
A Computer Simulation of SARS-CoV-2 Mutation Spectra for Empirical Data Characterization and Analysis

 ,
,
Abstract
:1. Introduction
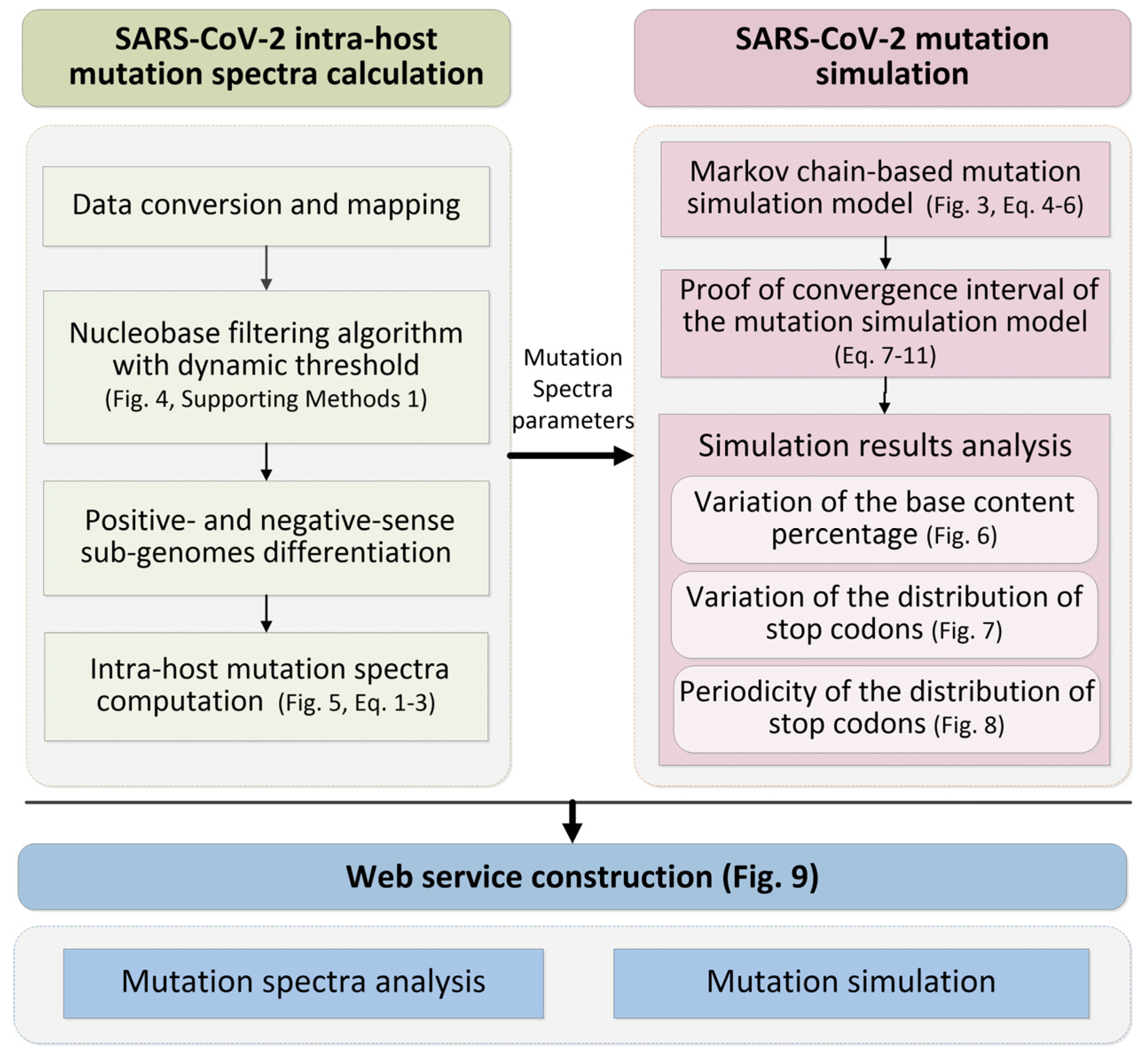
2. Materials and Methods
2.1. SARS-CoV-2 Intra-Host Mutation Spectra Computation
2.1.1. Data Conversion and Mapping
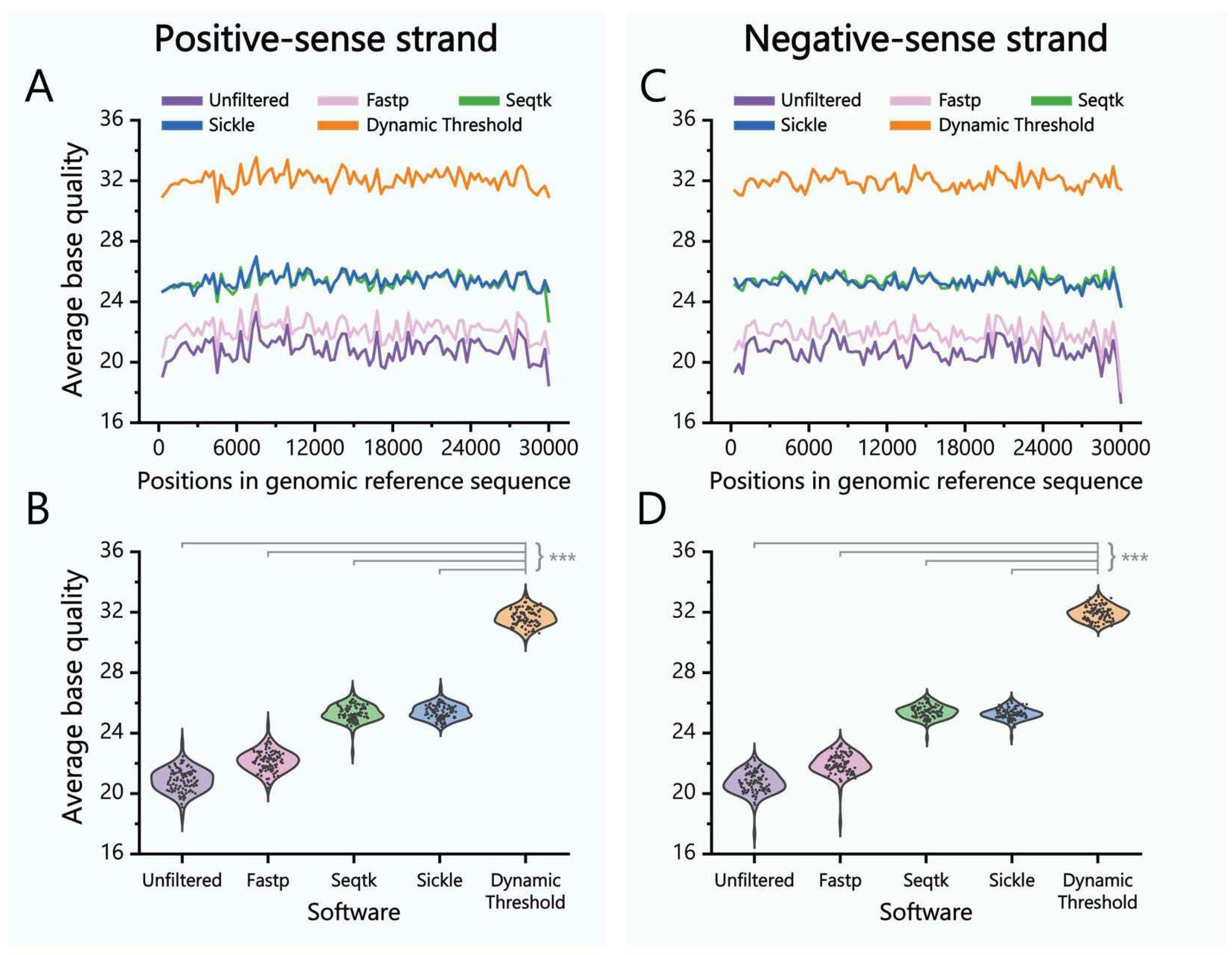
2.1.2. Nucleobase Filtering Algorithm with Dynamic Threshold
2.1.3. Positive- and Negative-Sense Sub-Genomes Differentiation
2.1.4. Intra-Host Mutation Spectra Computation
2.2. SARS-CoV-2 Mutation simulation
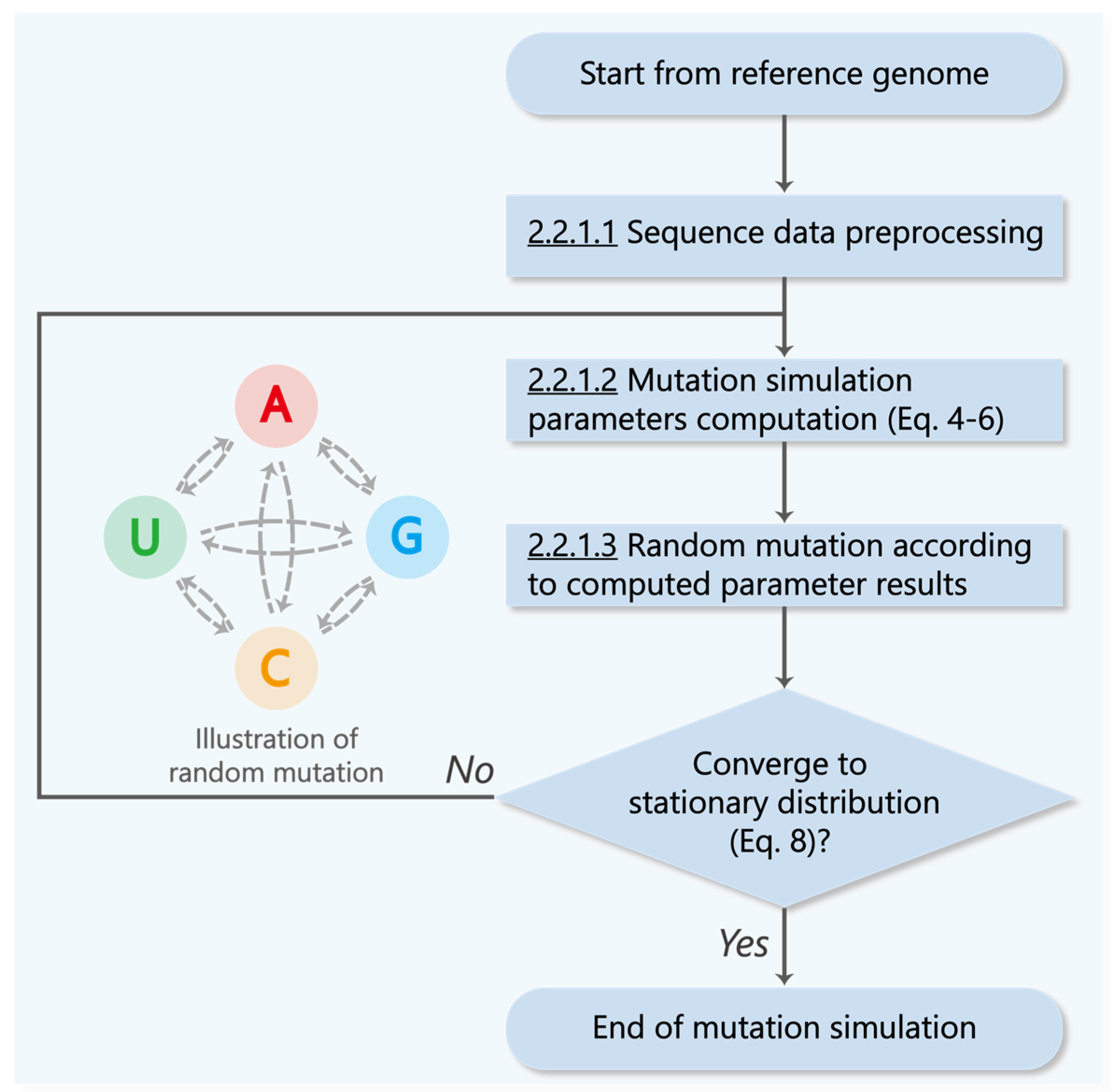
2.2.1. Markov Chain-Based Mutation Simulation Model
2.2.1.1. Sequences Data Preprocessing
2.2.1.2. Mutation Simulation Parameters Computation
2.2.1.3. Random Mutation according to Computed Parameter Results
2.2.2. Proof of Convergence Interval of the Mutation Simulation Model
3. Results
3.1. SARS-CoV-2 Intra-Host Mutation Spectra Computation
3.1.1. Nucleobase Filtering Algorithm with Dynamic Threshold
3.1.2. Intra-Host Mutation Spectra Computation
3.2. SARS-CoV-2 Mutation Simulation
