.; Kucharski, A.J.; Gibbs, H.; Clifford, S.; et al. Age-dependent effects in the transmission and control of COVID-19 epidemics. Nat. Med. 2020, 26, 1205–1211. [Google Scholar] [CrossRef]
- Chaw, L.; Koh, W.C.; Jamaludin, S.A.; Naing, L.; Alikhan, M.F.; Wong, J. SARS-CoV-2 transmission in different settings: Analysis of cases and close contacts from the Tablighi cluster in Brunei Darussalam. MedRxiv 2020. [Google Scholar] [CrossRef]
- Ding, Z.; Qian, H.; Xu, B.; Huang, Y.; Miao, T.; Yen, H.-L.; Xiao, S.; Cui, L.; Wu, X.; Shao, W.; et al. Toilets dominate environmental detection of SARS-CoV-2 virus in a hospital. MedRxiv 2020. [Google Scholar] [CrossRef]
- Oreshkova, N.; Molenaar, R.-J.; Vreman, S.; Harders, F.; Munnink, B.B.O.; Honing, R.W.H.; Gerhards, N.; Tolsma, P.; Bouwstra, R.; Sikkema, R.; et al. SARS-CoV2 infection in farmed mink, Netherlands, April 2020. Biorxiv 2020. [Google Scholar] [CrossRef]
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.; Di Napoli, R. Features, Evaluation and Treatment Coronavirus (COVID-19); Statpearls: Treasure Island, LS, USA, 2020. [Google Scholar]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 6736, 1–9. [Google Scholar] [CrossRef]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, C.; Huang, F.; Yang, Y.; Wang, F.; Yuan, J.; Zhang, Z.; Qin, Y.; Li, X.; Zhao, D.; et al. Elevated plasma levels of selective cytokines in COVID-19 patients reflect viral load and lung injury. Natl. Sci. Rev. 2020, 7, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Matthay, M.; Calfee, C. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern. Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef]
- Grimes, J.M.; Grimes, K.V. p38 MAPK inhibition: A promising therapeutic approach for COVID-19. J. Mol. Cell. Cardiol. 2020, 144, 63–65. [Google Scholar] [CrossRef]
- Wehbe, Z.; Hammoud, S.; Soudani, N.; Zaraket, H.; El-Yazbi, A.; Eid, A.H. Molecular Insights Into SARS COV-2 Interaction with Cardiovascular Disease: Role of RAAS and MAPK Signaling. Front. Pharmacol. 2020, 11, 836. [Google Scholar] [CrossRef]
- Park, J.K.; Fischer, R.; Dechend, R.; Shagdarsuren, E.; Gapeljuk, A.; Wellner, M.; Meiners, S.; Gratze, P.; Al-Saadi, N.; Feldt, S.; et al. p38 mitogen-activated protein kinase inhibition ameliorates angiotensin II-induced target organ damage. Hypertension 2007, 49, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Cui, L.; Hou, F.; Liu, X.; Wang, Y.; Wen, Y.; Chi, C.; Li, C.; Liu, R.; Yin, C. Angiotensin-converting enzyme 2-angiotensin (1-7)-Mas axis prevents pancreatic acinar cell inflammatory response via inhibition of the p38 mitogen-activated protein kinase/nuclear factor-?B pathway. Int. J. Mol. Med. 2018, 41, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 2, 271–280. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Palese, P. 7a Protein of Severe Acute Respiratory Syndrome Coronavirus Inhibits Cellular Protein Synthesis and Activates p38 Mitogen-Activated Protein Kinase. J. Virol. 2006, 80, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Börgeling, Y.; Schmolke, M.; Viemann, D.; Nordhoff, C.; Roth, J.; Ludwig, S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J. Biol. Chem. 2014, 289, 13–27. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Guardeño, J.M.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Enjuanes, L. The PDZ-Binding Motif of Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Is a Determinant of Viral Pathogenesis. PLoS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef] [Green Version]
