
Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors



Abstract
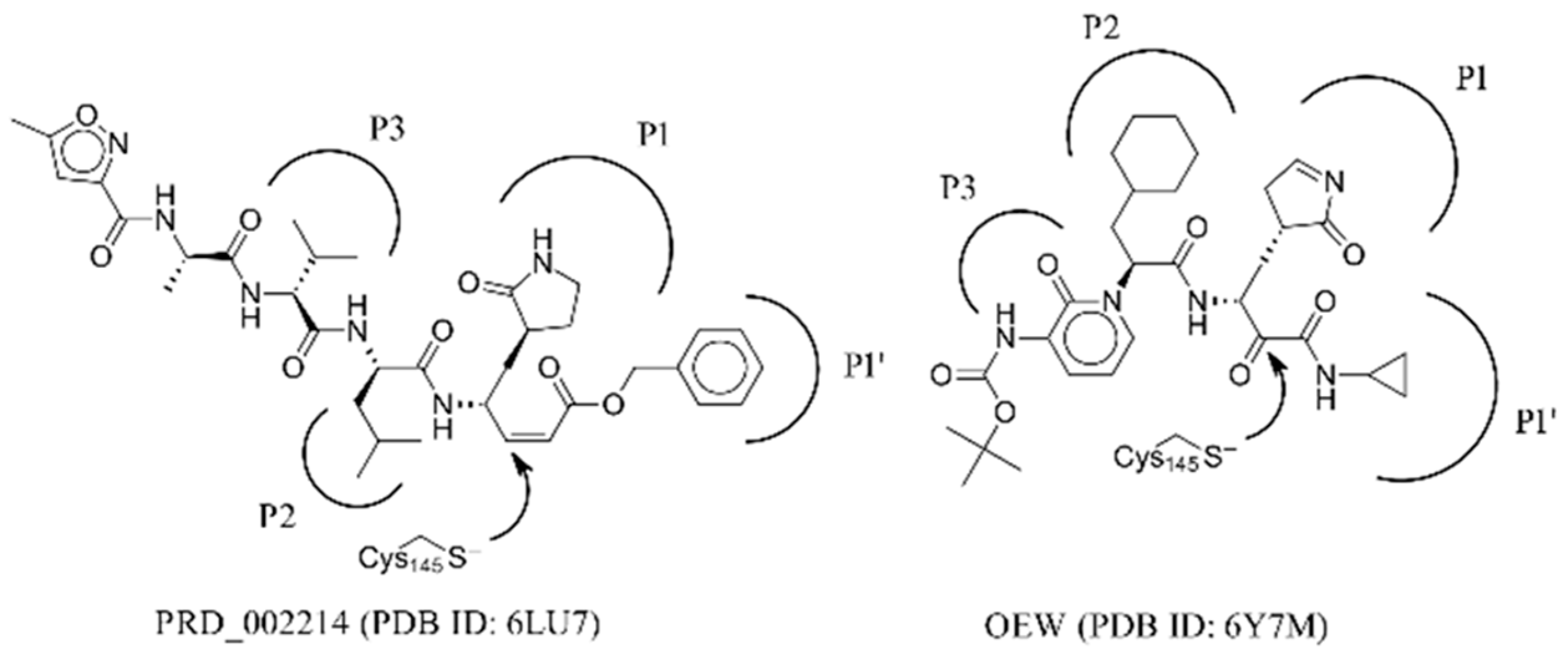
:1. Introduction
2. Results and Discussion
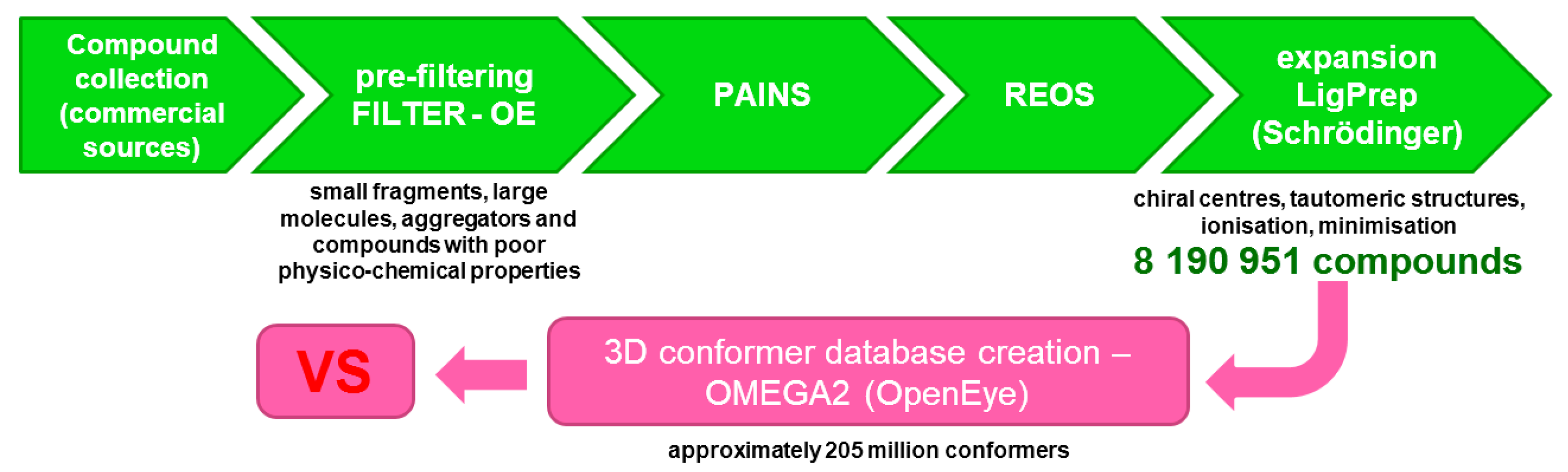
2.1. Database Preparation
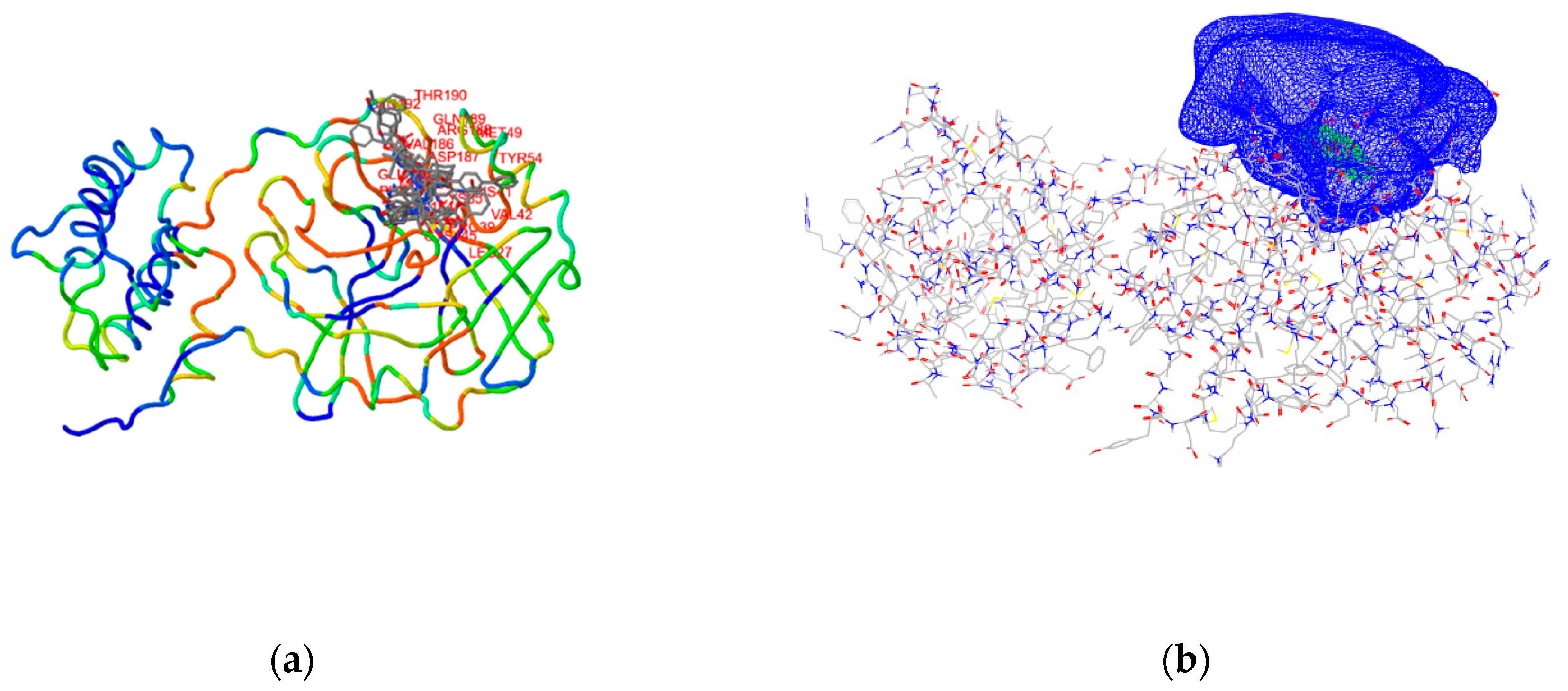
2.2. Target Preparation
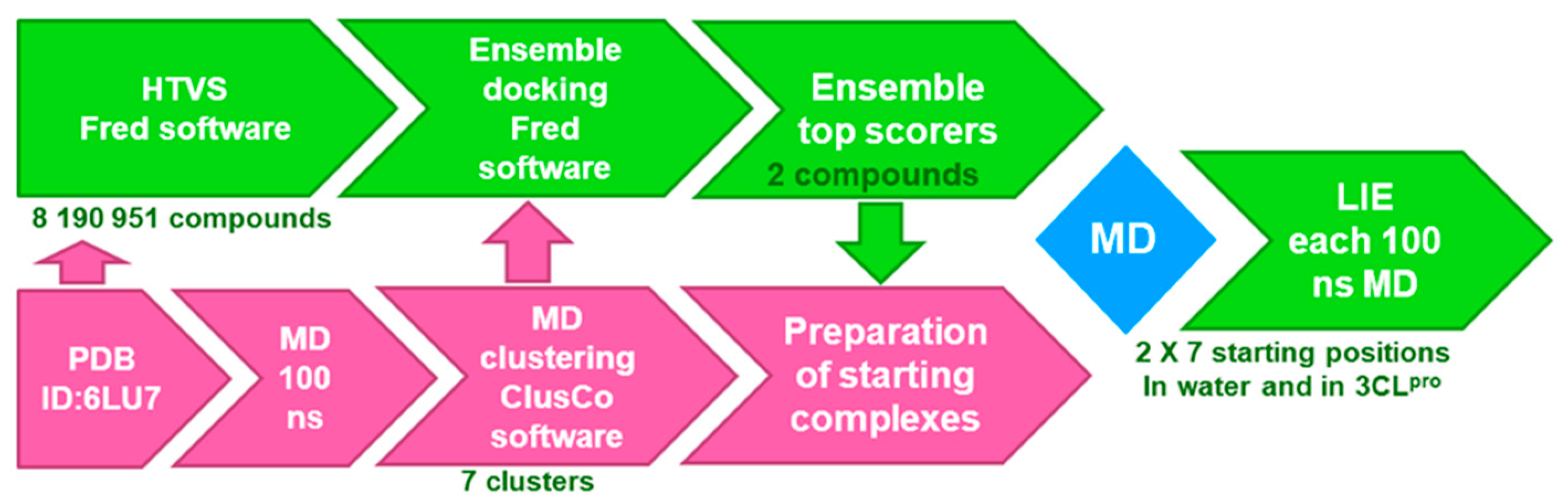
2.3. Ensemble Docking
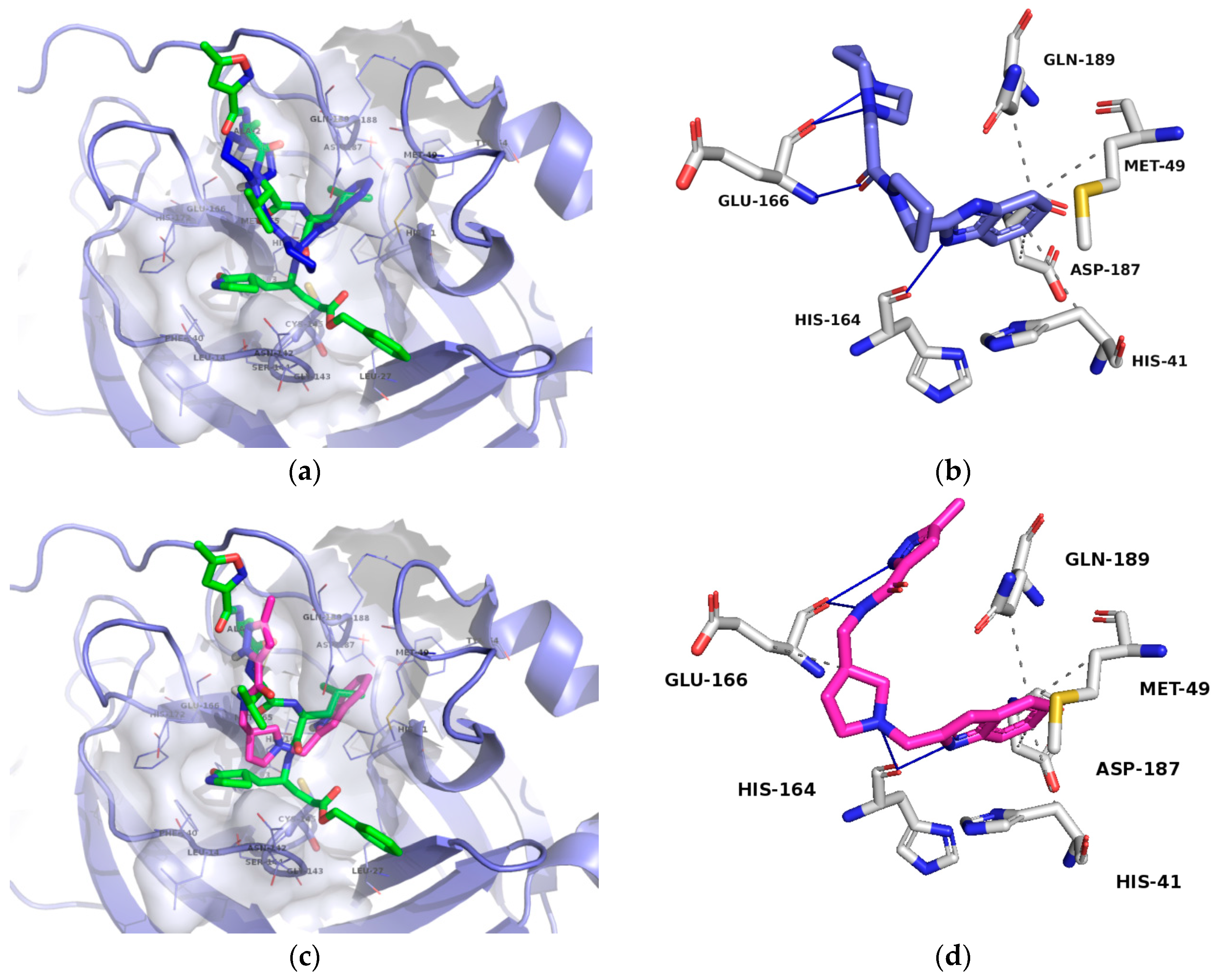
2.4. Free-Energy Calculations and Contact Analysis
3. Materials and Methods
3.1. MD and Ensemble Docking
3.2. Free Energy Calculation Using LIE Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HTVS | High-Throughput Virtual Screening |
| LIE | Linear Interaction Energy |
| MD | Molecular Dynamics |
| Mpro | Main Protease |
| PAINS | Pan-assay interference compounds |
| REOS | Rapid elimination of swill filter |
| VS | Virtual Screening |
References
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Grottesi, A.; Bešker, N.; Emerson, A.; Manelfi, C.; Beccari, A.R.; Frigerio, F.; Lindahl, E.; Cerchia, C.; Talarico, C. Computational Studies of SARS-CoV-2 3CLpro: Insights from MD Simulations. Int. J. Mol. Sci. 2020, 21, 5346. [Google Scholar] [CrossRef]
- Shi, J.; Sivaraman, J.; Song, J. Mechanism for Controlling the Dimer-Monomer Switch and Coupling Dimerization to Catalysis of the Severe Acute Respiratory Syndrome Coronavirus 3C-Like Protease. J. Virol. 