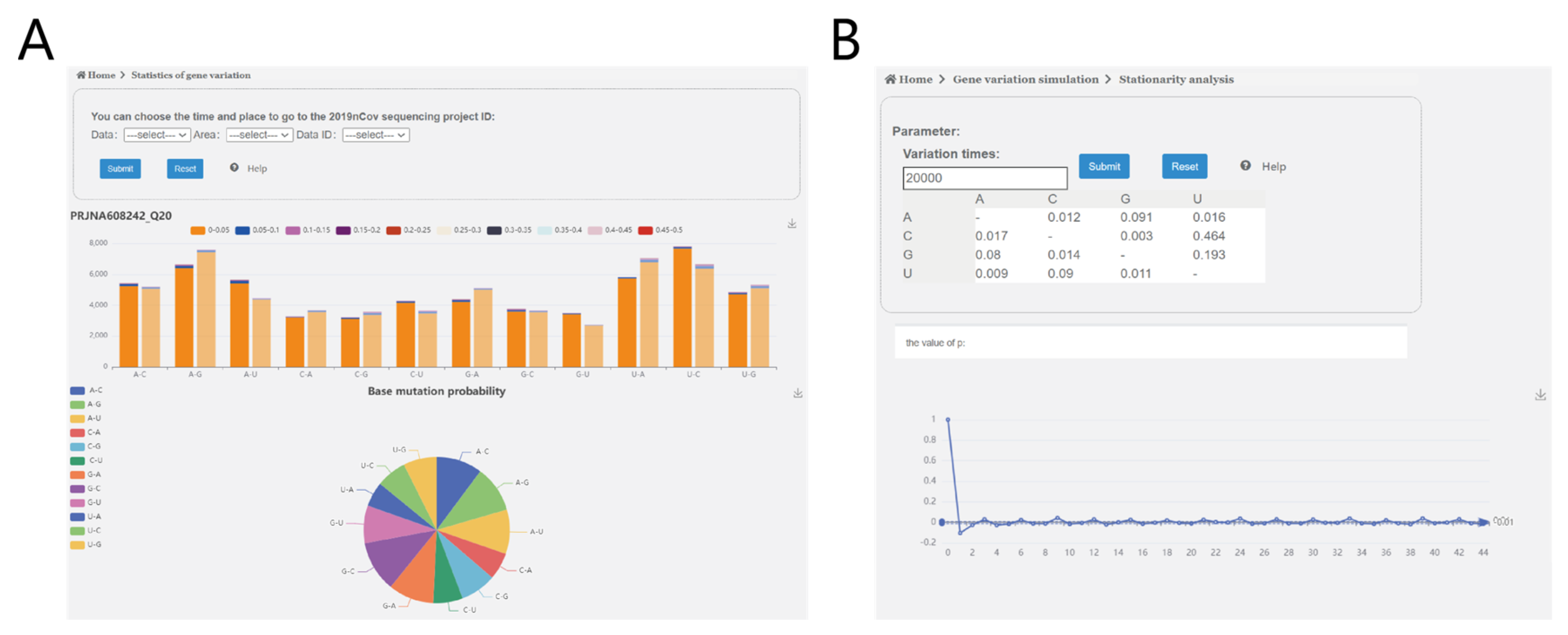
3.2.1. Proof of Convergence Interval of the Mutation Simulation Model
3.2.2. Variation of the Base Content Percentage
3.2.3. Variation of the Distribution of Stop Codons
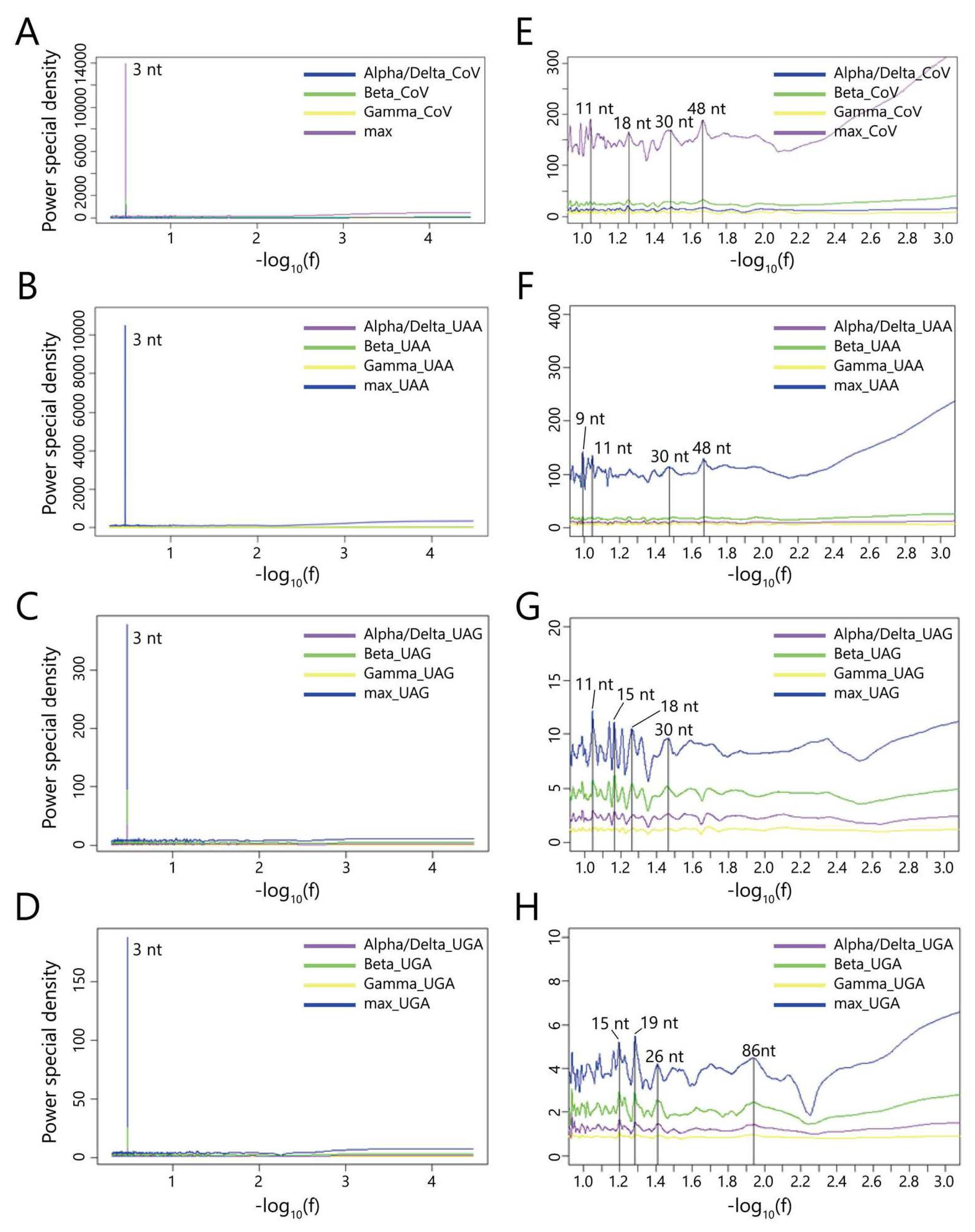
3.2.4. Periodicity of the Distribution of Stop Codons
3.3. Web Service Construction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Day, T.; Gandon, S.; Lion, S.; Otto, S.P. On the evolutionary epidemiology of SARS-CoV-2. Curr. Biol. 2020, 30, R849–R857. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kearney, J.; Mahmoud, M.; Kille, B.; Sedlazeck, F.J.; Treangen, T.J. Rescuing low frequency variants within intra-host viral populations directly from Oxford Nanopore sequencing data. Nat. Commun. 2022, 13, 1321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dai, Z.; Yu, J.; Xiao, M. CpG-island-based annotation and analysis of human housekeeping genes. Brief. Bioinform. 2021, 22, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yu, J. The Pendulum Model for Genome Compositional Dynamics: From the Four Nucleotides to the Twenty Amino Acids. Genom. Proteom. Bioinform. 