- Yokota, T.; Wang, Y. P38 MAP kinases in the heart. Gene 2016, 575, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Kim, N.J. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [Green Version]
- Grant, S.K. Therapeutic Protein Kinase Inhibitors. Cell. Mol. Life Sci. 2009, 66, 1163–1177. [Google Scholar] [CrossRef]
- Sharun, K.; Tiwari, R.; Dhama, J.; Dhama, K. Dexamethasone to combat cytokine storm in COVID-19: Clinical trials and preliminary evidence. Int. J. Surg. 2020, 82, 179–181. [Google Scholar] [CrossRef]
- Thorlund, K.; Dron, L.; Park, J.; Hsu, G.; Forrest, J.I.; Mills, E.J. A real-time dashboard of clinical trials for COVID-19. Lancet Digit. Health 2020, 2, e286–e287. [Google Scholar] [CrossRef]
- Ciliberto, G.; Cardone, L. Boosting the arsenal against COVID-19 through computational drug repurposing. Drug Discov. Today 2020, 25, 946–948. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Broni, E.; Asiedu, S.O.; Kwarko, G.B.; Dankwa, B.; Enninful, K.S.; Tiburu, E.K.; Wilson, M.D. Cheminformatics-Based Identification of Potential Novel Anti-SARS-CoV-2 Natural Compounds of African Origin. Molecules 2021, 26, 406. [Google Scholar] [CrossRef]
- Tian, D.; Liu, Y.; Liang, C.; Xin, L.; Xie, X.; Zhang, D.; Wan, M.; Li, H.; Fu, X.; Liu, H.; et al. An update review of emerging small-molecule therapeutic options for COVID-19. Biomed. Pharmacother. 2021, 137, 111313. [Google Scholar] [CrossRef]
- Gaudêncio, S.P.; Pereira, F. A Computer-Aided Drug Design Approach to Predict Marine Drug-Like Leads for SARS-CoV-2 Main Protease Inhibition. Mar. Drugs 2020, 18, 633. [Google Scholar] [CrossRef]
- Shehroz, M.; Zaheer, T.; Hussain, T. Computer-aided drug design against spike glycoprotein of SARS-CoV-2 to aid COVID-19 treatment. Heliyon 2020, 6, e05278. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Dhagat, S.; Eswari, J.S. Emerging strategies on in silico drug development against COVID-19: Challenges and opportunities. Eur. J. Pharm. Sci. 2020, 155, 105522. [Google Scholar] [CrossRef] [PubMed]
- Boozari, M.; Hosseinzadeh, H. Natural products for COVID-19 prevention and treatment regarding to previous coronavirus infections and novel studies. Phyther. Res. 2021, 35, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Nugraha, R.V.; Ridwansyah, H.; Ghozali, M.; Khairani, A.F.; Atik, N. Traditional Herbal Medicine Candidates as Complementary Treatments for COVID-19: A Review of Their Mechanisms, Pros and Cons. Evid. Based Complement. Altern. Med. 2020, 2020, 2560645. [Google Scholar] [CrossRef]
- Akindele, A.J.; Agunbiade, F.O.; Sofidiya, M.O.; Awodele, O.; Sowemimo, A.; Ade-Ademilua, O.; Akinleye, M.O.; Ishola, I.O.; Orabueze, I.; Salu, O.B.; et al. COVID-19 Pandemic: A Case for Phytomedicines. Nat. Prod. Commun. 2020, 15, 1934578X2094508. [Google Scholar] [CrossRef]
- Huang, J.; Tao, G.; Liu, J.; Cai, J.; Huang, Z.; Chen, J. Current Prevention of COVID-19: Natural Products and Herbal Medicine. Front. Pharmacol. 2020, 11, 1635. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.-F.; Li, X.-J.; Zhang, H.-Y. Natural products and drug discovery. Can thousands of years of ancient medical knowledge lead us to new and powerful drug combinations in the fight against cancer and dementia? EMBO Rep. 2009, 10, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.-C.C. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening In Silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [Green Version]
