
Biomedical Text Link Prediction for Drug Discovery: A Case Study with COVID-19

 , , and
, , and
Abstract
:1. Introduction
2. Link Prediction Methods
2.1. Knowledge Graph Definition
2.2. Link Prediction Task
- Initialize entity and relation embeddings randomly.
- Generate negative training triples by replacing either the head entity or tail entity of a positive triple with an entity picked randomly from E. Such a process is often referred to as negative sampling.
- Iterate over the positive and negative triples and update embeddings by optimizing over a loss function that maximizes the score for positive triples and minimizes the score for negative triples. The optimization is completed using a gradient descent algorithm.
2.2.1. TransE
2.2.2. RotatE
2.2.3. ComplEx
2.3. Evaluation Metrics
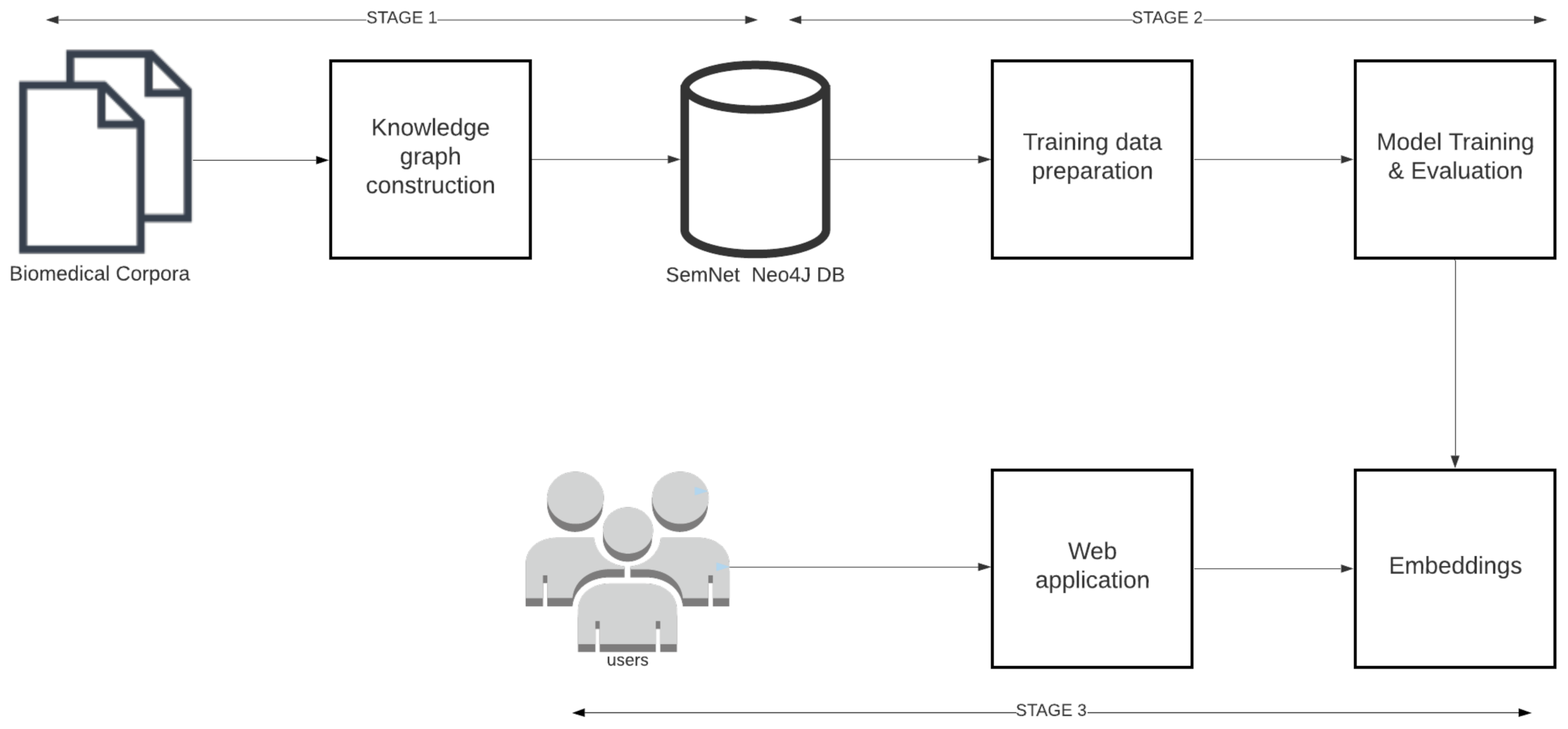
3. Link Prediction Pipeline
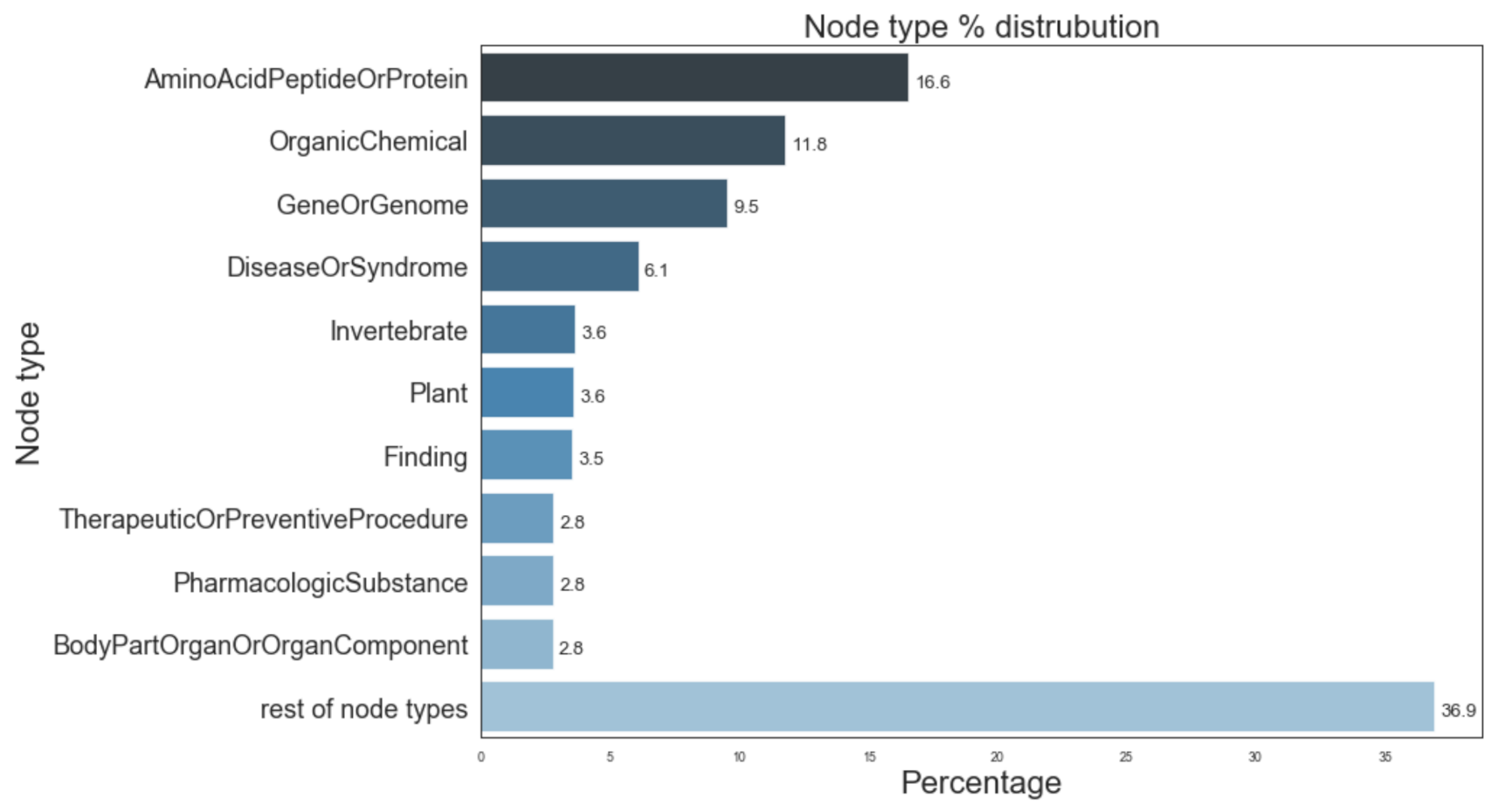
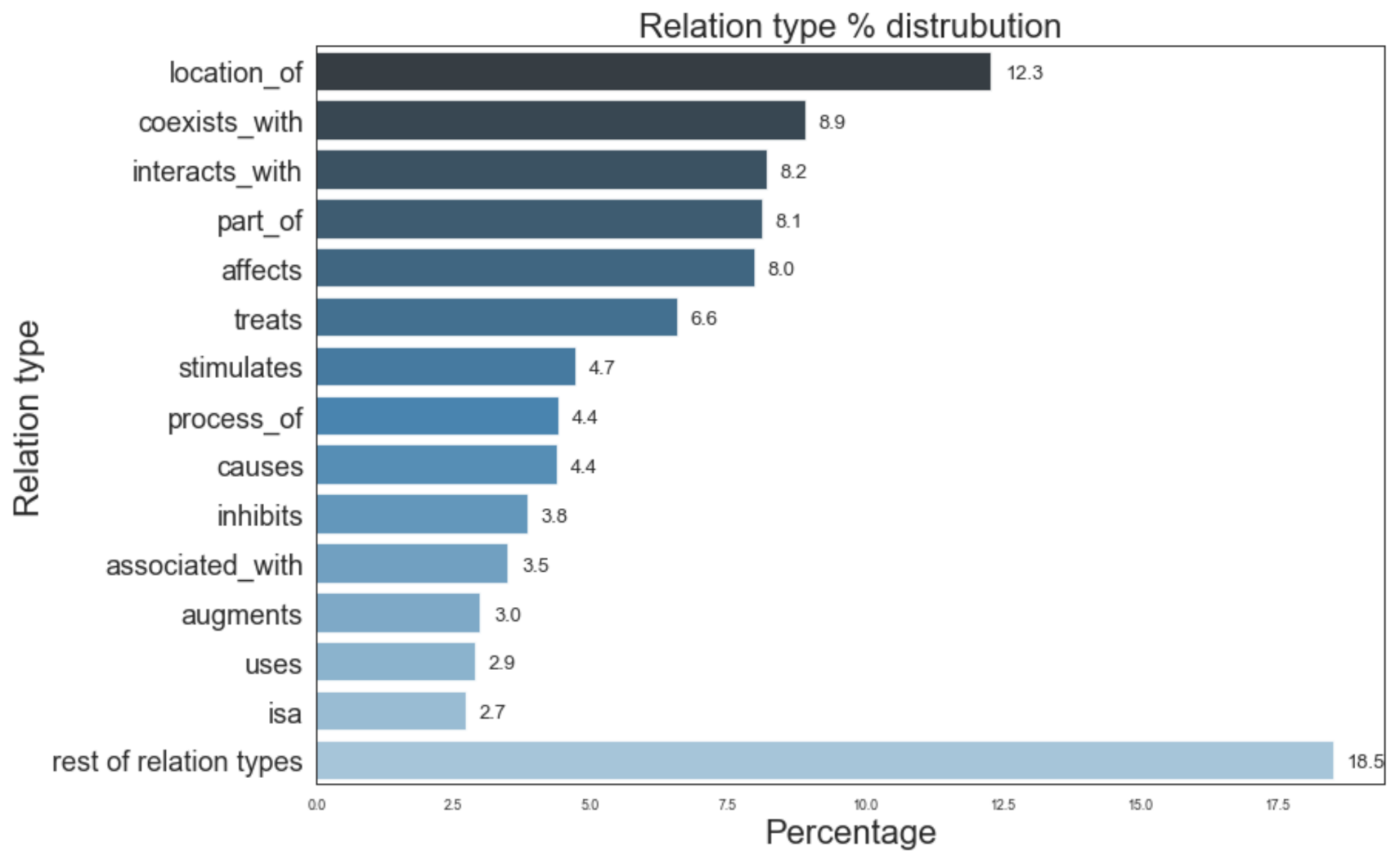
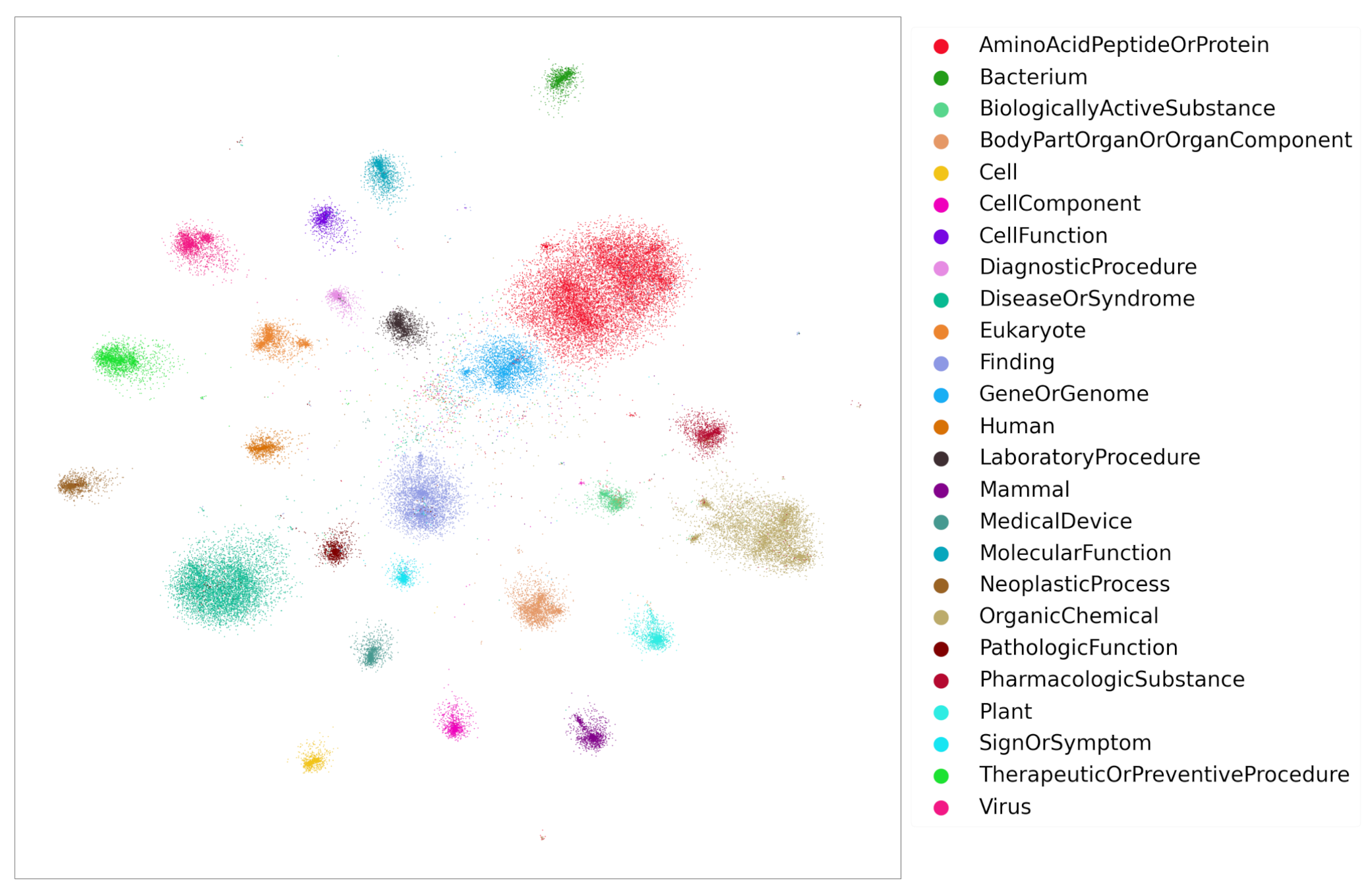
4. Knowledge Graph Construction
5. Model Training
5.1. Training Data Preparation
5.2. Implementation
6. Model Deployment
6.1. Embeddings
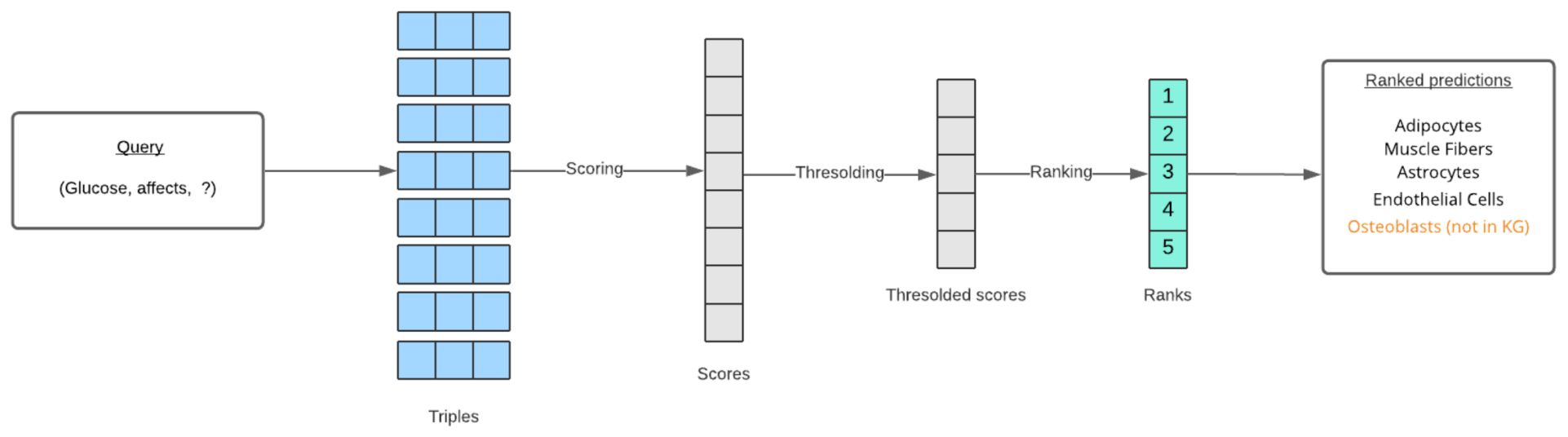
6.2. Ranking Predictions
6.3. REST APIs and User Interface
6.3.1. Embeddings API
6.3.2. Entity Prediction API
6.3.3. User Interface
6.4. Locality Sensitive Hashing
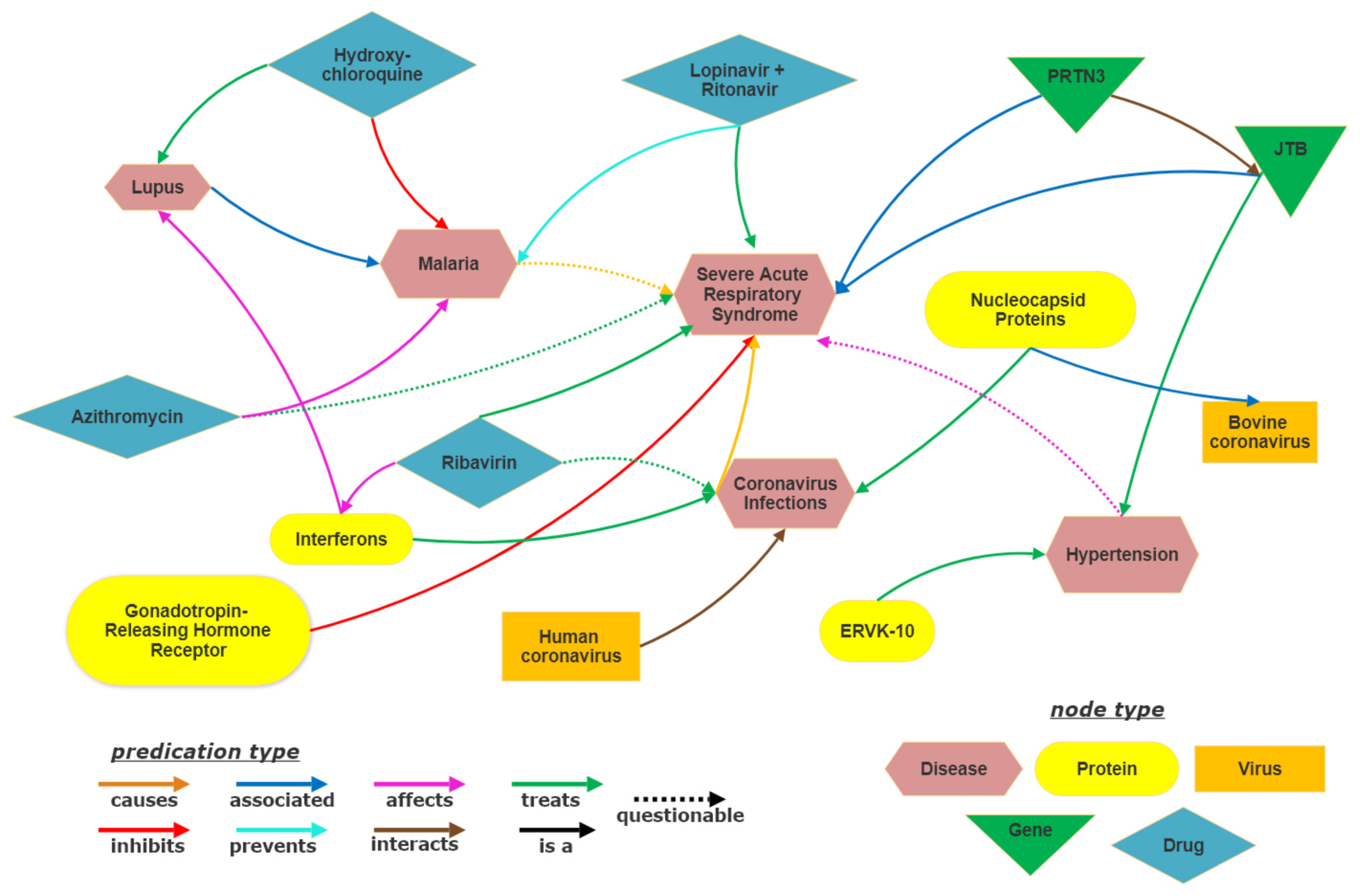
7. COVID-19 Case Study
7.1. Case Study Background
7.2. Case Study Methods
7.2.1. Link Prediction Validation Label Assignment
7.2.2. Pharmacokinetic Label Assignment
7.3. Case Study Results
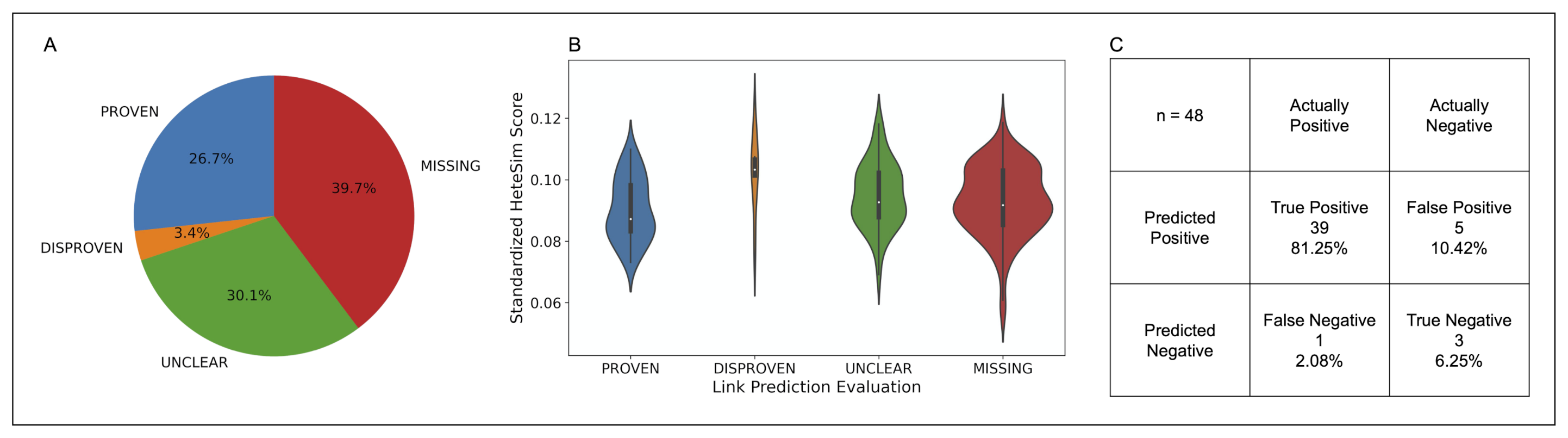
7.3.1. HeteSim Score Distributions for Link Prediction Validation Labels