2012, 10, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Lythgoe, K.; Hall, M.; Ferretti, L.; Cesare, M.D.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.; Moore, N.; et al. SARS-CoV-2 within-host diversity and transmission. Science 2021, 372, eabg0821. [Google Scholar] [CrossRef]
- Braun, K.; Moreno, G.; Wagner, C.; Accola, M.; Rehrauer, W.; Baker, D.; Koelle, K.; O’Connor, D.; Bedford, T.; Friedrich, T.; et al. Acute SARS-CoV-2 infections harbor limited within-host diversity and transmit via tight transmission bottlenecks. PLoS Path. 2021, 17, e1009849. [Google Scholar] [CrossRef]
- Islam, R.; Raju, R.S.; Tasnim, N.; Shihab, I.H.; Bhuiyan, M.A.; Araf, Y.; Islam, T. Choice of assemblers has a critical impact on de novo assembly of SARS-CoV-2 genome and characterizing variants. Brief. Bioinform. 2021, 22, bbab102. [Google Scholar] [CrossRef]
- Rice, A.M.; Morales, A.C.; Ho, A.T.; Mordstein, C.; Mühlhausen, S.; Watson, S.; Cano, L.; Young, B.; Kudla, G.; Hurst, L.D. Evidence for Strong Mutation Bias toward, and Selection against, U Content in SARS-CoV-2: Implications for Vaccine Design. Mol. Biol. Evol. 2021, 38, 67–83. [Google Scholar] [CrossRef]
