
From Innate Immunity to Inflammation: A Primer on Multiple Facets of NF-κB Signaling in COVID-19


1
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA
2
Molecular, Cellular and Developmental Biology Department, University of California Santa Barbara, Santa Barbara, CA 93106, USA
*
Author to whom correspondence should be addressed.
Physiologia 2022, 2(2), 34-45; https://doi.org/10.3390/physiologia2020004
Submission received: 24 May 2022
/
Revised: 15 June 2022
/
Accepted: 16 June 2022
/
Published: 17 June 2022
(This article belongs to the Special Issue Feature Papers in Human Physiology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Nuclear factor-kappa B (NF-κB) induces the expression of many pro-inflammatory genes, including cytokines and chemokines. In the past decades, a wealth of clinical as well as animal model-based studies have demonstrated the association of the deregulated NF-κB signaling pathway with the progression of various inflammatory diseases, including inflammatory bowel disease (IBD), multiple sclerosis (MS), and chronic obstructive pulmonary disease (COPD). Given the conserved role of the NF-κB pathway as the pivotal regulator of pro-inflammatory gene expression, different components of the NF-κB pathway are proposed as major therapeutic targets against these diseases. The ongoing coronavirus disease of 2019 (COVID-19) has posed a significant public health crisis regarding inflammation-related diseases. A robust inflammatory response is associated with COVID-19-infection-related complications, including muti-organ failure and death. This review summarizes the past and current state of knowledge on the role of the NF-κB signaling pathway in the innate immune response and inflammatory diseases with the objective of potential therapeutic use in developing effective treatment options for COVID-19.
Keywords:
innate immunity; inflammation; NF-κB; SARS-Co-V2; cytokine storm; lymphopenia; multi-organ failure; ROS; COVID-191. Introduction
Inflammatory responses are the key defense mechanisms against pathogenic encounters. Misregulation of the inflammatory response is associated with many human diseases, including inflammatory bowel disease (IBD) and rheumatoid arthritis. The expression of many genes involved in the inflammatory immune response is regulated by one inducible transcription factor family, NF-κB. All five NF-κB family proteins, i.e., NF-κB1(p50), NF-κB2(p52), RelA(p65), RelB, and c-Rel bind to the kB enhancer DNA segment to induce the expression of targeted genes [1,2]. The NF-κB signaling pathway is employed in various cellular functions, including redox maintenance and innate immune development. Deregulation of NF-κB signaling is associated with the pathogenesis of various immunological diseases, and thus the NF-κB pathway is a promising therapeutic target. NF-κB signaling in response to infections is depicted in Figure 1.
The ongoing COVID-19 pandemic started a little over two years ago. It is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). According to the Johns Hopkins Coronavirus Resource Center, 2022 data, the number of confirmed cases worldwide is over 500 million, while the number of deaths is over 5 million (https://coronavirus.jhu.edu/ (accessed on 16 May 2022). Relentless efforts from different research labs facilitated the development of SARS-CoV-2 vaccines, which helped meet the urgent need for survival against COVID-19 complications, including death. Emerging reports on persistent clinical symptoms, even in vaccinated individuals, and reoccurrence of clinical manifestations post COVID-19 recovery and post-vaccination warrant further research for effective treatment options.
In the SARS-CoV-2 infected tissue, viral components cause overactivation of inflammatory responses in the innate immune cells, e.g., macrophages, neutrophils, and monocytes [3]. The innate immune inflammatory response is an antiviral defense mechanism against viral infection. However, the overabundance of immune response regarding the immoderate increase in inflammatory cytokines and chemokines upon SARS-CoV-2 infection is detrimental to airways and epithelial and vascular endothelial cells [4]. In line with this, reports on the clinical manifestations of COVID-19 infection documented increased titers of different inflammatory molecules, including IL-2, IL-6, IL-8, IFNγ TNFα, MIP1α, MCP1, and IP-10, in the blood samples of COVID-19 patients [3,5,6,7]. Notably, studies have found a correlation between the increased expression of IL-6 and TNF-α and the severity of disease and mortality in COVID-19 patients [3,8].