2008, 82, 4620–4629. [Google Scholar] [CrossRef] [Green Version]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weißbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and Enzymes Involved in SARS Coronavirus Genome Expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- Malcolm, B.A. The picornaviral 3C proteinases: Cysteine nucleophiles in serine proteinase folds. Prot. Sci. 1995, 4, 1439–1445. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Feng, S.; Qi, F.; Bao, L.; Du, L.; et al. Inhibition of SARS-CoV-2 infection (previously 2019-nCoV) by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell. Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Shanker, A.; Bhanu, D.; Alluri, A. Analysis of Whole Genome Sequences and Homology Modelling of a 3C Like Peptidase and a Non-Structural Protein of the Novel Coronavirus COVID-19 Shows Protein Ligand Interaction with an Aza-Peptide and a Noncovalent Lead Inhibitor with Possible Antiviral Properties. ChemRxiv 2020, 1–39. [Google Scholar] [CrossRef]
- Goetz, D.H.; Choe, Y.; Hansell, E.; Chen, Y.T.; McDowell, M.; Jonsson, C.B.; Roush, W.R.; McKerrow, J.; Craik, C.S. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry 2007, 46, 8744–8752. [Google Scholar] [CrossRef]
- Chuck, C.P.; Chong, L.T.; Chen, C.; Chow, H.F.; Wan, D.C.C.; Wong, K.B. Profiling of substrate specificity of SARS-CoV 3CLpro. PLoS ONE 2010, 5, e13197. [Google Scholar] [CrossRef] [Green Version]
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, L.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004, 279, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Sellner, M.; Neranjan, S.; Smieško, M.; Lill, M.A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. [Google Scholar] [CrossRef]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Disc. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef]
