
SARS-CoV-2 Envelope (E) Protein Binds and Activates TLR2 Pathway: A Novel Molecular Target for COVID-19 Interventions

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cells
2.3. Virus Inactivation and Purification
2.4. Isolation of Human Monocytes
2.5. Generation of Monocyte-Derived Macrophages
2.6. Chemical Products, Proteins and Antibodies
2.7. Interaction of E Protein with TLR2 in a Solid-Phase Assay
2.8. Inhibition Assay of E–TLR2 Interaction
2.9. Flow Cytometry Analysis
2.10. Microscopy Analysis
2.11. Cell-Based Biological Assays
2.12. Phosphorylation Analysis of NF-kB P65 Subunit and Western Blot Analysis
2.13. NF-kB Assay Using HEK-Blue and HEK-TLR2-Blue
2.14. CXCL8 Quantification by ELISA
2.15. Statistical Analyses
3. Results
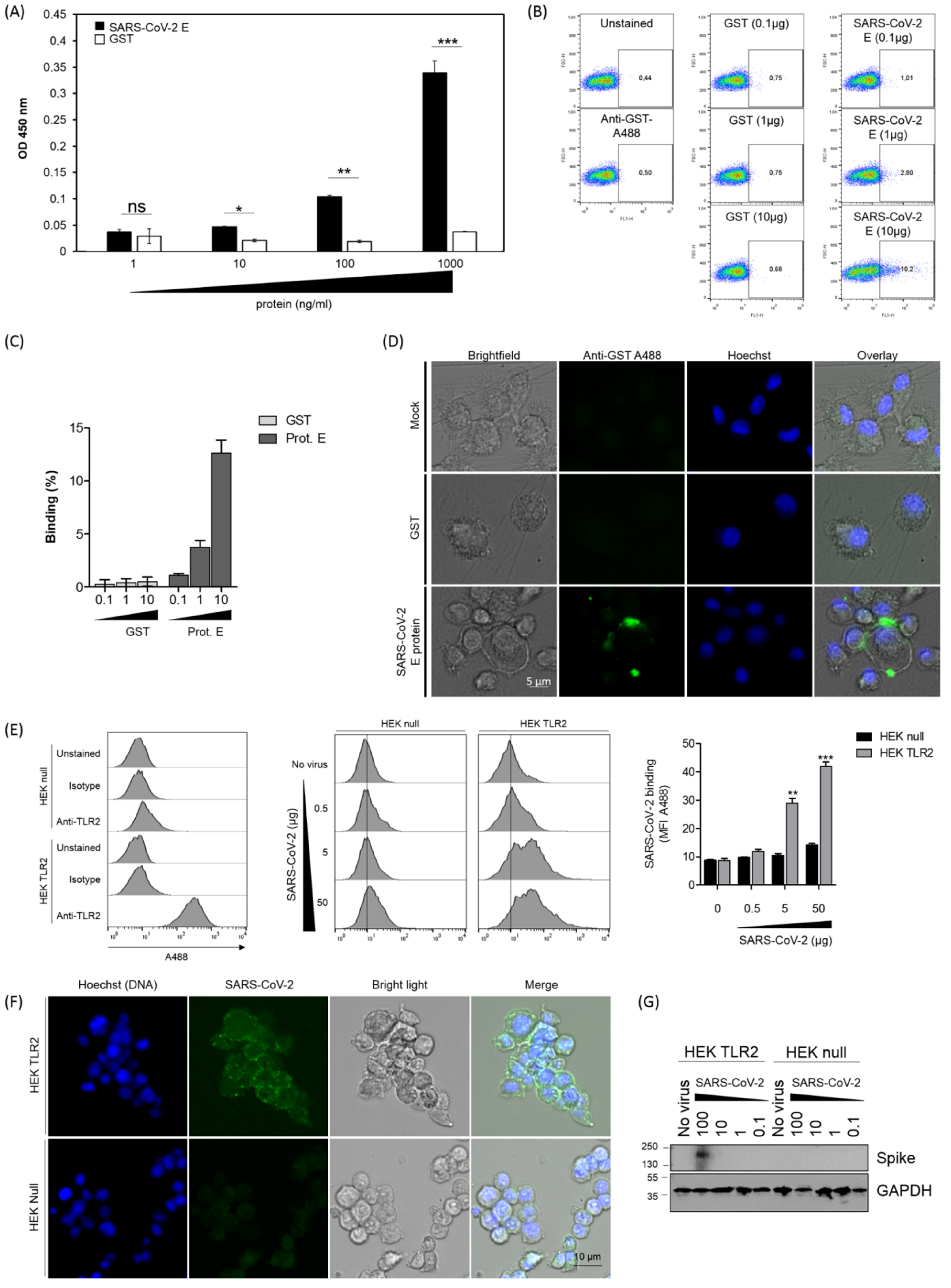
3.1. SARS-CoV-2 Envelope (E) Protein Interacts Directly and Physically with TLR2
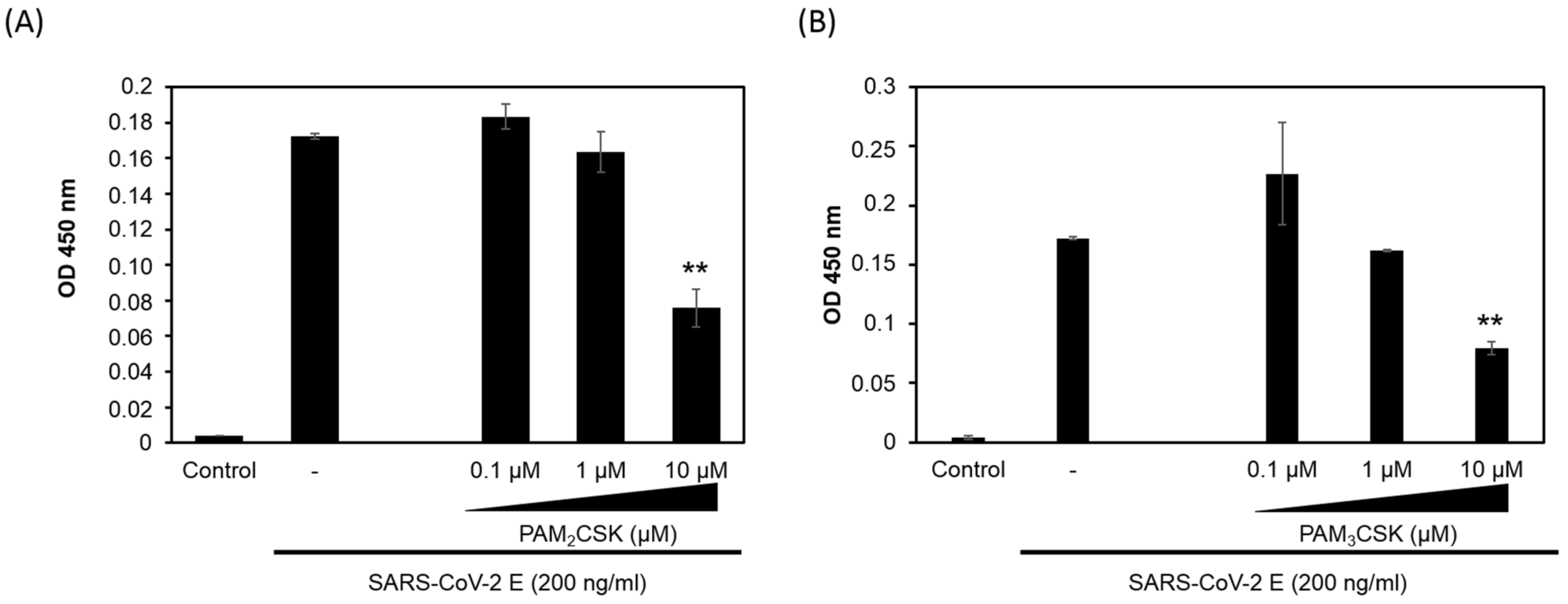
3.2. PAM2CSK4 and PAM3CSK4 Antagonize SARS-CoV-2 E Protein Binding to TLR2
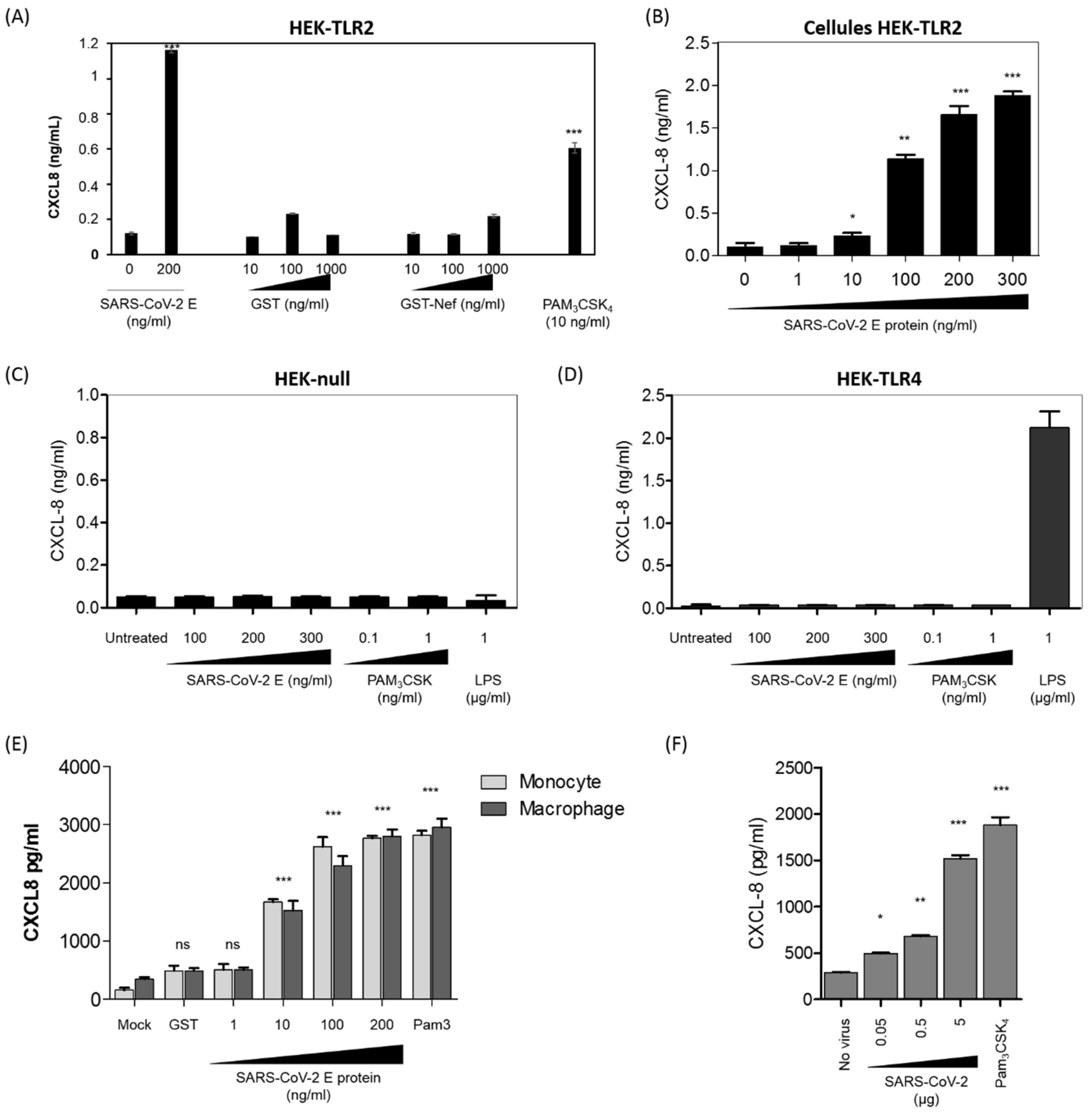
3.3. SARS-CoV-2 E Protein and Inactivated SARS-CoV-2 Stimulate the Production of CXCL8 Inflammatory Chemokine by Recruiting TLR2 Pathway
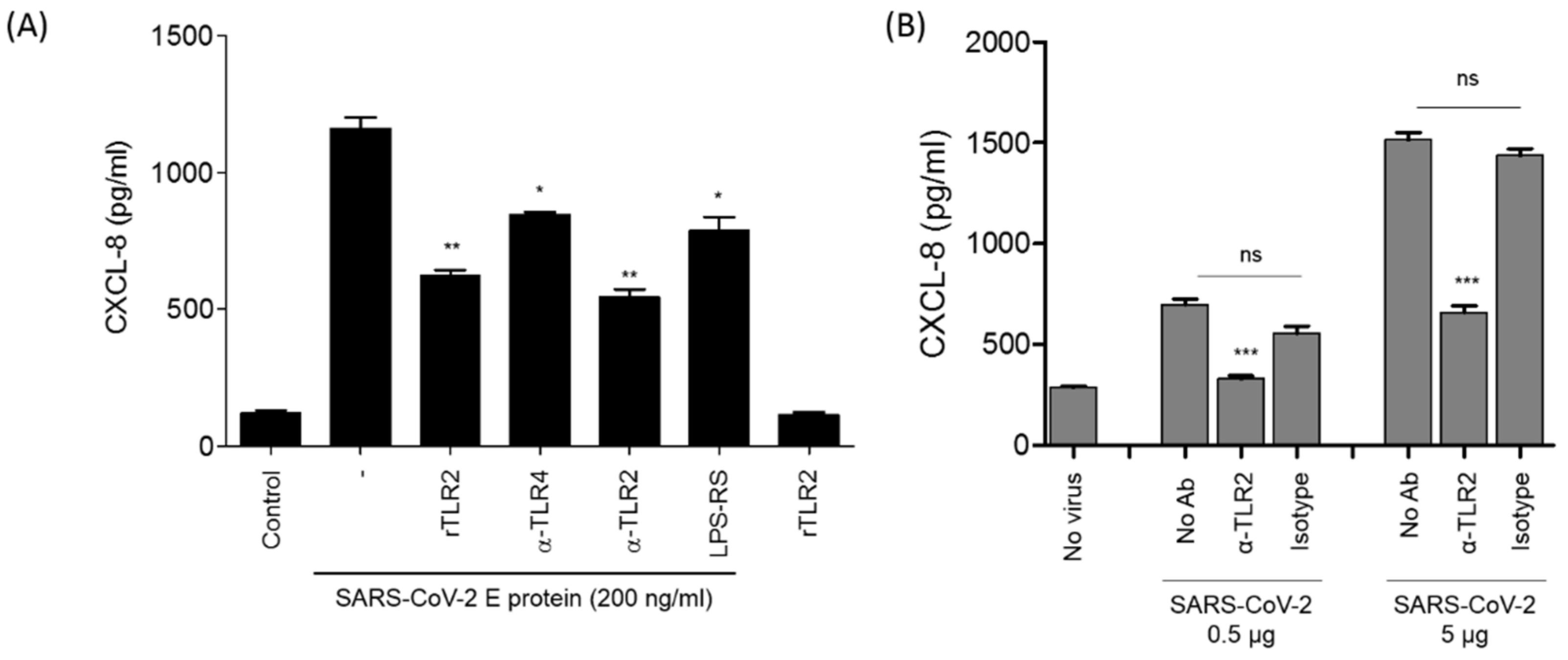
3.4. The Stimulation of CXCL8 Production by SARS-CoV-2 E Protein or SARS-CoV-2 Viral Particles Is Reversed by Soluble rTLR2 and Anti-TLR2 Antibodies
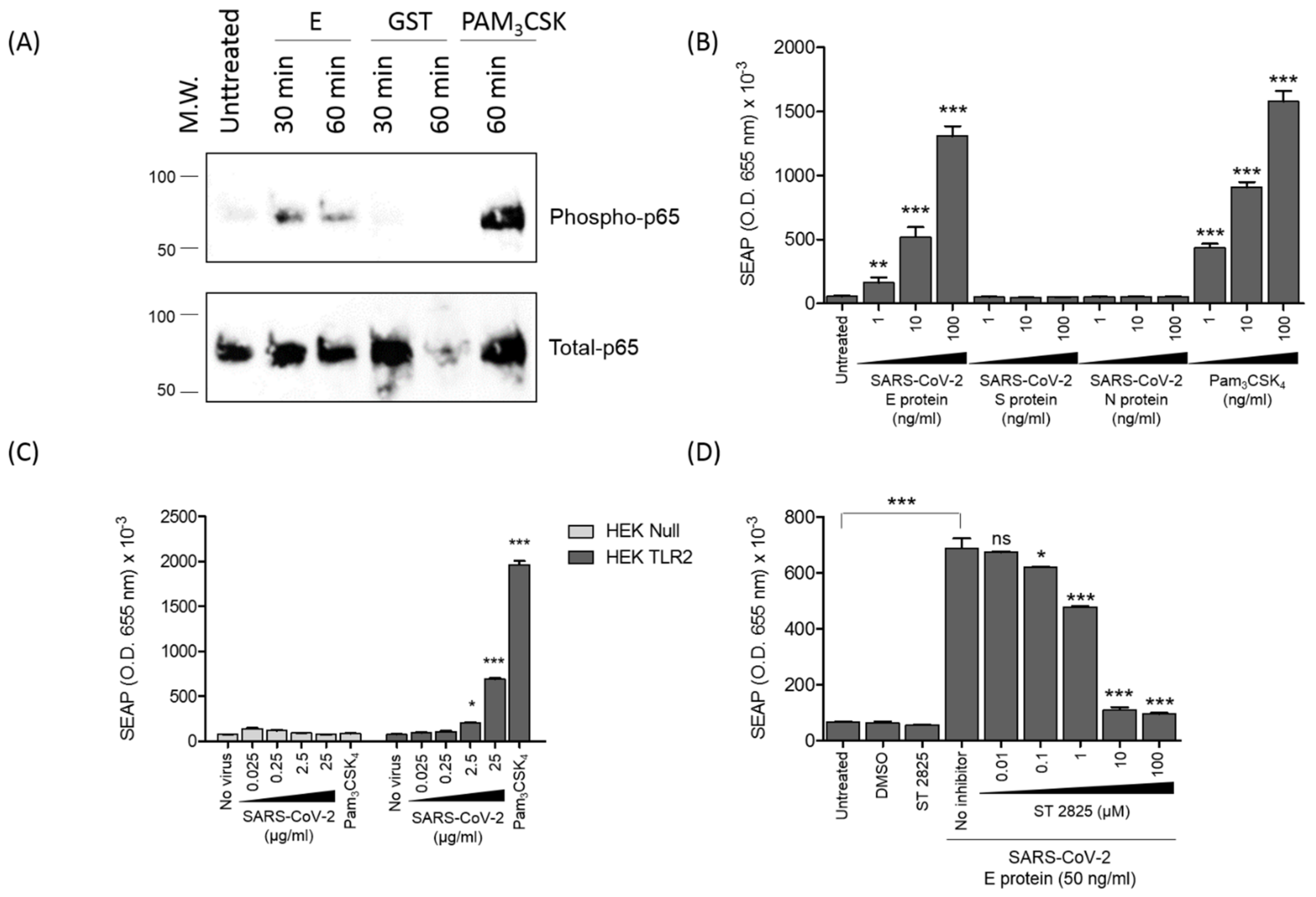
3.5. SARS-CoV-2 E Protein and Inactivated SARS-CoV-2 Activate NF-kB as a Signature of the Recruitment of TLR2 Pathway
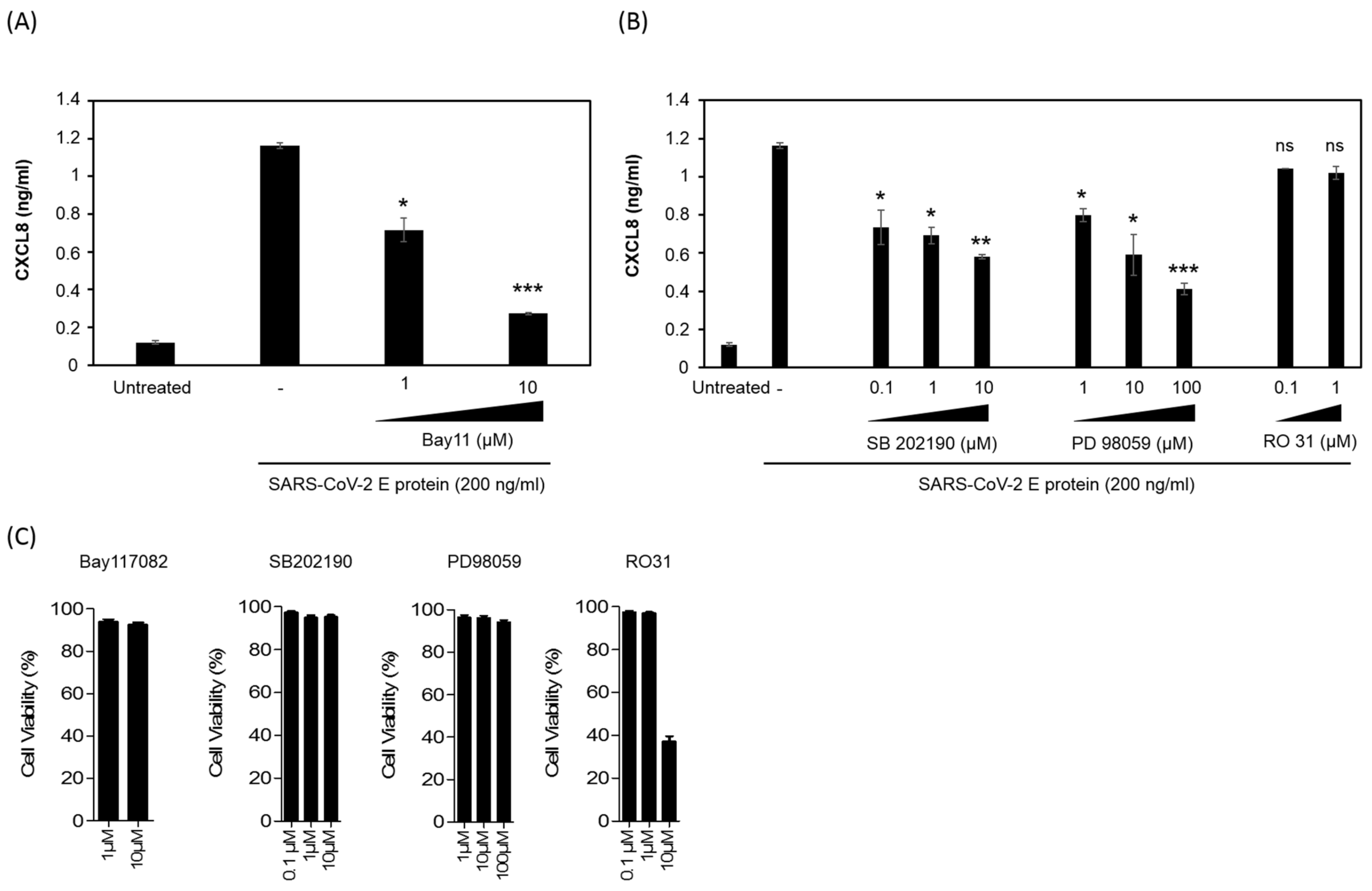