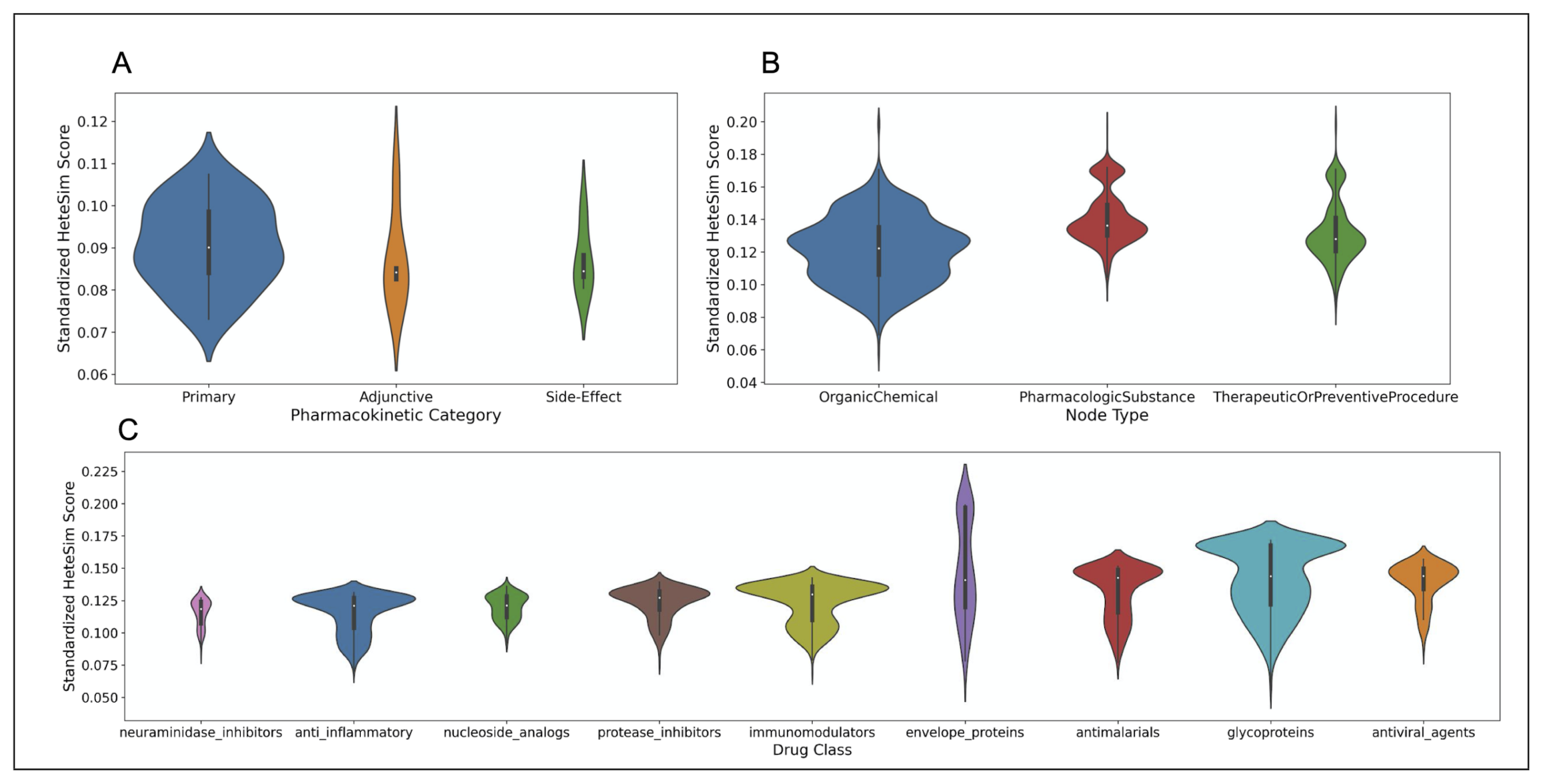
7.3.2. HeteSim Score Distributions for Pharmacokinetic Labels
7.3.3. HeteSim Score Distributions for Different Drug Classes
7.3.4. HeteSim Score Distributions for Different Node Types
7.4. COVID-19 Results Interpretation and Discussion
7.4.1. Value of Link Prediction for Emergent Diseases
7.4.2. HeteSim Score Distributions for Labels assigned through Human in the Loop Validation
7.4.3. HeteSim Score Distributions for Pharmacokinetic Labels
7.4.4. HeteSim Score Distributions for Different Drug Classes
7.4.5. HeteSim Score Distributions for Different Node Types
7.5. Comparison to Existing Drug Repurposing Efforts
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Q.; Allot, A.; Lu, Z. Keep up with the latest coronavirus research. Nature 2020, 579, 193. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.L.; Lo, K.; Chandrasekhar, Y.; Reas, R.; Yang, J.; Eide, D.; Funk, K.; Kinney, R.; Liu, Z.; Merrill, W.; et al. CORD-19: The Covid-19 Open Research Dataset. arXiv 2020, arXiv:2004.10706. [Google Scholar]
- Wilcke, X.; Bloem, P.; De Boer, V. The knowledge graph as the default data model for learning on heterogeneous knowledge. Data Sci. 2017, 1, 39–57. [Google Scholar] [CrossRef] [Green Version]
- Bordes, A.; Usunier, N.; Garcia-Duran, A.; Weston, J.; Yakhnenko, O. Translating embeddings for modeling multi-relational data. In Proceedings of the Advances in Neural Information Processing Systems, Lake Tahoe, NV, USA, 5–8 December 2013; pp. 2787–2795. [Google Scholar]