- Baillie, T.A. Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Gong, G.; Grum-Tokars, V.; Mulhearn, D.C.; Baker, S.C.; Coughlin, M.; Prabhakar, B.S.; Sleeman, K.; Johnson, M.E.; Mesecar, A.D. Design, synthesis and antiviral efficacy of a series of potent chloropyridyl ester-derived SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 5684–5688. [Google Scholar] [CrossRef]
- Shoichet, B.K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Saubern, S.; Guha, R.; Baell, J.B. KNIME workflow to assess PAINS filters in SMARTS format. Comparison of RDKit and Indigo cheminformatics libraries. Mol. Inform. 2011, 30, 847–850. [Google Scholar] [CrossRef]
- Walters, W.P.; Stahl, M.T.; Murcko, M.A. Virtual screening—An overview. Drug Disc. Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Hevener, K.E. Hit identification and optimization in virtual screening: Practical recommendations based on a critical literature analysis: Miniperspective. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aid. Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aid. Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Su, H.X.; Yao, S.; Zhao, W.F.; Li, M.J.; Liu, J.; Shang, W.J.; Yu, K.Q. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef]
- Konc, J.; Miller, B.T.; Stular, T.; Lesnik, S.; Woodcock, H.L.; Brooks, B.R.; Janežič, D. ProBiS-CHARMMing: Web interface for prediction and optimization of ligands in protein binding sites. J. Chem. Inf. Model. 2015, 55, 2308–2314. [Google Scholar] [CrossRef]