- Levinstein Hallak, K.; Rosset, S. Statistical modeling of SARS-CoV-2 substitution processes: Predicting the next variant. Commun. Biol. 2022, 5, 285. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-M.; Song, S.-H.; Chen, M.-L.; Zou, D.; Ma, L.-N.; Ma, Y.-K.; Li, R.-J.; Hao, L.-L.; Li, C.-P.; Tian, D.-M. The 2019 novel coronavirus resource. Yi Chuan Hered. 2020, 42, 212–221. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Bull, R.; Adikari, T.; Ferguson, J.; Hammond, J.; Stevanovski, I.; Beukers, A.; Naing, Z.; Yeang, M.; Verich, A.; Gamaarachchi, H.; et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat. Commun. 2020, 11, 6272. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Yan, S.; Zhang, J.; Wang, Y.; Wang, L.; Wang, Y.; Zhang, S.; Du, X.; Zhang, P.; Chen, H.-Y. Nanopore sequencing accurately identifies the cisplatin adduct on DNA. ACS Sens. 2021, 6, 3082–3092. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Bar-On, Y.M.; Gleizer, S.; Bernshtein, B.; Flamholz, A.; Phillips, R.; Milo, R. The total number and mass of SARS-CoV-2 virions. Proc. Natl. Acad. Sci. USA 2021, 118, e2024815118. [Google Scholar] [CrossRef]
- Park, W.B.; Kwon, N.-J.; Choi, S.-J.; Kang, C.K.; Choe, P.G.; Kim, J.Y.; Yun, J.; Lee, G.-W.; Seong, M.-W.; Kim, N.J. Virus isolation from the first patient with SARS-CoV-2 in Korea. J. Korean Med. Sci. 2020, 35, e84. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Kodama, Y.; Shumway, M.; Leinonen, R. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.N.; Piro, R.M.; Jager, M. Computational Exome and Genome Analysis; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [Green Version]
- Setliff, I.; Shiakolas, A.R.; Pilewski, K.A.; Murji, A.A.; Mapengo, R.E.; Janowska, K.; Richardson, S.; Oosthuysen, C.; Raju, N.; Ronsard, L. High-throughput mapping of B cell receptor sequences to antigen specificity. Cell 2019, 179, 1636–1646.e15. [Google Scholar] [CrossRef] [PubMed]