- Yue, X.; Wang, Z.; Huang, J.; Parthasarathy, S.; Moosavinasab, S.; Huang, Y.; Lin, S.M.; Zhang, W.; Zhang, P.; Sun, H. Graph embedding on biomedical networks: Methods, applications and evaluations. Bioinformatics 2020, 36, 1241–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Zhang, J.; Li, D.; Li, P. Knowledge graph embedding based question answering. In Proceedings of the Twelfth ACM International Conference on Web Search and Data Mining, Houston, TX, USA, 5–9 February 2019; pp. 105–113. [Google Scholar]
- Nicholson, D.N.; Greene, C.S. Constructing knowledge graphs and their biomedical applications. Comput. Struct. Biotechnol. J. 2020, 18, 1414. [Google Scholar] [CrossRef]
- Rossi, A.; Firmani, D.; Matinata, A.; Merialdo, P.; Barbosa, D. Knowledge Graph Embedding for Link Prediction: A Comparative Analysis. arXiv 2020, arXiv:2002.00819. [Google Scholar]
- Yang, B.; Yih, W.T.; He, X.; Gao, J.; Deng, L. Embedding entities and relations for learning and inference in knowledge bases. arXiv 2014, arXiv:1412.6575. [Google Scholar]
- Trouillon, T.; Welbl, J.; Riedel, S.; Gaussier, É.; Bouchard, G. Complex embeddings for simple link prediction. In Proceedings of the International Conference on Machine Learning (ICML), New York, NY, USA, 19–24 June 2016. [Google Scholar]
- Kazemi, S.M.; Poole, D. Simple embedding for link prediction in knowledge graphs. In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Canada, 3–8 December 2018; pp. 4284–4295. [Google Scholar]
- Sun, Z.; Deng, Z.H.; Nie, J.Y.; Tang, J. Rotate: Knowledge graph embedding by relational rotation in complex space. arXiv 2019, arXiv:1902.10197. [Google Scholar]