3.6. SARS-CoV-2 E Protein Activation of CXCL8 Production Is Dependent on NF-kB Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, L.; Hurst, K.R.; Masters, P.S. Exceptional flexibility in the sequence requirements for coronavirus small envelope protein function. J. Virol. 2007, 81, 2249–2262. [Google Scholar] [CrossRef] [Green Version]
- Baudoux, P.; Carrat, C.; Besnardeau, L.; Charley, B.; Laude, H. Coronavirus pseudoparticles formed with recombinant M and E proteins induce alpha interferon synthesis by leukocytes. J. Virol. 1998, 72, 8636–8643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatagopalan, P.; Daskalova, S.M.; Lopez, L.A.; Dolezal, K.A.; Hogue, B.G. Coronavirus envelope (E) protein remains at the site of assembly. Virology 2015, 478, 75–85. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Alvarez, E.; Almazan, F.; Rejas, M.T.; Lamirande, E.; Roberts, A.; Shieh, W.J.; Zaki, S.R.; Subbarao, K.; Enjuanes, L. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 2007, 81, 1701–1713. [Google Scholar] [CrossRef] [Green Version]
- Ortego, J.; Ceriani, J.E.; Patino, C.; Plana, J.; Enjuanes, L. Absence of E protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 2007, 368, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Netland, J.; DeDiego, M.L.; Zhao, J.; Fett, C.; Alvarez, E.; Nieto-Torres, J.L.; Enjuanes, L.; Perlman, S. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 2010, 399, 120–128. [Google Scholar] [CrossRef]
- Lim, K.P.; Liu, D.X. The missing link in coronavirus assembly. Retention of the avian coronavirus infectious bronchitis virus envelope protein in the pre-Golgi compartments and physical interaction between the envelope and membrane proteins. J. Biol. Chem. 2001, 276, 17515–17523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corse, E.; Machamer, C.E. Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J. Virol. 2000, 74, 4319–4326. [Google Scholar] [CrossRef] [Green Version]