- Braun, K.M.; Haddock, L.A.; Crooks, C.M.; Barry, G.L.; Lalli, J.; Neumann, G.; Watanabe, T.; Imai, M.; Yamayoshi, S.; Ito, M.; et al. Avian H7N9 influenza viruses are evolutionarily constrained by stochastic processes during replication and transmission in mammals. bioRxiv 2022, 4, 1–40. [Google Scholar] [CrossRef]
- Legebeke, J.; Lord, J.; Penrice-Randal, R.; Vallejo, A.F.; Poole, S.; Brendish, N.J.; Dong, X.; Hartley, C.; Holloway, J.W.; Lucas, J.S. Evaluating the immune response in treatment-naive hospitalised patients with influenza and COVID-19. Front. Immunol. 2022, 13, 853265. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Teng, X.; Li, Q.; Li, Z.; Zhang, Y.; Niu, G.; Xiao, J.; Yu, J.; Zhang, Z.; Song, S. Compositional variability and mutation spectra of monophyletic SARS-CoV-2 clades. Genom. Proteom. Bioinform. 2020, 18, 648–663. [Google Scholar] [CrossRef]
- Ross, S.M. Simulation; Academic Press: Salt Lake City, UT, USA, 2022. [Google Scholar]
- Spade, D.A. Markov chain Monte Carlo methods: Theory and practice. In Handbook of Statistics; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Andrews, S. FastQC A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 August 2018).