- Mohamed, S.K.; Nováček, V.; Nounu, A. Discovering protein drug targets using knowledge graph embeddings. Bioinformatics 2020, 36, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Sedler, A.R.; Mitchell, C.S. SemNet: Using local features to navigate the biomedical concept graph. Front. Bioeng. Biotechnol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Dettmers, T.; Minervini, P.; Stenetorp, P.; Riedel, S. Convolutional 2d knowledge graph embeddings. arXiv 2017, arXiv:1707.01476. [Google Scholar]
- Vu, T.; Nguyen, T.D.; Nguyen, D.Q.; Phung, D. A capsule network-based embedding model for knowledge graph completion and search personalization. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, (Long and Short Papers). Minneapolis, MN, USA, 2–7 June 2019; Volume 1, pp. 2180–2189. [Google Scholar]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.S.; Dean, J. Distributed representations of words and phrases and their compositionality. In Proceedings of the Advances in Neural Information Processing Systems, Lake Tahoe, NV, USA, 5–8 December 2013; pp. 3111–3119. [Google Scholar]
- Bodenreider, O. The unified medical language system (UMLS): Integrating biomedical terminology. Nucleic Acids Res. 2004, 32, D267–D270. [Google Scholar] [CrossRef] [Green Version]
- Kilicoglu, H.; Shin, D.; Fiszman, M.; Rosemblat, G.; Rindflesch, T.C. SemMedDB: A PubMed-scale repository of biomedical semantic predications. Bioinformatics 2012, 28, 3158–3160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, S.K.; Nounu, A.; Nováček, V. Drug target discovery using knowledge graph embeddings. In Proceedings of the 34th ACM/SIGAPP Symposium on Applied Computing, Limassol, Cyprus, 8–12 April 2019; pp. 11–18. [Google Scholar]