The single-stranded RNA of SARS-CoV-2 encodes four proteins for the spike, the membrane, the nucleocapsid, and the envelope [9]. The binding of viral spike proteins to the angiotensin-converting enzyme 2 (ACE2) receptors on the surface of human epithelial cells facilitates the invasion of the virus into the host cell, where it replicates. Upon recognizing the viral proteins, macrophages are activated and pro-inflammatory proteins, such as cytokines and chemokines, are released to clear the viral particles from the infected cells [10]. Notably, a deregulated pro-inflammatory response may lead to a robust release of these immune response proteins and a satiation called a cytokine storm, which can be detrimental to the host cells [11,12]. A recent report by Khan and colleagues showed that the robust inflammatory responses of the macrophages and the epithelial cells are caused by the spike proteins of SARC-CoV-2 [13]. In this study, the authors demonstrated that one of the Toll-like receptor proteins (TLRs), TLR2, binds the viral spike proteins and induces the pro-inflammatory protein through MyD88/NF-κB pathway activation. Interestingly, another study by Zheng and colleagues documented the association of COVID-19 severity with TLR2 and MyD88 expression [14]. These studies strongly suggest that the robust increase in the inflammatory response and cytokine storm are significant contributors to the detrimental outcomes of COVID-19, and TLR2 and its downstream molecule (possibly NF-κB related) may have therapeutic potential against the cytokine storm in COVID-19 infection. NF-κB signaling in COVID-19 infection is illustrated in Figure 2.
Overall, a robust inflammatory response and cytokine storm are associated with COVID-19-infection-related complications, including multi-organ failure and death. This review aims to summarize the past and current state of knowledge on the role of the NF-κB signaling pathway in the innate immune response and inflammatory diseases with the objective of potential therapeutic use in developing effective treatment options for COVID-19.
2. The Modulatory Role of NF-κB in the Innate Immune Response: Inflammation and ROS Production
Innate immunity and inflammation employ different immune cells, including neutrophils, macrophages, and dendritic cells. Innate immune response activation is mediated by a family of pattern-recognition receptors (PRRs) which can recognize pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) [15]. Different families of PRRs include Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), Nod-like receptors (NLRs), and cytosolic DNA sensors [16]. Activation of the canonical NF-κB pathway is a common downstream signaling cascade of the PRRs and leads to the induction of pro-inflammatory molecules, including cytokines and chemokines. NF-κB proteins are located in the cytosol and remain associated with the inhibitory protein called IkBs. Various NF-κB stimuli include viral or bacterial infections, antigen (e.g., TLRs) activation, cytokines (e.g., TNFα and IL-1), and genotoxicity induced by UV radiation or oxidative stress [1].
Upon the activation by the inducers, the inhibitor of the NF-κB (IκB) proteins becomes phosphorylated and subsequently undergoes ubiquitylation and proteasome degradation. Dissociation of IκB leads to the translocation of NF-κB proteins from the cytosol to the nucleus and induces the transcription of target genes, including cytokines, chemokines, and antimicrobial peptides. The activity of the NF-κB signaling pathway is essential for the survival and activation of lymphocyte cells and the induction of immune responses [17]. In response to infections or injury, an inflammatory immune response is one of the defense mechanisms, and its deregulation of inflammation can lead to the onset of tissue damage or chronic inflammatory diseases. Studies have shown evidence that NF-κB signaling plays pro- and anti-inflammatory roles in inflammation [18].
Another important cellular entity, reactive oxygen species (ROS), is well-documented for its roles in the immune system’s development and the execution of immune responses. NF-κB regulates cellular ROS levels by increasing the expression of antioxidant-encoding genes. Different NF-κB-targeted antioxidants include copper–zinc superoxide dismutase (Cu,Zn-SOD/SOD1), manganese superoxide dismutase (Mn-SOD/SOD2), ferritin heavy chains (FHC), catalase, thioredoxin-1 (Trx1), thioredoxin-2 (Trx2), glutathione-s-transferase (GST), metallothionein-3 (MT3), NADPH dehydrogenase quinone 1 (NQO1), heme oxygenase 1 (HO1), glutathione peroxidase 1 (Gpx1), and dihydrodiol dehydrogenase (DDH1) [19]. Out of these, the superoxide dismutases and ferritin heavy chains are the most common antioxidant employed by NF-κB signaling to regulate ROS levels. In the context of inflammation, NF-κB regulates different enzymes in immune cells. Majorly documented enzymes are NADPH oxidase NOX2, xanthine oxidase/dehydrogenase (XOR), inducible nitric oxide synthase (iNOS), neuronal nitric oxide synthase (nNOS), cyclooxygenase-2 (COX2), and cytochrome p450 (Cyp450).