- Mortola, E.; Roy, P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004, 576, 174–178. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Usera, F.; Enjuanes, L. Coronavirus virulence genes with main focus on SARS-CoV envelope gene. Virus Res. 2014, 194, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kindler, E.; Thiel, V. SARS-CoV and IFN: Too Little, Too Late. Cell Host. Microbe. 2016, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.M.; Shin, E.C. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 2021, 53, 750–760. [Google Scholar] [CrossRef]
- Vanderbeke, L.; van Mol, P.; van Herck, Y.; de Smet, F.; Humblet-Baron, S.; Martinod, K.; Antoranz, A.; Arijs, I.; Boeckx, B.; Bosisio, F.M.; et al. Monocyte-driven atypical cytokine storm and aberrant neutrophil activation as key mediators of COVID-19 disease severity. Nat. Commun. 2021, 12, 4117. [Google Scholar] [CrossRef]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168. [Google Scholar] [CrossRef]
- Jose, R.J.; Manuel, A. COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet Respir. Med. 2020, 8, e46–e47. [Google Scholar] [CrossRef]
- Odak, I.; Barros-Martins, J.; Bosnjak, B.; Stahl, K.; David, S.; Wiesner, O.; Busch, M.; Hoeper, M.M.; Pink, I.; Welte, T.; et al. Reappearance of effector T cells is associated with recovery from COVID-19. EBioMedicine 2020, 57, 102885. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Wang, G.; Yuan, Z.