- Hagberg, A.; Swart, P.; Chult, D.S. Exploring Network Structure, Dynamics, and Function Using NetworkX; Technical Report; Los Alamos National Lab. (LANL): Los Alamos, NM, USA, 2008. [Google Scholar]
- Zheng, D.; Song, X.; Ma, C.; Tan, Z.; Ye, Z.; Dong, J.; Xiong, H.; Zhang, Z.; Karypis, G. Dgl-ke: Training knowledge graph embeddings at scale. arXiv 2020, arXiv:2004.08532. [Google Scholar]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient estimation of word representations in vector space. arXiv 2013, arXiv:1301.3781. [Google Scholar]
- Pennington, J.; Socher, R.; Manning, C.D. Glove: Global vectors for word representation. In Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP), Doha, Qatar, 25–29 October 2014; pp. 1532–1543. [Google Scholar]
- Peters, M.E.; Neumann, M.; Iyyer, M.; Gardner, M.; Clark, C.; Lee, K.; Zettlemoyer, L. Deep contextualized word representations. arXiv 2018, arXiv:1802.05365. [Google Scholar]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Zhu, Z.; Xu, S.; Qu, M.; Tang, J. GraphVite: A High-Performance CPU-GPU Hybrid System for Node Embedding. In The World Wide Web Conference; ACM: New York, NY, USA, 2019; pp. 2494–2504. [Google Scholar]
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Corrado, G.S.; Davis, A.; Dean, J.; Devin, M.; et al. Tensorflow: Large-scale machine learning on heterogeneous distributed systems. arXiv 2016, arXiv:1603.04467. [Google Scholar]
- Truchon, M. Borda and the maximum likelihood approach to vote aggregation. Math. Soc. Sci. 2008, 55, 96–102. [Google Scholar] [CrossRef]
- Sternlicht, D.; Oreli Levi, J.S. RESTool. 2019. Available online: https://https://github.com/dsternlicht/RESTool/ (accessed on 16 January 2021).