Regulation of ROS levels by NF-κB signaling is well established, but it is to be noted that cellular ROS also regulate the activity of the NF-κB pathway. ROS include various radical and non-radical reactive molecules and can target the NF-κB signaling pathway at different steps. One of the well-studied mechanisms of NF-κB regulation by ROS is the oxidation of cysteine residue leading to the functional inactivation of protein [20]. Through the oxidation of cysteine residue, ROS can also regulate NF-κB by suppressing its DNA binding ability [21]. ROS also regulate NF-κB signaling through upstream activators. One of the most common non-radical forms of ROS, hydrogen peroxide (H2O2), can differentially affect the upstream regulators of NF-κB signaling, including IkBα, IKK, MEKK1, and NIK [19]. The schematic in Figure 3 depicts the role of ROS in regulating inflammation in the context of the NF-κB signaling pathway. Overall, in the intricate crosstalk between ROS and the NF-κB pathway, the expression of NF-κB-targeted genes affects ROS levels, and ROS regulates the NF-κB signaling pathways. However, the outcome of regulation, whether inhibition or activation of signaling, is highly context dependent. ROS are implicated in the progression of various inflammatory disease conditions.
3. NF-κB in Inflammatory Diseases
Given the role of NF-κB in the induction of cytokines, chemokines, cell adhesion molecules, cyclooxygenases 2, and matrix metalloproteinases and in the development of innate immune cells and inflammatory T cells, the deregulation of NF-κB signaling has been incriminated in the pathogenesis of various inflammatory diseases, including multiple sclerosis, inflammatory bowel disease (IBD), rheumatoid arthritis, lupus erythematosus, and type I diabetes [22,23].
To discuss a few of these, the pathogenesis of multiple sclerosis involves the central nervous system-specific CD4+ T cells, e.g., Th1 and Th17 cells [24]. The genome-wide association studies noise reduction method (GWAS-NR) has identified the NF-κB pathway associated molecules, including RelA, IkBα, IkBz, and MALT1, and Bcl10, as susceptible candidates in multiple sclerosis [25]. IBD involves pathogenic action in intestinal epithelial cells and innate immune cells, including macrophages, neutrophils, and lymphoid cells [26].
Mutations in NF-κB-signaling-associated genes, including IL-12, IL-23, and NOD2, are casually linked with human IBD [27]. Studies on animal models have shown that genetic deficiencies in deubiquitinases CYLD and A20 (negative regulators of NF-κB signaling) induce colonic inflammation [28]. In corroboration of this, NF-κB decoy oligonucleotides targeting the DNA-binding activity of NF-κB proteins improved chemically induced IBD phenotypes in a murine model [29]. These reports are in line with the induction of pro-inflammatory cytokines by NF-κB in innate immune cells and the differentiation of inflammatory T cells.
In addition to the above discussed inflammatory diseases related to the nervous system and gastrointestinal system, NF-κB signaling has been documented to play essential roles in the two inflammatory airway/lung disorders, e.g., chronic obstructive pulmonary diseases (COPD) and asthma [30,31]. Oxidative stress and redox imbalance–induced airway inflammation are common manifestations in both diseases. Both asthmatics and COPD patients exhibit increased NF-κB activation [32,33].
In two allergic asthma models, deletion of TLR2 and TLR4 genes attenuated the NF-κB overreaction, suggesting that increased NF-κB signaling is contributed to by the innate immune system [34,35]. The anti-inflammatory activity of glucocorticoids offers them as treatment options for these diseases. Ligand-bound glucocorticoid receptors bind NF-κB and, subsequently, the expression of target inflammatory genes is inhibited [36]. Understanding the intricate NF-κB signaling pathway and its target genes and processes has led to the identification of different inhibitor molecules for potential therapeutic purposes against inflammatory diseases, including asthma and COPD. The NF-κB pathway upstream kinases, such as IKK1, IKK2, and MEKK3, and the downstream effectors, including IκB ubiquitin E3 ligase, are promising targets for the selective regulation of NF-κB functions [31,37]. In addition, the components of the TNF and IL-1 signaling pathways, e.g., TRADD, TRAF2, TRAF6, and IRAK, as well as PI3K and PKC isoforms, also offer their candidature as potential NF-κB inhibitory targets.