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. AfroDb: A Select Highly Potent and Diverse Natural Product Library from African Medicinal Plants. PLoS ONE 2013, 8, e78085. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Telukunta, K.K.; Döring, K.; Simoben, C.V.; Moumbock, A.F.A.; Malange, Y.I.; Njume, L.E.; Yong, J.N.; Sippl, W.; Günther, S. NANPDB: A Resource for Natural Products from Northern African Sources. J. Nat. Prod. 2017, 80, 2067–2076. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2017; Volume 1607, pp. 627–641. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, R.; Van Zeeland, M.; Raaijmakers, H.; Kazemier, B.; De Vlieg, J.; Oubrie, A. X-ray structure of p38 bound to TAK-715: Comparison with three classic inhibitors. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1041–1050. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. PyMOL: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Chan, H.C.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef]
- Gajula, M.; Kumar, A.; Ijaq, J. Protocol for Molecular Dynamics Simulations of Proteins. Bio-Protocol 2016, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Berweger, C.D.; van Gunsteren, W.F.; Müller-Plathe, F. Force field parametrization by weak coupling. Re-engineering SPC water. Chem. Phys. Lett. 1995, 232, 429–436. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Doytchinova, I.; Atanasova, M.; Valkova, I.; Stavrakov, G.; Philipova, I.; Zhivkova, Z.; Zheleva-Dimitrova, D.; Konstantinov, S.; Dimitrov, I. Novel hits for acetylcholinesterase inhibition derived by docking-based screening on ZINC database. J. Enzyme Inhib. Med. Chem. 2018, 33, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Kwofie, S.K.; Broni, E.; Teye, J.; Quansah, E.; Issah, I.; Wilson, M.D.; Miller, W.A.; Tiburu, E.K.; Bonney, J.H.K.K. Pharmacoinformatics-based identification of potential bioactive compounds against Ebola virus protein VP24. Comput. Biol. Med. 2019, 113, 103414. [Google Scholar] [CrossRef] [PubMed]
- Goksuluk, D.; Korkmaz, S.; Zararsiz, G.; Karaagaoglu, A.E. easyROC: An interactive web-tool for ROC curve analysis using R language environment. R J. 2016, 8, 213–230. [Google Scholar] [CrossRef] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, EfficientOptimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V.; Brooks, C.L.; Huang, R. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Parasuraman, S. Prediction of activity spectra for substances. J. Pharmacol. Pharmacother. 2011, 2, 52–53. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- van Gunsteren, W.F. Biomolecular Simulation: The GROMOS96 Manual and User Guide; Biomos: Zürich, Switzerland, 1996; ISBN 9783728124227. [Google Scholar]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa—A GROMACS tool for MM-PBSA and its optimization for high-throughput binding energy calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Cuenda, A.; Rousseau, S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta—Mol. Cell Res. 2007, 1773, 1358–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Li, Z.; Tao, Y.; Wang, Q.; Lai, Y.; Wu, W.; Peng, S.; Guo, Z.; Huang, H. Discovering novel P38α inhibitors for the treatment of prostate cancer through virtual screening methods. Future Med. Chem. 2019, 11, 3125–3137. [Google Scholar] [CrossRef]