- You, Y.; Lai, X.; Pan, Y.; Zheng, H.; Vera, J.; Liu, S.; Deng, S.; Zhang, L. Artificial intelligence in cancer target identification and drug discovery. Signal Transduct. Target. Ther. 2022, 7, 156. [Google Scholar] [CrossRef]
- Liu, G.-D.; Li, Y.-C.; Zhang, W.; Zhang, L. A Brief Review of Artificial Intelligence Applications and Algorithms for Psychiatric Disorders. Engineering 2020, 6, 462–467. [Google Scholar] [CrossRef]
- Liu, S.; You, Y.; Tong, Z.; Zhang, L. Developing an Embedding, Koopman and Autoencoder Technologies-Based Multi-Omics Time Series Predictive Model (EKATP) for Systems Biology research. Front. Genet. 2021, 12, 761629. [Google Scholar] [CrossRef]
- Song, H.; Chen, L.; Cui, Y.; Li, Q.; Wang, Q.; Fan, J.; Yang, J.; Zhang, L. Denoising of MR and CT images using cascaded multi-supervision convolutional neural networks with progressive training. Neurocomputing 2022, 469, 354–365. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, X.; Yu, J.; Zhang, L. CGIDLA:Developing the Web Server for CpG Island Related Density and LAUPs (Lineage-Associated Underrepresented Permutations) Study. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 2148–2154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bai, W.; Yuan, N.; Du, Z. Comprehensively benchmarking applications for detecting copy number variation. PLoS Comp. Biol. 2019, 15, e1007069. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lv, J.; Xiao, M.; Yang, L.; Zhang, L. Exploring the underlying mechanism of action of a traditional Chinese medicine formula, Youdujing ointment, for cervical cancer treatment. Quant. Biol. 2021, 9, 292–302. [Google Scholar] [CrossRef]
- Gao, J.; Liu, P.; Liu, G.-D.; Zhang, L. Robust Needle Localization and Enhancement Algorithm for Ultrasound by Deep Learning and Beam Steering Methods. J. Comput. Sci. Technol. 2021, 36, 334–346. [Google Scholar] [CrossRef]
- Fotopoulos, S.B. Probability and Random Processes. Technometrics 2007, 49, 365. [Google Scholar] [CrossRef]
- Lee, C.; Ozdaglar, A.; Shah, D. Computing the Stationary Distribution Locally. In Advances in Neural Information Processing Systems; Curran Associates, Inc.: Montreal, QC, Canada, 2014; pp. 1376–1384. [Google Scholar]
- Zhang, L.; Zheng, C.Q.; Li, T.; Xing, L.; Zeng, H.; Li, T.T.; Yang, H.; Cao, J.; Chen, B.D.; Zhou, Z.Y. Building Up a Robust Risk Mathematical Platform to Predict Colorectal Cancer. Complexity 2017, 2017, 8917258. [Google Scholar] [CrossRef] [Green Version]
- Santangelo, T.J.; Artsimovitch, I. Termination and antitermination: RNA polymerase runs a stop sign. Nat. Rev. Microbiol. 2011, 9, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Yu, J. From Mutation Signature to Molecular Mechanism in the RNA World: A Case of SARS-CoV-2. Genom. Proteom. Bioinform. 2020, 18, 627–639. [Google Scholar] [CrossRef]