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zhao, C.; Dong, Q.; Zhuang, H.; Song, S.; Peng, G.; Dwyer, D.E. Effects of severe acute respiratory syndrome (SARS) coronavirus infection on peripheral blood lymphocytes and their subsets. Int. J. Infect. Dis. 2005, 9, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Kieser, K.J.; Kagan, J.C. Multi-receptor detection of individual bacterial products by the innate immune system. Nat. Rev. Immunol. 2017, 17, 376–390. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129. [Google Scholar] [CrossRef]
- Bauernfried, S.; Scherr, M.J.; Pichlmair, A.; Duderstadt, K.E.; Hornung, V. Human NLRP1 is a sensor for double-stranded RNA. Science 2021, 371, 6528. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; Hertoghs, N.; Kaptein, T.M.; Zijlstra-Willems, E.M.; Sarrami-Forooshani, R.; Sprokholt, J.K.; van Teijlingen, N.H.; Kootstra, N.A.; Booiman, T.; van Dort, K.A.; et al. HIV-1 blocks the signaling adaptor MAVS to evade antiviral host defense after sensing of abortive HIV-1 RNA by the host helicase DDX3. Nat. Immunol. 2017, 18, 225–235. [Google Scholar] [CrossRef]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Cenac, C.; Ducatez, M.F.; Guery, J.C. Hydroxychloroquine inhibits proteolytic processing of endogenous TLR7 protein in human primary plasmacytoid dendritic cells. Eur. J. Immunol. 2021, 52, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Planes, R.; BenMohamed, L.; Leghmari, K.; Delobel, P.; Izopet, J.; Bahraoui, E. HIV-1 Tat protein induces PD-L1 (B7-H1) expression on dendritic cells through tumor necrosis factor alpha- and toll-like receptor 4-mediated mechanisms. J. Virol. 