- Wang, Z.; Harkins, P.C.; Ulevitch, R.J.; Han, J.; Cobb, M.H.; Goldsmith, E.J. The structure of mitogen-activated protein kinase p38 at 2.1-Å resolution. Proc. Natl. Acad. Sci. USA 1997, 94, 2327–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Empereur-mot, C.; Guillemain, H.; Latouche, A.; Zagury, J.-F.; Viallon, V.; Montes, M. Predictiveness curves in virtual screening. J. Cheminform. 2015, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Jiang, J.H.; Li, R.Y.; Deng, P. Docking-based virtual screening of TβR1 inhibitors: Evaluation of pose prediction and scoring functions. BMC Chem. 2020, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, J.N. Receiver Operating Characteristic Curve in Diagnostic Test Assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsin, K.Y.; Matsuoka, Y.; Asai, Y.; Kamiyoshi, K.; Watanabe, T.; Kawaoka, Y.; Kitano, H. systemsDock: A web server for network pharmacology-based prediction and analysis. Nucleic Acids Res. 2016, 44, W507–W513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, S.T.; Tam, N.M.; Pham, M.Q.; Nguyen, T.H. Benchmark of Popular Free Energy Approaches Revealing the Inhibitors Binding to SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2021. [Google Scholar] [CrossRef]
- Mishra, A.; Dey, S. Molecular docking studies of a cyclic octapeptide-cyclosaplin from sandalwood. Biomolecules 2019, 9, 740. [Google Scholar] [CrossRef] [Green Version]
- Santana, I.V.; Côrtes Filho, A.B.; Pinheiro, A.A.F.; dos Santos, É.G.; Lima, D.M.; Barreto, M.M.; Lima, E.R.; Valasques Junior, G.L. In silico screening of compounds from the Bahia semiarid region for identification of potential inhibitors of the p38 MAPK protein. Res. Soc. Dev. 2020, 9, e4439108723. [Google Scholar] [CrossRef]
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties. In Dosage Form Design Parameters; Elsevier: Amsterdam, The Netherlands, 2018; Volume 2, pp. 731–755. ISBN 9780128144220. [Google Scholar]
- Elmeliegy, M.; Vourvahis, M.; Guo, C.; Wang, D.D. Effect of P-glycoprotein (P-gp) Inducers on Exposure of P-gp Substrates: Review of Clinical Drug–Drug Interaction Studies. Clin. Pharmacokinet. 2020, 59, 699–714. [Google Scholar] [CrossRef] [Green Version]
- Kwon, H.; Lionberger, R.A.; Yu, L.X. Impact of P-glycoprotein-mediated intestinal efflux kinetics on oral bioavailability of P-glycoprotein substrates. Mol. Pharm. 2004, 1, 455–465. [Google Scholar] [CrossRef]
- Klein, K.; Zanger, U.M. Pharmacogenomics of Cytochrome P450 3A4: Recent Progress toward the “Missing Heritability” Problem. Front. Genet. 2013, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Wang, Z.; Canagarajah, B.J.; Boehm, J.C.; Kassisà, S.; Cobb, M.H.; Young, P.R.; Abdel-Meguid, S.; Adams, J.L.; Goldsmith, E.J. Structural basis of inhibitor selectivity in MAP kinases. Structure 1998, 6, 1117–1128. [Google Scholar] [CrossRef] [Green Version]
- Tong, L.; Pav, S.; White, D.M.; Rogers, S.; Crane, K.M.; Cywin, C.L.; Brown, M.L.; Pargellis, C.A. A highly specific inhibitor of human p38 MAP kinase binds in the ATP pocket. Nat. Struct. Biol. 1997, 4, 311–316. [Google Scholar] [CrossRef]
- Bukhtiyarova, M.; Karpusas, M.; Northrop, K.; Namboodiri, H.V.M.; Springman, E.B. Mutagenesis of p38α MAP kinase establishes key roles of Phe169 in function and structural dynamics and reveals a novel DFG-OUT state. Biochemistry 2007, 46, 5687–5696. [Google Scholar] [CrossRef]
- Jamkhande, P.G.; Barde, S.R. Evaluation of anthelmintic activity and in silico PASS assisted prediction of Cordia dichotoma (Forst.) root extract. Anc. Sci. Life 2014, 34, 39–43. [Google Scholar] [CrossRef]