- Konc, J.; Janežič, D. ProBiS tools (algorithm, database, and web servers) for predicting and modeling of biologically interesting proteins. Prog. Biophys. Mol. Biol. 2017, 128, 24–32. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Jamroz, M.; Kolinski, A. ClusCo: Clustering and comparison of protein models. BMC Bioinform. 2013, 14, 1–6. [Google Scholar] [CrossRef] [Green Version]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aid. Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Gan, J.L.; Kumar, D.; Chen, C.; Taylor, B.C.; Jagger, B.R.; Amaro, R.E.; Lee, C.T. Benchmarking ensemble docking methods as a scientific outreach project. bioRxiv 2020, 1–16. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; KVassilatis, D.; Cournia, Z. Structure-based virtual screening for drug discovery: Principles, applications and recent advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Bleylevens, I.W.; Bitorina, A.V.; Wichapong, K.; Nicolaes, G.A. Optimization of Compound Ranking for Structure-Based Virtual Ligand Screening Using an Established FRED–Surflex Consensus Approach. Chem. Biol. Drug. Des. 2014, 83, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Stjernschantz, E.; Oostenbrink, C. Improved ligand-protein binding affinity predictions using multiple binding modes. Biophys. J. 2010, 98, 2682–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, T.; Marelius, J.; Åqvist, J. Ligand binding affinity prediction by linear interaction energy