- Pauleve, L.; Herve Jegou, L.A. Locality sensitive hashing: A comparison of hash function types and querying mechanisms. Pattern Recognit. Lett. 2010. [Google Scholar] [CrossRef] [Green Version]
- Bernhardsson, E. Annoy. 2019. Available online: https://github.com/spotify/annoy (accessed on 16 January 2021).
- Li, R.; Pei, S.; Chen, B.; Song, Y.; Zhang, T.; Yang, W.; Shaman, J. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, A.; Caccuri, F.; Bugatti, A.; Zani, A.; Vanoni, M.; Bonfanti, P.; Cazzaniga, M.E.; Perno, C.F.; Messa, C.; Alberghina, L. Methotrexate inhibits SARS-CoV-2 virus replication “in vitro”. J. Med Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Firpo, M.R.; Mastrodomenico, V.; Hawkins, G.M.; Prot, M.; Levillayer, L.; Gallagher, T.; Simon-Loriere, E.; Mounce, B.C. Targeting Polyamines Inhibits Coronavirus Infection by Reducing Cellular Attachment and Entry. ACS Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Parnham, M.J. Potential therapeutic use of ebselen for COVID-19 and other respiratory viral infections. Free. Radic. Biol. Med. 2020. [Google Scholar] [CrossRef]
- Weber, A.; Chau, A.; Egeblad, M.; Barnes, B.; Janowitz, T. Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: A case series. medRxiv Prepr. Serv. Health Sci. 2020. [Google Scholar] [CrossRef]
- Meini, S.; Zini, C.; Passaleva, M.T.; Frullini, A.; Fusco, F.; Carpi, R.; Piani, F. Pneumatosis intestinalis in COVID-19. BMJ Open Gastroenterol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chang, C.K.; Chang, Y.W.; Sue, S.C.; Bai, H.I.; Riang, L.; Hsiao, C.D.; Huang, T.H. Structure of the SARS Coronavirus Nucleocapsid Protein RNA-binding Dimerization Domain Suggests a Mechanism for Helical Packaging of Viral RNA. J. Mol. Biol. 2007. [Google Scholar] [CrossRef]