- Patil, K.R.; Mohapatra, P.; Patel, H.M.; Goyal, S.N.; Ojha, S.; Kundu, C.N.; Patil, C.R. Pentacyclic triterpenoids inhibit ikkβmediated activation of nf-κb pathway: In silico and in vitro evidences. PLoS ONE 2015, 10, e0125709. [Google Scholar] [CrossRef] [Green Version]
- Emon, N.U.; Jahan, I.; Sayeed, M.A. Investigation of antinociceptive, anti-inflammatory and thrombolytic activity of Caesalpinia digyna (Rottl.) leaves by experimental and computational approaches. Adv. Tradit. Med. 2020, 20, 451–459. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [Green Version]
- Levi, M.; Thachil, J. Coronavirus Disease 2019 Coagulopathy: Disseminated Intravascular Coagulation and Thrombotic Microangiopathy—Either, Neither, or Both. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2020. [Google Scholar] [CrossRef]
- Yoshioka, H.; Mizuno, Y.; Yamaguchi, T.; Ichimaru, Y.; Takeya, K.; Hitotsuyanagi, Y.; Nonogaki, T.; Aoyagi, Y. Methyl dehydroabietate counters high fat diet-induced insulin resistance and hepatic steatosis by modulating peroxisome proliferator-activated receptor signaling in mice. Biomed. Pharmacother. 2018, 99, 214–219. [Google Scholar] [CrossRef]
- Hughes, J.P.; Rees, S.S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Ma, J.; Hwang, Y.K.; Cho, W.H.; Han, S.H.; Hwang, J.K.; Han, J.S. Macelignan attenuates activations of mitogen-activated protein kinases and nuclear factor kappa B induced by lipopolysaccharide in microglial cells. Biol. Pharm. Bull. 2009, 32, 1085–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaoud, T.S.; Park, H.; Mitra, S.; Yan, C.; Tseng, C.C.; Shi, Y.; Jose, J.; Taliaferro, J.M.; Lee, K.; Ren, P.; et al. Manipulating JNK signaling with (-)-zuonin A. ACS Chem. Biol. 2012, 7, 1873–1883. [Google Scholar] [CrossRef] [PubMed]
- Maruca, A.; Ambrosio, F.A.; Lupia, A.; Romeo, I.; Rocca, R.; Moraca, F.; Talarico, C.; Bagetta, D.; Catalano, R.; Costa, G.; et al. Computer-based techniques for lead identification and optimization i: Basics. Phys. Sci. Rev. 2019, 4. [Google Scholar] [CrossRef]
- Zhou, F.H.; Foster, B.K.; Zhou, X.-F.; Cowin, A.J.; Xian, C.J. TNF-alpha mediates p38 MAP kinase activation and negatively regulates bone formation at the injured growth plate in rats. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2006, 21, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Song, C.-H.; Kim, N.; Kim, D.-H.; Lee, H.-N.; Surh, Y.-J. 17-β estradiol exerts anti-inflammatory effects through activation of Nrf2 in mouse embryonic fibroblasts. PLoS ONE 2019, 14, e0221650. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Pillay, T.S. Identification of promising anti-DNA gyrase antibacterial compounds using de novo design, molecular docking and molecular dynamics studies. J. Biomol. Struct. Dyn. 2019, 6, 1798–1809. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Reynolds, R.C. Group Additivity in Ligand Binding Affinity: An Alternative Approach to Ligand Efficiency. J. Chem. Inf. Model. 2017, 57, 3086–3093. [Google Scholar] [CrossRef]
- Kumar, S.; McDonnell, P.C.; Gum, R.J.; Hand, A.T.; Lee, J.C.; Young, P.R. Novel homologues of CSBP/p38 MAP kinase: Activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem. Biophys. Res. Commun. 1997, 235, 533–538. [Google Scholar] [CrossRef]
- Fehr, S.; Unger, A.; Schaeffeler, E.; Herrmann, S.; Laufer, S.; Schwab, M.; Albrecht, W. Impact of p38 MAP kinase inhibitors on LPS-induced release of TNF-α in whole blood and primary cells from different species. Cell. Physiol. Biochem. 2015, 36, 2237–2249. [Google Scholar] [CrossRef]