In the context of inflammatory lung disorders/abnormalities, the ongoing coronavirus disease (COVID-19) outbreak is of major concern. The causative pathogen of COVID-19 is severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), and the clinical manifestations of the disease include pulmonary, e.g., acute respiratory distress, respiratory failure, and pneumonia, as well as extrapulmonary, e.g., arrhythmia, acute coronary syndrome, elevated liver enzymes, acute kidney failure, and neuropathy (Johnson et al., 2020/PMID: 32903492). Recurrent clinical occurrences of pulmonary abnormalities and persistent COVID-19 manifestations are well documented in COVID-19 patients [38,39,40,41]. As inflammation is majorly employed in COVID-19-associated multi-organ dysfunction and is well regulated by NF-κB signaling, the following sections discuss NF-κB signaling in the context of cytokine storm, multiple organ failure, comorbidities, and NF-κB signaling modulation as a therapeutic measure for COVID-19.
4. NF-κB Activation in Cytokine Storm Syndrome and Multiple Organ Failure during
Some of the major consequences of severe cases of COVID-19 include cytokine storm syndrome and multiple organ failure. These events are related to complications such as high levels of pro-inflammatory cytokines and chemokines, such as TNF-α, IL-1, IL-6, IL-8, IFN-γ, MCP-1, etc. ‘Cytokine storm’ was originally coined to define certain medical conditions in response to the use of muromonab-CD3 (OKT3) for allograft rejection [42]. Increased levels of pro-inflammatory cytokines can result in a systemic inflammatory response leading to tissue and organ damage and eventually multiple organ failure. A number of infections can cause these systemic immune responses, and it is becoming increasingly evident that severe cases of COVID-19 are one of the leading causes of fatalities in the recent past owing to these complications. The cause and manifestation of cytokine storm can vary depending on the type of infection, and thus the treatment options should be considered accordingly. A clear understanding of the mechanism of cytokine storm in COVID-19 is rapidly evolving, and this can provide useful insights into the development of specific diagnoses and effective treatment strategies.
One of the reasons for the increase in pro-inflammatory cytokines in COVID-19 is the activation of NF-κB by viral spike protein [6]. The most potent manifestation of the cytokine storm can result from the activation of both CD4+ T and effector CD8+ T cells. In addition, other immune cells such as monocytes, macrophages, dendritic cells, and mast cells are also hyperactivated [42]. The spike protein subunit 1 (CoV2-S1) of the SARS-CoV-2 has a strong binding affinity for the ACE2 receptor and is a key inducer of full-blown pro-inflammatory cytokine storm [43]. Once NF-κB is activated by the binding of the spike protein onto the ACE2 receptor, multiple cytokines, chemokines, and signaling intermediates are upregulated, resulting in a self-activating NF-κB pathway loop and leading to hyperimmune responses and fatalities
Depending on the extent of the cytokine storm syndrome, the clinical and laboratory manifestations can range from symptoms such as high-grade fever, anorexia, arthralgia, myalgia, and systemic intravascular coagulation to complex pulmonary symptoms such as cardiomyopathy and acute renal and hepatic failure [42]. In addition, COVID-19 long haulers also show neurological and neuropsychiatric symptoms and neurotoxicity syndrome. Hence, the extent, presentation, and degree of the manifestations of cytokine storm syndrome are directly associated with the extent of multiple organ dysfunction and failure. A detailed description of the clinical features and laboratory findings are illustrated in Figure 4.
5. NF-κB in Crosstalk between COVID-19 and Associated Comorbidities
One of the main reasons for the diagnostic and treatment challenges in COVID-19 is the risk of associated comorbidities. These comorbid conditions include lifestyle factors, such as obesity, diabetes, hypertension, and other diseases, such as cardiovascular/cerebrovascular diseases, chronic renal disease, tuberculosis, and chronic obstructive pulmonary disease (COPD), among others [44,45,46,47,48]. Complete knowledge of the associated risk factors and their role in personalized diagnostics and treatment is now considered necessary. Several groups are now undertaking various strategies such as CORD-19 database analysis [48], pathway analysis [44], and literature-based meta-analysis [46,47] to understand pathway-based associated risk factors in COVID-19.