2014, 88, 6672–6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahraoui, E.; Serrero, M.; Planes, R. HIV-1 Tat-TLR4/MD2 interaction drives the expression of IDO-1 in monocytes derived dendritic cells through NF-kappaB dependent pathway. Sci. Rep. 2020, 10, 8177. [Google Scholar] [CrossRef]
- Schwandner, R.; Dziarski, R.; Wesche, H.; Rothe, M.; Kirschning, C.J. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J. Biol. Chem. 1999, 274, 17406–17409. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Lien, E.; Ingalls, R.R.; Tuomanen, E.; Dziarski, R.; Golenbock, D. Cutting edge: Recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J. Immunol. 1999, 163, 1–5. [Google Scholar]
- Brightbill, H.D.; Libraty, D.H.; Krutzik, S.R.; Yang, R.B.; Belisle, J.T.; Bleharski, J.R.; Maitland, M.; Norgard, M.V.; Plevy, S.E.; Smale, S.T.; et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 1999, 285, 732–736. [Google Scholar] [CrossRef] [Green Version]
- Lien, E.; Sellati, T.J.; Yoshimura, A.; Flo, T.H.; Rawadi, G.; Finberg, R.W.; Carroll, J.D.; Espevik, T.; Ingalls, R.R.; Radolf, J.D.; et al. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J. Biol. Chem. 1999, 274, 33419–33425. [Google Scholar] [CrossRef] [Green Version]
- Serrero, M.; Planes, R.; Bahraoui, E. PKC-delta isoform plays a crucial role in Tat-TLR4 signalling pathway to activate NF-kappaB and CXCL8 production. Sci. Rep. 2017, 7, 2384. [Google Scholar] [CrossRef]