- Koeberle, S.C.; Romir, J.; Fischer, S.; Koeberle, A.; Schattel, V.; Albrecht, W.; Grütter, C.; Werz, O.; Rauh, D.; Stehle, T.; et al. Skepinone-L is a selective p38 mitogen-activated protein kinase inhibitor. Nat. Chem. Biol. 2012, 8, 141–143. [Google Scholar] [CrossRef]
- Goldstein, D.M.; Kuglstatter, A.; Lou, Y.; Soth, M.J. Selective p38α inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J. Med. Chem. 2010, 53, 2345–2353. [Google Scholar] [CrossRef]
- Hope, H.R.; Anderson, G.D.; Burnette, B.L.; Compton, R.P.; Devraj, R.V.; Hirsch, J.L.; Keith, R.H.; Li, X.; Mbalaviele, G.; Messing, D.M.; et al. Anti-inflammatory properties of a novel N-phenyl pyridinone inhibitor of p38 mitogen-activated protein kinase: Preclinical-to-clinical translation. J. Pharmacol. Exp. Ther. 2009, 331, 882–895. [Google Scholar] [CrossRef] [Green Version]
- Mazumder, M.; Ponnan, P.; Das, U.; Gourinath, S.; Khan, H.A.; Yang, J.; Sakharkar, M.K. Investigations on Binding Pattern of Kinase Inhibitors with PPAR γ: Molecular Docking, Molecular Dynamic Simulations, and Free Energy Calculation Studies. PPAR Res. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Musyoka, T.M.; Kanzi, A.M.; Lobb, K.A.; Tastan Bishop, Ö. Structure Based Docking and Molecular Dynamic Studies of Plasmodial Cysteine Proteases against a South African Natural Compound and its Analogs. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Shen, Y.; Liu, H.; Yao, X. Molecular dynamics simulation and free energy calculation studies of the binding mechanism of allosteric inhibitors with p38α MAP kinase. J. Chem. Inf. Model. 2011, 51, 3235–3246. [Google Scholar] [CrossRef]
- Khan, M.F.; Verma, G.; Alam, P.; Akhter, M.; Bakht, M.A.; Hasan, S.M.; Shaquiquzzaman, M.; Alam, M.M. Dibenzepinones, dibenzoxepines and benzosuberones based p38α MAP kinase inhibitors: Their pharmacophore modelling, 3D-QSAR and docking studies. Comput. Biol. Med. 2019, 110, 175–185. [Google Scholar] [CrossRef]
- Suplatov, D.; Kopylov, K.; Sharapova, Y.; Švedas, V. Human p38α mitogen-activated protein kinase in the Asp168-Phe169-Gly170-in (DFG-in) state can bind allosteric inhibitor Doramapimod. J. Biomol. Struct. Dyn. 2019, 37, 2049–2060. [Google Scholar] [CrossRef]
- Tabassum, H.; Ahmad, I.Z. Molecular Docking and Dynamics Simulation Analysis of Thymoquinone and Thymol Compounds from Nigella sativa L. that Inhibits P38 Protein: Probable Remedies for Hepatocellular Carcinoma. Med. Chem. 2019, 16, 350–357. [Google Scholar] [CrossRef]
- Lobanov, M.I.; Bogatyreva, N.S.; Galzitskaia, O. V Radius of gyration is indicator of compactness of protein structure. Mol. Biol. 2008, 42, 701–706. [Google Scholar] [CrossRef]
- Boateng, R.A.; Tastan Bishop, Ö.; Musyoka, T.M. Characterisation of plasmodial transketolases and identification of potential inhibitors: An in silico study. Malar. J. 2020, 19, 1–19. [Google Scholar] [CrossRef]
- Wen, C.C.; Kuo, Y.H.; Jan, J.T.; Liang, P.H.; Wang, S.Y.; Liu, H.G.; Lee, C.K.; Chang, S.T.; Kuo, C.J.; Lee, S.S.; et al. Specific plant terpenoids and lignoids possess potent antiviral activities against severe acute respiratory syndrome coronavirus. J. Med. Chem. 2007, 50, 4087–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Lynn, A.M.; Gupta, V. Standardization of virtual-screening and post-processing protocols relevant to in-silico drug discovery. 3 Biotech 2018, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Macchiagodena, M.; Pagliai, M.; Karrenbrock, M.; Guarnieri, G.; Iannone, F.; Procacci, P. Virtual Double-System Single-Box: A Nonequilibrium Alchemical Technique for Absolute Binding Free Energy Calculations: Application to Ligands of the SARS-CoV-2 Main Protease. J. Chem. Theory Comput. 2020, 16, 7160–7172. [Google Scholar] [CrossRef]