These studies have underpinned the involvement of NF-κB signaling-associated crosstalk in several comorbid conditions such as cancers, chronic renal disease, and cardiovascular disease [44]. Lifestyle factors such as obesity and insulin resistance are characterized by chronic low-grade inflammation [49]. The non-canonical NF-κB pathway is reported to be involved in β-cell failure in diabetes [50]. A recent report also proposed that some of these comorbid conditions can result from viral–bacterial interactions initiated by bacterial products such as lipopolysaccharide (LPS) [51]. Increased levels of LPS in circulation are reported in obesity and diabetes, and gut dysbiosis is also involved in the pathogenesis of insulin resistance. An interaction between the viral S protein and bacterial LPS can cause pro-inflammatory NF-κB activation in such conditions. Interestingly, blood microbiota and circulating microbial metabolites are also linked to the etiology and progression of cardiovascular disease [52]. Hence, a systematic correlation between COVID-19 progression and pro-inflammatory NF-κB activation with these comorbid conditions can provide novel insights into the severity associated with COVID-19 and also treatment strategies. Efforts are underway to identify shared pathway-based targets for drug repurposing and the treating comorbidities associated with severe COVID-19.
In addition to many cancers, accumulating evidence suggests that sustained activation of the NF-κB pathway is also observed in many neurodegenerative disorders, such as multiple sclerosis (MS) and Alzheimer’s disease [53,54,55]. In fact, severe COVID-19 is associated with many neurological symptoms such as demyelination, seizures, anosmia, ageusia, encephalitis, Guillain-Barré Syndrome (GBS), and long-term cognitive deficits [53]. The term ‘neuro-COVID-19′ was introduced to define the symptoms associated with severe and prolonged COVID-19. Many hospitals inducted neuro-COVID-19 wards during the pandemic to treat and study the neurological symptoms associated with COVID-19. Several patients manifested stroke, delirium, seizures, encephalitis, brain fog, fatigue, and cognitive impairments associated with COVID-19 [53]. The risk association of neurological disorders with the severity of COVID-19 is not yet well cataloged. Pathway and meta-analysis-based approaches are required to identify NF-κB-dependent, shared drug targets to treat COVID-19 long haulers suffering from prolonged neurological symptoms.
6. Therapeutic Use of NF-κB Modulators in COVID-19
Earlier studies suggested that inhibition of NF-κB-mediated inflammation in severe acute respiratory syndrome coronavirus–infected mice increased survival [56]. Severe cases of COVID-19 also have lymphopenia, a condition observed by decreased lymphocyte count, making patients more susceptible to infections [57,58] The abnormal activation of T cells followed by their depletion has been proposed as one of the vital steps in many immunological responses [59]. Furthermore, it is proposed that the SARS-CoV-2 virus may infect T cells, interfere with T-cell expansion, and result in T-cell exhaustion [58,60] NF-κB-induced cellular proptosis is another proposed mechanism of lymphopenia [42]. This may explain the observed lymphopenia in some patients. It is also hypothesized that inflammatory cytokine storm is likely a key factor behind the lymphopenia observed [58]. Since multiple cytokines and chemokines are involved in NF-κB-dependent cytokine storm, a reasonable approach to mitigate severity in some cases of COVID-19 is to prevent NF-κB activation, thereby preventing inflammation and cytokine storm.
One approach is to repurpose antiviral drugs to treat COVID-19 symptoms concurrent with NF-κB activation. Many antiviral drugs that suppress activation of the NF-κB pathway, such as lopinavir and ritonavir, are being repurposed to treat COVID-19 [61,62]. In addition, tocilizumab, which is used to treat rheumatoid arthritis, also suppresses NF-κB and has been repurposed for the treatment of COVID-19 [53]. The NF-κB inhibitor digoxin, an approved drug, has been proposed in one of the pathway-based target identification studies as a promising candidate for COVID-19 patients with comorbid conditions such as diabetes and cancer [44]. Although the therapeutic effects of chloroquine and hydroxychloroquine in the treatment of COVID-19 remain controversial, their immunosuppressive effects are dependent on the reduction of NF-κB [63].
One of the key steps in the activation of NF-κB signaling is the phosphorylation of IKK-β, followed by its degradation. Hence, blocking the phosphorylation of IKKβ is another approach to blocking the NF-κB pathway. Many pharmacological phosphorylation inhibitors of IKK-β, such as vinpocetine, resveratrol, N-acetylcysteine (NAC), or the proposed inhibitor botulinum neurotoxin (BoNT), may be other candidates of interest to prevent NF-κB pathway activation [59].
Common NSAIDs, such as acetylsalicylic acid and aspirin, and a synthetic form of glucocorticoid, dexamethasone, have shown varying degrees of clinical efficacy in patients with COVID-19 [59,64]. A novel proteasome inhibitor, VL-01, has shown inhibition of NF-κB in both virus-induced and LPS-induced cytokine storm and may be a promising therapeutic candidate [64].