- Wu, Y.C.; Chen, C.S.; Chan, Y.J. The outbreak of COVID-19: An overview. J. Chin. Med Assoc. 2020, 83, 217–220. [Google Scholar] [CrossRef]
- Cinatl, J.; Morgenstern, B.; Bauer, G.; Chandra, P.; Rabenau, H.; Doerr, H.W. Treatment of SARS with human interferons. Lancet 2003, 362, 293–294. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020. [Google Scholar] [CrossRef]
- Stower, H. Lopinavir-ritonavir in severe COVID-19. Nat. Med. 2020. [Google Scholar] [CrossRef] [Green Version]
- Seyed Hosseini, E.; Riahi Kashani, N.; Nikzad, H.; Azadbakht, J.; Hassani Bafrani, H.; Haddad Kashani, H. The novel coronavirus Disease-2019 (COVID-19): Mechanism of action, detection and recent therapeutic strategies. Virology 2020. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asselah, T.; Durantel, D.; Pasmant, E.; Lau, G.; Schinazi, R.F. COVID-19: Discovery, diagnostics and drug development. J. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.S.; Hsueh, P.R. Old and re-purposed drugs for the treatment of COVID-19. Expert Rev. Anti Infect. Ther. 2020, 18, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Al-Nazawi, M. Virtual screening and repurposing of FDA approved drugs against COVID-19 main protease. Life Sci. 2020. [Google Scholar] [CrossRef]
- NCI Thesaurus, Edetic Acid. Available online: https://ncithesaurus.nci.nih.gov/ncitbrowser/ConceptReport.jsp?dictionary=NCI_Thesaurus&ns=ncit&code=C61742 (accessed on 27 April 2021).