- Deng, N.J.; Zhang, P.; Cieplak, P.; Lai, L. Elucidating the energetics of entropically driven protein-ligand association: Calculations of absolute binding free energy and entropy. J. Phys. Chem. B 2011, 115, 11902–11910. [Google Scholar] [CrossRef] [Green Version]
- Campanera, J.M.; Pouplana, R. MMPBSA decomposition of the binding energy throughout a molecular dynamics simulation of amyloid-beta (Aß10-35) aggregation. Molecules 2010, 15, 2730–2748. [Google Scholar] [CrossRef] [Green Version]
- Martins, L.C.; Torres, P.H.M.; de Oliveira, R.B.; Pascutti, P.G.; Cino, E.A.; Ferreira, R.S. Investigation of the binding mode of a novel cruzain inhibitor by docking, molecular dynamics, ab initio and MM/PBSA calculations. J. Comput. Aided. Mol. Des. 2018, 32, 591–605. [Google Scholar] [CrossRef]
- Medina-Enríquez, M.M.; Lopez-León, S.; Carlos-Escalante, J.A.; Aponte-Torres, Z.; Cuapio, A.; Wegman-Ostrosky, T. ACE2: The molecular doorway to SARS-CoV-2. Cell Biosci. 2020, 10, 1–17. [Google Scholar] [CrossRef]
- Mendoza-Pinto, C.; García-Carrasco, M.; Munguía Realpozo, P.; Méndez-Martínez, S. Therapeutic options for the management of severe COVID-19: A rheumatology perspective. Reumatol. Clín. 2020. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZINC ID/Drug Name | Binding Energy (kcal/mol). | Number of Hydrogen Bonds | Hydrogen Bond Residues | Hydrogen Bond Length (Å) | Hydrophobic Contacts |
|---|---|---|---|---|---|
| ZINC95486106 | −12.1 | 1 | Met109 | 3.03 | Val30, Ala51, Val38, Leu171, Leu108, Gly170, Lys53, Tyr35, Phe169 |
| ZINC95913720 | −11.8 | 1 | Lys53 | 3.26 | Val30, Ala51, Val38, Leu108, Gly170, Tyr35, Phe169 |
| ZINC33832090 | −11.8 | 1 | Lys53 | 3.18 | Ala51, Val38, Gly170, Tyr35, Phe169 |
| ZINC95919076 | −11.7 | 1 | Lys53 | 2.89 | Val30, Ala51, Val38, Leu108, Gly170, Tyr35, Phe169, Val138, Met109 |
| ZINC1691180 | −11.6 | 1 | Tyr35 | 3.17 | Val30, Ala51, Val38, Phe169, Glu71, Leu75, Leu104, Thr106, Gly31 |
| ZINC5519433 | −11.6 | Ala51, Val38, Gly170, Lys53, Tyr35, Phe169, Leu75, Leu86, Ile84, Leu104, Val105, Thr106 | |||
| ZINC4520996 | −11.6 | 1 | Tyr35 | 3.16 | Ala51, Val38, Val30, Lys53, Gly31, Glu71, Thr106 Phe169, Leu104, Thr106 |
| ZINC1531907 | −11.6 | Ala51, Val38, Gly170, Lys53, Tyr35, Phe169, Leu75, Leu86, Ile84, Leu104, Val105, Thr106 | |||
| ZINC4098804 | −11.6 | Ala51, Val38, Gly170, Lys53, Tyr35, Phe169, Leu75, Leu86, Ile84, Leu104, Val105, Thr106 | |||
| ZINC95919075 | −11.5 | Ala51, Val30, Gly170, Lys53, Tyr35, Phe169, Val38, Met109, Leu108 | |||
| ZINC13302897 | −11.4 | Ala51, Gly170, Lys53, Tyr35, Phe169, Val38, | |||
| ZINC4215683 | −11.2 | 3 | Glu71, Lys53 [2] | 2.81, 3.12, 2.90 | Gly170, Tyr35, Phe169, Val38, Leu86, Leu104, Leu171, Thr106, Leu75, Val105 |
| ZINC13302884 | −11.2 | Val30, Val38, Ala51, Thr106, Leu104, Glu71, Leu75, Lys53, Phe169, Tyr35 | |||
| ZINC13302890 | −11.2 | Leu171, Val38, Gly170, Tyr35, Lys53, Phe169, Ala51 | |||
| ZINC4023706 | −11.1 | 3 | Tyr35, Gly170 [2] | 3.22, 3.18, 3.17 | Phe169, Leu104, Leu75, Glu71, Thr106, Val38, Ala51, Lys53, Val20, Gly31 |
| ZINC5733756 | −11.1 | Leu75, Ile84, Leu104, Lys53, Phe169, Leu171, Tyr35, Val38, Leu171, Thr106 | |||
| ZINC70454959 | −11.1 | 1 | Lys53 | 2.95 | Tyr35, Gly170, Leu171, la51, Val138, Phe169 |