methods. J. Comput. Aid. Mol. Des. 1998, 12, 27–35. [Google Scholar] [CrossRef]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein–ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Huang, Y.Y.; Wu, Y.; Liu, R.; Zhou, L.; Lin, Y.; Wu, D.; Zhang, L.; Liu, H.; et al. Identify potent SARS-CoV-2 main protease inhibitors via accelerated free energy perturbation-based virtual screening of existing drugs. Proc. Natl. Acad. Sci. USA 2020, 117, 27381–27387. [Google Scholar] [CrossRef]
- Odhar, H.A.; Ahjel, S.W.; Albeer, A.A.M.A.; Hashim, A.F.; Rayshan, A.M.; Humadi, S.S. Molecular docking and dynamics simulation of FDA approved drugs with the main protease from 2019 novel coronavirus. Bioinformation 2020, 16, 236. [Google Scholar] [CrossRef] [Green Version]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Krieger, E.; Dunbrack, R.L.; Hooft, R.W.; Krieger, B. Assignment of protonation states in proteins and ligands: Combining pK a prediction with hydrogen bonding network optimization. Methods. Mol. Biol. 2012, 819, 405–421. [Google Scholar] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theobald, D.L.; Wuttke, D.S. THESEUS: Maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics 2006, 22, 2171–2172. [Google Scholar] [CrossRef] [Green Version]
- Aqvist, J.; Medina, C.; Samuelsson, J.E. A new method for predicting binding affinity in computer-aided drug design. Protein Eng. 1994, 7, 385–391. [Google Scholar] [CrossRef]
- Klvaňa, M.; Bren, U. Aflatoxin B1-Formamidopyrimidine DNA adducts: Relationships between structures, free energies, and melting temperatures. Molecules 2019, 24, 150. [Google Scholar] [CrossRef] [Green Version]