In addition, many studies have shown that blocking the NF-κB pathway with antioxidants could be an effective therapeutic approach. These antioxidants include vitamin A, vitamin C, vitamin E, glutathione, and zinc. Furthermore, vitamin D has neuroprotective roles and can provide prophylactic protection for COVID-19 long haulers suffering from neurological symptoms of neuro-COVID-19 [53]. Figure 5 represents a schematic showing the targets of the NF-κB pathway used by some of the inhibitor drugs for the treatment of COVID-19. In a parallel approach, utilization of some natural compounds including propolis [65], astaxanthin [66], and palmitoylethanolamide [67] in regulating NF-κB-dependent immune response during COVID-19 is also being investigated by many laboratories.
7. Conclusions
The ongoing COVID-19 pandemic has caused severe public and community health issues that need to be dealt with effectively to mitigate the risk of COVID-19-associated long-term problems. The severity of the symptoms depends on the degree of inflammation and overactivation of the immune response. The unregulated pro-inflammatory response results in cytokine storm syndrome and multiple organ failure leading to fatalities. NF-κB pathway activation is one of the leading causes of these severe symptoms, and comorbidities increase the risk of these symptoms and causalities several fold. Hence, when designing comprehensive treatment strategies, the NF-κB activation associated with comorbidities should be considered. Several groups have undertaken multiple approaches to effectively formulate and repurpose NF-κB modulators for treating severe symptoms of COVID-19, and future studies are warranted to expand the repertoire of these modulators.
Author Contributions
Conceptualization, A.P. and A.K.M.; writing—original draft preparation, A.P. and A.K.M.; writing—review and editing, A.P. and A.K.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to acknowledge all those researchers whose work could not be cited in the present article due to space limitations. The study utilized Google scholar and PubMed search engines for literature mining.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB Family of Transcription Factors and Its Regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C.; Chang, J.-H.; Jin, J. Regulation of nuclear factor-kappaB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; on behalf of theHLH Across Speciality Collaboration, UK. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Wang, W.; Ye, L.; Ye, L.; Li, B.; Gao, B.; Zeng, Y.; Kong, L.; Fang, X.; Zheng, H.; Wu, Z.; et al. Up-regulation of IL-6 and TNF-alphaα induced by SARS-coronavirus spike protein in murine macrophages via NF-kappaκB pathway. Virus Res. 2007, 128, 1–8. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus—Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in Innate Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Verma, I.M. NF-kappaκB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The Nuclear Factor NF-kappa B Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-kappaκB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, C.; Carroll, K.S. Orchestrating Redox Signaling Networks through Regulatory Cysteine Switches. ACS Chem. Biol. 2010, 5, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledano, M.B.; Leonard, W.J. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 4328–4332. [Google Scholar] [CrossRef] [Green Version]
- Pai, S.; Thomas, R. Immune deficiency or hyperactivity-Nf-kappaκb illuminates autoimmunity. J. Autoimmun. 2008, 31, 245–251. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaκB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussman, J.; Beecham, A.H.; Schmidt, M.; Martin, E.R.; McCauley, J.L.; Vance, J.; Haines, J.L.; Pericak-Vance, M.A. GWAS analysis implicates NF-κB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun. 2016, 17, 305–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, K.L.; Zheng, L.B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Cho, J.H. Inflammatory Bowel Disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stirling, B.; Temmerman, S.T.; Ma, C.A.; Fuss, I.J.; Derry, J.M.J.; Jain, A. Impaired regulation of NF-κB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J. Clin. Investig. 