| ZINC85993836 | −11.1 | Tyr35, Val38, Ala51, Lys53, Leu75, Ile84, Val105, Leu104, Thr106, Phe169, Gly170 | |||
| SB 202190 | −11.0 | Tyr35, Val38, Ala51, Lys53, Leu104, Thr106, Leu108, Met109, Phe169, Gly170, Leu171 | |||
| SB 203580 | −10.9 | Tyr35, Val38, Ala51, Lys53, Leu104, Thr106, Leu108, Met109, Val30, Phe169, Gly170, Leu171 |
| Ligand ID | Common/IUPAC Name | 2D Structure |
|---|---|---|
| ZINC5519433 | Zuihonin A |  |
| ZINC5733756 | 8,11,13-Abietatriene-3beta-ol |  |
| ZINC95486106 | (1S,2aS,2bR,4aS,5R,8aS,8bR,10aR)-1,5,8a-trimethyl-hexadecahydrocyclobuta[a]phenanthrene-5-carboxylic acid |  |
| ZINC1691180 | Methyl dehydroabietate |  |
| ZINC4520996 | Methyl (1S,4aR,10aS)-1,4a-dimethyl-7-propan-2-yl-2,3,4,9,10,10a-hexahydrophenanthrene-1-carboxylate |  |
| Name | Electrostatic Energy (kJ/mol) | Van Der Waal Energy (kJ/mol) | Polar solvation Energy (kJ/mol) | Non-Polar Solvation Energy (kJ/mol) | Binding Energy (kJ/mol) |
|---|---|---|---|---|---|
| ZINC5733756 | −15.833 ± 16.423 | −118.111 ± 77.765 | 47.787 ± 28.117 | −9.689 ± 8.856 | −95.846 ± 74.930 |
| ZINC5519433 | −2.575 ± 7.169 | −222.685 ± 10.572 | 58.332 ± 15.055 | −18.194 ± 0.535 | −185.122 ± 21.347 |
| ZINC95486106 | 75.738 ± 77.290 | −86.044 ± 50.930 | 49.672 ± 75.935 | −8.747 ± 4.846 | 30.620 ± 42.755 |
| ZINC1691180 | −12.086 ± 5.07 | −180.593 ± 17.415 | 63.084 ± 16.371 | −16.413 ± 0.870 | −146.008 ± 17.297 |
| ZINC4520996 | 2.277 ± 5.466 | −203.698 ± 17.665 | 66.835 ± 11.789 | −16.976 ± 0.738 | −151.561 ± 22.622 |
| SB 202190 | −2.041 ± 3.990 | −209.281 ± 15.503 | 63.370 ± 16.798 | 18.417 ± 0.895 | −166.369 ± 19.355 |
| SB 203580 | −5.094 ± 3.533 | −280.578 ± 10.532 | 70.328 ± 22.075 | −20.831 ± 1.546 | −236.175 ± 26.555 |
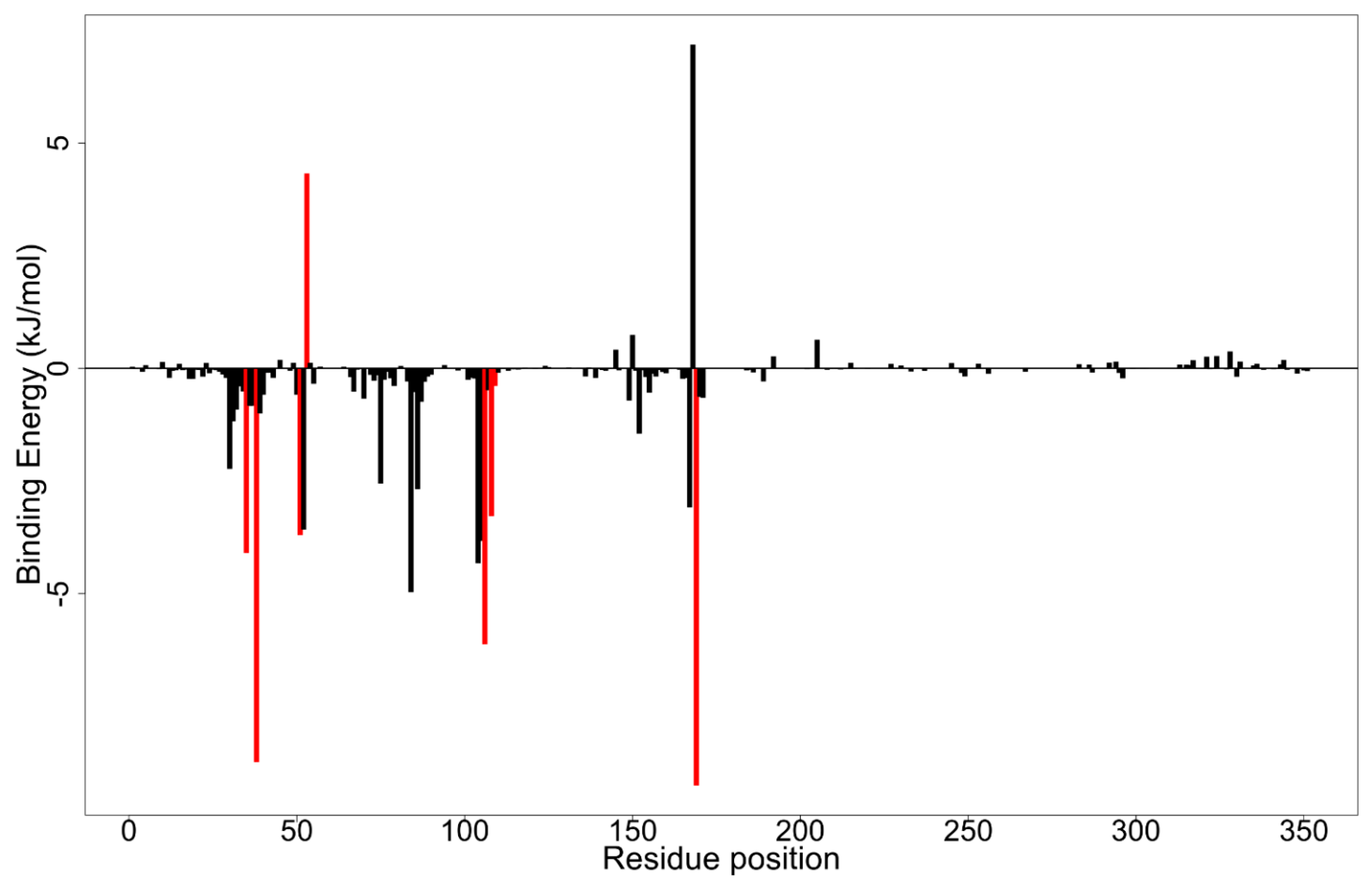
| Residue | ZINC5733756 | ZINC5519433 | ZINC95486106 | ZINC1691180 | ZINC4520996 | SB 202190 | SB 203580 |
|---|---|---|---|---|---|---|---|
| Tyr35 | −0.0810 | −4.0969 | −0.0881 | −1.9196 | −0.5451 | −1.6995 | −3.0867 |
| Val38 | −3.8266 | −8.7247 | −0.7025 | −7.6582 | −4.7471 | −11.3682 | −4.3341 |
| Ala51 | −1.7431 | −3.5809 | −0.6868 | −2.7066 | −4.0012 | −5.1339 | −2.7369 |
| Lys53 | 4.9439 | 4.3252 | −12.3692 | 8.4199 | 13.3187 | 12.708 | 14.7836 |
| Thr106 | −0.6584 | −6.1224 | 0.0030 | 0.5693 | −0.1483 | −2.5140 | −6.7417 |
| Leu108 | −3.1269 | −3.2785 | −0.7509 | −4.0640 | −7.2590 | −3.8154 | −0.0608 |
| Met109 | −4.2382 | −0.3931 | −2.1912 | −0.2787 | −1.8900 | −0.4842 | −0.7980 |
| Phe169 | −5.8140 | −9.2470 | −1.3285 | −14.5954 | −6.8001 | −14.9162 | −10.2842 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asiedu, S.O.; Kwofie, S.K.; Broni, E.; Wilson, M.D. Computational Identification of Potential Anti-Inflammatory Natural Compounds Targeting the p38 Mitogen-Activated Protein Kinase (MAPK): Implications for COVID-19-Induced Cytokine Storm. Biomolecules 2021, 11, 653. https://doi.org/10.3390/biom11050653
Asiedu SO, Kwofie SK, Broni E, Wilson MD. Computational Identification of Potential Anti-Inflammatory Natural Compounds Targeting the p38 Mitogen-Activated Protein Kinase (MAPK): Implications for COVID-19-Induced Cytokine Storm. Biomolecules. 2021; 11(5):653. https://doi.org/10.3390/biom11050653
Chicago/Turabian StyleAsiedu, Seth O., Samuel K. Kwofie, Emmanuel Broni, and Michael D. Wilson. 2021. "Computational Identification of Potential Anti-Inflammatory Natural Compounds Targeting the p38 Mitogen-Activated Protein Kinase (MAPK): Implications for COVID-19-Induced Cytokine Storm" Biomolecules 11, no. 5: 653. https://doi.org/10.3390/biom11050653