- Rifai, E.A.; Van Dijk, M.; Vermeulen, N.P.; Yanuar, A.; Geerke, D.P. A Comparative Linear Interaction Energy and MM/PBSA Study on SIRT1–Ligand Binding Free Energy Calculation. J. Chem. Inf. Model. 2019, 59, 4018–4033. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Structure | Mr (g/mol) | Binding Mode | Fred Docking Score 1 |
|---|---|---|---|---|
| 1 |  | 343.5 | P2–P3 | −14.5 |
| 2 |  | 338.4 | P2–P3 | −13.5 |
| 3 |  | 309.3 | P2–P3 | −13.0 |
| 4 |  | 293.4 | P1′–P2 | −13.0 |
| 5 |  | 380.5 | P1′–P2 | −13.0 |
| Compound | Free VdW (kcal/mol) | Free Coulomb (kcal/mol) | Complex VdW Weighted Sum (kcal/mol) | Complex Coulomb Weighted Sum (kcal/mol) | (kcal/mol) |
|---|---|---|---|---|---|
| 1 | −16.2 ± 0.2 | −32.3 ± 0.1 | −22.0 ± 1.4 | −37.3 ± 2.4 | −8.2 ± 1.9 |
| 2 | −14.7 ± 0.2 | −19.0 ± 0.1 | −22.5 ± 2.4 | −18.7 ± 2.6 | −3.5 ± 1.7 |
Sample Availability: Not available. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jukič, M.; Janežič, D.; Bren, U. Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors. Molecules 2020, 25, 5808. https://doi.org/10.3390/molecules25245808
Jukič M, Janežič D, Bren U. Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors. Molecules. 2020; 25(24):5808. https://doi.org/10.3390/molecules25245808
Chicago/Turabian StyleJukič, Marko, Dušanka Janežič, and Urban Bren. 2020. "Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors" Molecules 25, no. 24: 5808. https://doi.org/10.3390/molecules25245808