- Kozak, J.J.; Gray, H.B.; Garza-lópez, R.A. Structural stability of the SARS-CoV-2 main protease: Can metal ions affect function? J. Inorg. Biochem. 2020. [Google Scholar] [CrossRef]
- Kuroishi, T. Regulation of immunological and inflammatory functions by biotin. Can. J. Physiol. Pharmacol. 2015, 93, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- NCI Thesaurus, Fluoroquinolones. Available online: https://ncithesaurus.nci.nih.gov/ncitbrowser/ConceptReport.jsp?dictionary=NCI_Thesaurus&ns=ncit&code=C126712 (accessed on 27 April 2021).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Statistics |
|---|---|
| Entities | 74,086 |
| Triples | 8,928,797 |
| Entity types | 121 |
| Relation types | 61 |
| Appended relation types | 25,341 |
| Training triples | 8,828,797 |
| Validation triples | 50,000 |
| Test triples | 50,000 |
| Query: SARS Coronavirus | Query: Chloroquine | Query: Cyclosporine |
|---|---|---|
| Middle East Respiratory Syndrome Coronavirus | Hydroxychloroquine | Calcineurin |
| Genus: Coronavirus | Polymyxin B Sulfate | rituximab |
| SARS coronavirus Urbani | Aminoquinoline | infliximab |
| Beluga Whale coronavirus SW1 | Bauxite | Calcitonin Receptor |
| SARS-related bat coronavirus | Mefloquine | Neoral |
| Query | Top Ranked Relation |
|---|---|
| (Chloroquine, ?, SARS Coronavirus) | treats |
| (SARS Coronavirus, ?, Chloroquine) | location_of |
| (Dexamethasone, ?, SARS Coronavirus) | treats |
| (Albuterol, ?, Dornase Alfa) | uses |
| (Nucelocapsid, ?, SARS Coronavirus) | part_of |
| Query: (?, treats, SARS Coronavirus) | Query: (Chloroquine, Chloroquine, ?) |
|---|---|
| Chloroquine | Ebola Virus |
| Duration | Virus |
| Glycyrrhizic Acid | SARS coronavirus |
| Ritonavir | Zika Virus |
| Octanoic acid | HIV |
| Node | Drug Class | Node Type | Standardized HeteSim Score | Predicted Link | Pharmacokinetics |
|---|---|---|---|---|---|
| Top 5 PROVEN Nodes | |||||
| Chloroquine | glycoproteins big | OrganicChemical | 0.073 | treats | Primary |
| Glycyrrhizic Acid | anti-inflammatory | OrganicChemical | 0.074 | treats | Primary |
| Quinine | anti-inflammatory | OrganicChemical | 0.077 | treats | Primary |
| Chloroquine | antimalarial | OrganicChemical | 0.077 | treats | Primary |
| Fluoroquinolones | antimalarial | OrganicChemical | 0.077 | treats | Adjunctive |
| Top 5 DISPROVEN Nodes | |||||
| Polyamines | antimalarial | OrganicChemical | 0.080 | treats | Other |
| Complement System Proteins | neuraminidase inhibitors | ImmunologicFactor | 0.101 | prevents | Other |
| Dopamine Receptor | neuraminidase inhibitors | Receptor | 0.103 | treats | Other |
| Chemokine (C-C Motif) Receptor 5|CCR5 | envelope protein | Receptor | 0.104 | treats | Other |
| Antiviral prophylaxis | nucleoside analogs | TherapeuticOr PreventiveProcedure | 0.107 | neg treats | Primary |
| Top 5 UNCLEAR Nodes | |||||
| small molecule | immunomodulators | OrganicChemical | 0.069 | prevents | N/A |
| ebselen | anti-inflammatory | OrganicChemical | 0.077 | treats | N/A |
| Fluticasone propionate | anti-inflammatory | OrganicChemical | 0.079 | treats | N/A |
| Quinolone Antibacterial Agents | anti-inflammatory | OrganicChemical | 0.082 | prevents | N/A |
| Morphine | anti-inflammatory | OrganicChemical | 0.084 | treats | N/A |
| Top 5 MISSING Nodes | |||||
| small molecule | glycoproteins big | OrganicChemical | 0.056 | prevents | N/A |
| RABBIT SERUM | glycoproteins big | OrganicChemical | 0.061 | N/A | N/A |
| Esters | anti-inflammatory | OrganicChemical | 0.070 | N/A | N/A |
| Edetic Acid | glycoproteins big | OrganicChemical | 0.074 | treats | N/A |
| small molecule | protease inhibitors | OrganicChemical | 0.075 | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCoy, K.; Gudapati, S.; He, L.; Horlander, E.; Kartchner, D.; Kulkarni, S.; Mehra, N.; Prakash, J.; Thenot, H.; Vanga, S.V.; et al. Biomedical Text Link Prediction for Drug Discovery: A Case Study with COVID-19. Pharmaceutics 2021, 13, 794. https://doi.org/10.3390/pharmaceutics13060794
McCoy K, Gudapati S, He L, Horlander E, Kartchner D, Kulkarni S, Mehra N, Prakash J, Thenot H, Vanga SV, et al. Biomedical Text Link Prediction for Drug Discovery: A Case Study with COVID-19. Pharmaceutics. 2021; 13(6):794. https://doi.org/10.3390/pharmaceutics13060794
Chicago/Turabian StyleMcCoy, Kevin, Sateesh Gudapati, Lawrence He, Elaina Horlander, David Kartchner, Soham Kulkarni, Nidhi Mehra, Jayant Prakash, Helena Thenot, Sri Vivek Vanga, and et al. 2021. "Biomedical Text Link Prediction for Drug Discovery: A Case Study with COVID-19" Pharmaceutics 13, no. 6: 794. https://doi.org/10.3390/pharmaceutics13060794