- Badou, A.; Bennasser, Y.; Moreau, M.; Leclerc, C.; Benkirane, M.; Bahraoui, E. Tat protein of human immunodeficiency virus type 1 induces interleukin-10 in human peripheral blood monocytes: Implication of protein kinase C-dependent pathway. J. Virol. 2000, 74, 10551–10562. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.G.; Lee, H.; Lee, J.O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.; Wang, J.; Parthasarathy, K.; Liu, D.X. The transmembrane oligomers of coronavirus protein E. Biophys. J. 2005, 88, 1283–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pervushin, K.; Tan, E.; Parthasarathy, K.; Lin, X.; Jiang, F.L.; Yu, D.; Vararattanavech, A.; Soong, T.W.; Liu, D.X.; Torres, J. Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. 2009, 5, e1000511. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Baltimore, D. NF-kappa B: Ten years after. Cell 1996, 87, 13–20. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Mukaida, N.; Okamoto, S.; Ishikawa, Y.; Matsushima, K. Molecular mechanism of interleukin-8 gene expression. J. Leukoc. Biol. 1994, 56, 554–558. [Google Scholar] [CrossRef]
- Hoffmann, E.; Dittrich-Breiholz, O.; Holtmann, H.; Kracht, M. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 2002, 72, 847–855. [Google Scholar]
- Vij, N.; Amoako, M.O.; Mazur, S.; Zeitlin, P.L. CHOP transcription factor mediates IL-8 signaling in cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Khanmohammadi, S.; Rezaei, N. Role of Toll-like receptors in the pathogenesis of COVID-19. J. Med. Virol. 2021, 93, 2735–2739. [Google Scholar] [CrossRef]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Zust, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood 2007, 109, 1131–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onodi, F.; Bonnet-Madin, L.; Meertens, L.; Karpf, L.; Poirot, J.; Zhang, S.Y.; Picard, C.; Puel, A.; Jouanguy, E.; Zhang, Q.; et al. SARS-CoV-2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J. Exp. Med. 2021, 218, e20201387. [Google Scholar] [CrossRef]