2006, 116, 3042–3049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fichtner-Feigl, S.; Fuss, I.J.; Preiß, J.; Strober, W.; Kitani, A. Treatment of murine Th1- and Th2-mediated inflammatory bowel disease with NF-kappa B decoy oligonucleotides. J. Clin. Investig. 2005, 115, 3057–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.R.; Bartlett, N.W.; Clarke, D.; Birrell, M.; Belvisi, M.; Johnston, S.L. Targeting the NF-κB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol. Ther. 2009, 121, 1–13. [Google Scholar] [CrossRef]
- Schuliga, M. NF-kappaB Signaling in Chronic Inflammatory Airway Disease. Biomolecules 2015, 5, 1266–1283. [Google Scholar] [CrossRef] [PubMed]
- Caramori, G.; Romagnoli, M.; Casolari, P.; Bellettato, C.; Casoni, G.L.; Boschetto, P.; Chung, K.F.; Barnes, P.J.; Adcock, I.; Ciaccia, A.; et al. Nuclear localisation of p65 in sputum macrophages but not in sputum neutrophils during COPD exacerbations. Thorax 2003, 58, 348–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliardo, R.; Chanez, P.; Mathieu, M.; Bruno, A.; Costanzo, G.; Gougat, C.; Vachier, I.; Bousquet, J.; Bonsignore, G.; Vignola, A.M. Persistent Activation of Nuclear Factor–κB Signaling Pathway in Severe Uncontrolled Asthma. Am. J. Respir. Crit. Care Med. 2003, 168, 1190–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, D.; Ng, N.; Lee, S.; Batzer, G.; Horner, A.A. Airway House Dust Extract Exposures Modify Allergen-Induced Airway Hypersensitivity Responses by TLR4-Dependent and Independent Pathways. J. Immunol. 2008, 181, 2925–2932. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, Q.; Chu, C.; You, H.; Jin, M.; Zhao, X.; Zhu, X.; Zhou, W.; Ji, W. Ovalbumin-induced experimental allergic asthma is Toll-like receptor 2 dependent. Allergy Asthma Proc. 2014, 35, e15–e20. [Google Scholar] [CrossRef]
- Barnes, P.J. Anti-inflammatory Actions of Glucocorticoids: Molecular Mechanisms. Clin. Sci. 1998, 94, 557–572. [Google Scholar] [CrossRef] [Green Version]
- Swinney, D.C.; Xu, Y.-Z.; Scarafia, L.E.; Lee, I.; Mak, A.Y.; Gan, Q.-F.; Ramesha, C.S.; Mulkins, M.A.; Dunn, J.; So, O.-Y.; et al. A Small Molecule Ubiquitination Inhibitor Blocks NF-κB-dependent Cytokine Expression in Cells and Rats. J. Biol. Chem. 2002, 277, 23573–23581. [Google Scholar] [CrossRef] [Green Version]
- Caruso, D.; Guido, G.; Zerunian, M.; Polidori, T.; Lucertini, E.; Pucciarelli, F.; Polici, M.; Rucci, C.; Bracci, B.; Nicolai, M.; et al. Post-Acute Sequelae of COVID-19 Pneumonia: Six-month Chest CT Follow-up. Radiology 2021, 301, E396–E405. [Google Scholar] [CrossRef]
- Myall, K.J.; Mukherjee, B.; Castanheira, A.M.; Lam, J.L.; Benedetti, G.; Mak, S.M.; Preston, R.; Thillai, M.; Dewar, A.; Molyneaux, P.L.; et al. Persistent Post–COVID-19 Interstitial Lung Disease. An Observational Study of Corticosteroid Treatment. Ann. Am. Thorac. Soc. 2021, 18, 799–806. [Google Scholar] [CrossRef]
- Sahanic, S.; Tymoszuk, P.; Ausserhofer, D.; Rass, V.; Pizzini, A.; Nordmeyer, G.; Hüfner, K.; Kurz, K.; Weber, P.M.; Sonnweber, T.; et al. Phenotyping of Acute and Persistent Coronavirus Disease 2019 Features in the Outpatient Setting: Exploratory Analysis of an International Cross-sectional Online Survey. Clin. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Sonnweber, T.; Tymoszuk, P.; Sahanic, S.; Boehm, A.; Pizzini, A.; Luger, A.; Schwabl, C.; Nairz, M.; Grubwieser, P.; Kurz, K.; et al. Investigating phenotypes of pulmonary COVID-19 recovery: A longitudinal observational prospective multicenter trial. eLife 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Attiq, A.; Yao, L.J.; Afzal, S.; Khan, M.A. The triumvirate of NF-κB, inflammation and cytokine storm in COVID-19. Int. Immunopharmacol. 2021, 101, 108255. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Aljabali, A.A.; Tambuwala, M.M.; Tiwari, S.; Serrano-Aroca, A.; Alzahrani, K.J.; Silva Andrade, B.S.; Azevedo, V.; Ganguly, N.K.; Lundstrom, K. Predicting COVID-19—Comorbidity Pathway Crosstalk-Based Targets and Drugs: Towards Personalized COVID-19 Management. Biomedicines 2021, 9, 556. [Google Scholar] [CrossRef]