- Chen, K.; Meng, Q.; Ma, L.; Liu, Q.; Tang, P.; Chiu, C.; Hu, S.; Yu, J. A novel DNA sequence periodicity decodes nucleosome positioning. Nucleic Acids Res. 2008, 36, 6228–6236. [Google Scholar] [CrossRef] [Green Version]
- Yi, K.; Kim, S.Y.; Bleazard, T.; Kim, T.; Youk, J.; Ju, Y.S. Mutational spectrum of SARS-CoV-2 during the global pandemic. Exp. Mol. Med. 2021, 53, 1229–1237. [Google Scholar] [CrossRef]
- Padhi, A.K.; Tripathi, T. Can SARS-CoV-2 accumulate mutations in the S-protein to increase pathogenicity? ACS Pharmacol. Transl. Sci. 2020, 3, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, D.; Zhang, L.; Sun, W.; Zhang, Z.; Chen, W.; Zhu, A.; Huang, Y.; Xiao, F.; Yao, J.; et al. Intra-host variation and evolutionary dynamics of SARS-CoV-2 populations in COVID-19 patients. Genome Med. 2021, 13, 30. [Google Scholar] [CrossRef]
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Höbartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef] [PubMed]
- Hanusz, Z.; Tarasinska, J.; Zielinski, W. Shapiro-Wilk te st with known mean. REVSTAT-Stat. J. 2016, 14, 89–100. [Google Scholar]
- Schneider, J.W. Null hypothesis significance tests. A mix-up of two different theories: The basis for widespread confusion and numerous misinterpretations. Scientometrics 2015, 102, 411–432. [Google Scholar] [CrossRef] [Green Version]
- Boyce, E.G.; Nappi, J.M. Is there significance beyond the t-test. Drug Intell. Clin. Pharm. 1988, 22, 334–335. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
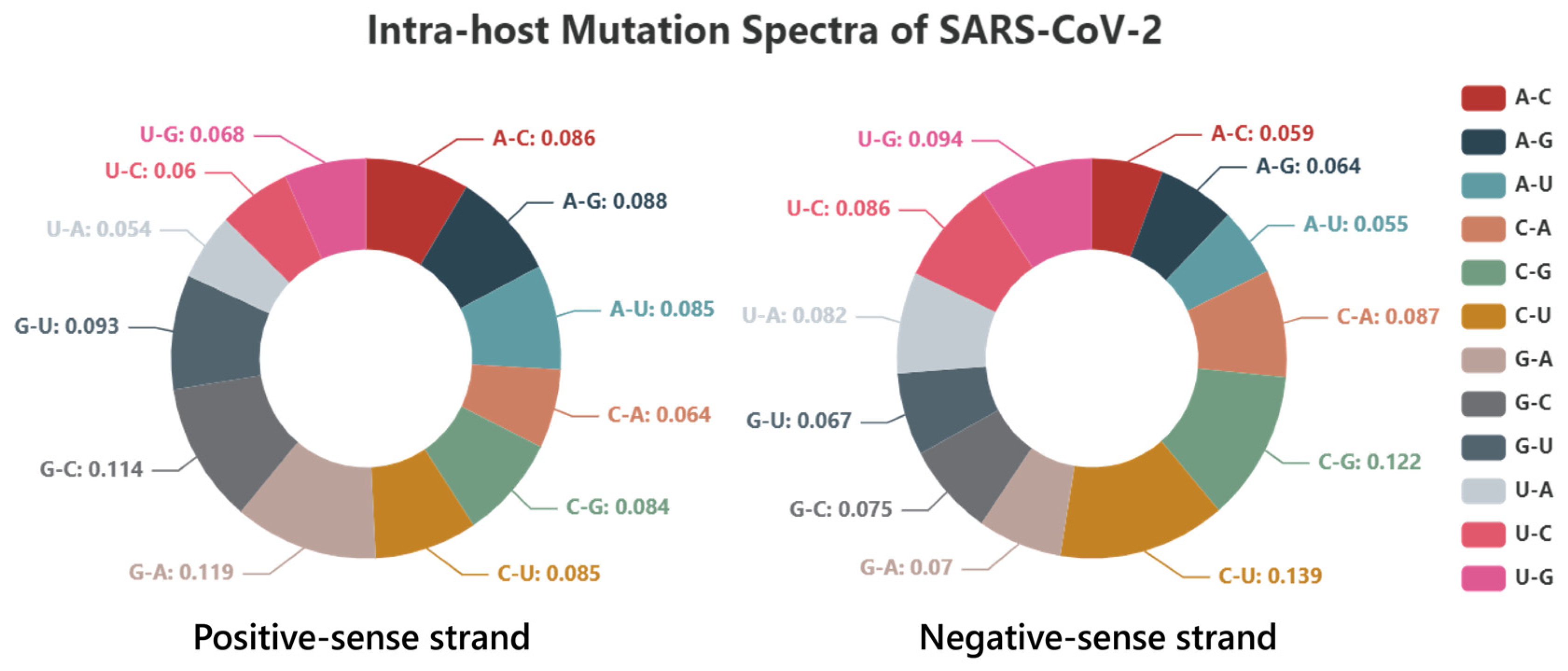{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stop Codons | The Length of Segment Corresponding to the Most Significant Peak Other Than 3 nt |
|---|---|
| UAA | 9/11/30/48 nt |
| UAG | 11/15/18/30 nt |
| UGA | 15/19/26/86 nt |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, M.; Ma, F.; Yu, J.; Xie, J.; Zhang, Q.; Liu, P.; Yu, F.; Jiang, Y.; Zhang, L. A Computer Simulation of SARS-CoV-2 Mutation Spectra for Empirical Data Characterization and Analysis. Biomolecules 2023, 13, 63. https://doi.org/10.3390/biom13010063
Xiao M, Ma F, Yu J, Xie J, Zhang Q, Liu P, Yu F, Jiang Y, Zhang L. A Computer Simulation of SARS-CoV-2 Mutation Spectra for Empirical Data Characterization and Analysis. Biomolecules. 2023; 13(1):63. https://doi.org/10.3390/biom13010063
Chicago/Turabian StyleXiao, Ming, Fubo Ma, Jun Yu, Jianghang Xie, Qiaozhen Zhang, Peng Liu, Fei Yu, Yuming Jiang, and Le Zhang. 2023. "A Computer Simulation of SARS-CoV-2 Mutation Spectra for Empirical Data Characterization and Analysis" Biomolecules 13, no. 1: 63. https://doi.org/10.3390/biom13010063