- Kasuga, Y.; Zhu, B.; Jang, K.J.; Yoo, J.S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 53, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, J.; Hume, A.J.; Muhlberger, E. Toll-like receptor 4 in acute viral infection: Too much of a good thing. PLoS Pathog. 2018, 14, e1007390. [Google Scholar] [CrossRef] [Green Version]
- Ben Haij, N.; Leghmari, K.; Planes, R.; Thieblemont, N.; Bahraoui, E. HIV-1 Tat protein binds to TLR4-MD2 and signals to induce TNF-alpha and IL-10. Retrovirology 2013, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Planes, R.; Bahraoui, E. HIV-1 Tat protein induces the production of IDO in human monocyte derived-dendritic cells through a direct mechanism: Effect on T cells proliferation. PLoS ONE 2013, 8, e74551. [Google Scholar] [CrossRef]
- Bieback, K.; Lien, E.; Klagge, I.M.; Avota, E.; Schneider-Schaulies, J.; Duprex, W.P.; Wagner, H.; Kirschning, C.J.; Ter Meulen, V.; Schneider-Schaulies, S. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J. Virol. 2002, 76, 8729–8736. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Hunter, C.A. Is IL-6 a key cytokine target for therapy in COVID-19? Nat. Rev. Immunol. 2021, 21, 337–339. [Google Scholar] [CrossRef]
- Rubin, E.J.; Longo, D.L.; Baden, L.R. Interleukin-6 Receptor Inhibition in COVID-19-Cooling the Inflammatory Soup. N. Engl. J. Med. 2021, 384, 1564–1565. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Rebendenne, A.; Valadao, A.L.C.; Tauziet, M.; Maarifi, G.; Bonaventure, B.; McKellar, J.; Planes, R.; Nisole, S.; Arnaud-Arnould, M.; Moncorge, O.; et al. SARS-CoV-2 triggers an MDA-5-dependent interferon response which is unable to control replication in lung epithelial cells. J. Virol. 2021, 95, e02415-20. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; To, R.K.; Hanna, J.; Spector, S.A. SARS-CoV-2, SARS-CoV-1, and HIV-1 derived ssRNA sequences activate the NLRP3 inflammasome in human macrophages through a non-classical pathway. iScience 2021, 24, 102295. [Google Scholar] [CrossRef] [PubMed]
- Patra, T.; Meyer, K.; Geerling, L.; Isbell, T.S.; Hoft, D.F.; Brien, J.; Pinto, A.K.; Ray, R.B.; Ray, R. SARS-CoV-2 spike protein promotes IL-6 trans-signaling by activation of angiotensin II receptor signaling in epithelial cells. PLoS Pathog. 2020, 16, e1009128. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients with Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Planès, R.; Bert, J.-B.; Tairi, S.; BenMohamed, L.; Bahraoui, E. SARS-CoV-2 Envelope (E) Protein Binds and Activates TLR2 Pathway: A Novel Molecular Target for COVID-19 Interventions. Viruses 2022, 14, 999. https://doi.org/10.3390/v14050999
Planès R, Bert J-B, Tairi S, BenMohamed L, Bahraoui E. SARS-CoV-2 Envelope (E) Protein Binds and Activates TLR2 Pathway: A Novel Molecular Target for COVID-19 Interventions. Viruses. 2022; 14(5):999. https://doi.org/10.3390/v14050999
Chicago/Turabian StylePlanès, Rémi, Jean-Baptiste Bert, Sofiane Tairi, Lbachir BenMohamed, and Elmostafa Bahraoui. 2022. "SARS-CoV-2 Envelope (E) Protein Binds and Activates TLR2 Pathway: A Novel Molecular Target for COVID-19 Interventions" Viruses 14, no. 5: 999. https://doi.org/10.3390/v14050999