- Guan, W.-J.; Liang, W.-H.; Zhao, Y.; Liang, H.-R.; Chen, Z.-S.; Li, Y.-M.; Liu, X.-Q.; Chen, R.-C.; Tang, C.-L.; Wang, T.; et al. Comorbidity and its impact on 1590 patients with COVID-19 in China: A nationwide analysis. Eur. Respir. J. 2020, 55, 2000547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Wang, Y. The Clinical Characteristics and Risk Factors of Severe COVID-19. Gerontology 2021, 67, 255–266. [Google Scholar] [CrossRef]
- Sanyaolu, A.; Okorie, C.; Marinkovic, A.; Patidar, R.; Younis, K.; Desai, P.; Hosein, Z.; Padda, I.; Mangat, J.; Altaf, M. Comorbidity and its Impact on Patients with COVID-19. SN Compr. Clin. Med. 2020, 2, 1069–1076. [Google Scholar] [CrossRef]
- Wolff, D.; Nee, S.; Hickey, N.S.; Marschollek, M. Risk factors for Covid-19 severity and fatality: A structured literature review. Infection 2021, 49, 15–28. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Meyerovich, K.; Ortis, F.; Cardozo, A.K. The non-canonical NF-κB pathway and its contribution to β-cell failure in diabetes. J. Mol. Endocrinol. 2018, 61, F1–F6. [Google Scholar] [CrossRef] [Green Version]
- Kruglikov, I.L.; Scherer, P.E. Preexisting and inducible endotoxemia as crucial contributors to the severity of COVID-19 outcomes. PLoS Pathog. 2021, 17, e1009306. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, G.; Dinakaran, V.; Rajendhran, J.; Swaminathan, K. Blood Microbiota and Circulating Microbial Metabolites in Diabetes and Cardiovascular Disease. Trends Endocrinol. Metab. 2020, 31, 835–847. [Google Scholar] [CrossRef]
- Davies, D.A.; Adlimoghaddam, A.; Albensi, B.C. The Effect of COVID-19 on NF-κB and Neurological Manifestations of Disease. Mol. Neurobiol. 2021, 58, 4178–4187. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Prescott, J.A.; Mitchell, J.P.; Cook, S.J. Inhibitory feedback control of NF-κB signalling in health and disease. Biochem. J. 2021, 478, 2619–2664. [Google Scholar] [CrossRef] [PubMed]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-Mediated Inflammation in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice Increases Survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, I.; Pranata, R. Lymphopenia in severe coronavirus disease-2019 (COVID-19): Systematic review and meta-analysis. J. Intensive Care 2020, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Tavakolpour, S.; Rakhshandehroo, T.; Wei, E.X.; Rashidian, M. Lymphopenia during the COVID-19 infection: What it shows and what can be learned. Immunol. Lett. 2020, 225, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, M. NF-κB signalling as a pharmacological target in COVID-19: Potential roles for IKKβ inhibitors. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 2021, 394, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef] [PubMed]
- Dewan, Z.; Tomita, M.; Katano, H.; Yamamoto, N.; Ahmed, S.; Yamamoto, M.; Sata, T.; Mori, N.; Yamamoto, N. An HIV protease inhibitor, ritonavir targets the nuclear factor-kappaB and inhibits the tumor growth and infiltration of EBV-positive lymphoblastoid B cells. Int. J. Cancer 2009, 124, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Kariya, R.; Taura, M.; Suzu, S.; Kai, H.; Katano, H.; Okada, S. HIV protease inhibitor Lopinavir induces apoptosis of primary effusion lymphoma cells via suppression of NF-κB pathway. Cancer Lett. 2014, 342, 52–59. [Google Scholar] [CrossRef]
- Liang, N.; Zhong, Y.; Zhou, J.; Liu, B.; Lu, R.; Guan, Y.; Wang, Q.; Liang, C.; He, Y.; Zhou, Y.; et al. Immunosuppressive effects of hydroxychloroquine and artemisinin combination therapy via the nuclear factor-κB signaling pathway in lupus nephritis mice. Exp. Ther. Med. 2018, 15, 2436–2442. [Google Scholar] [CrossRef] [Green Version]
- Kircheis, R.; Haasbach, E.; Lueftenegger, D.; Heyken, W.T.; Ocker, M.; Planz, O. NF-κB Pathway as a Potential Target for Treatment of Critical Stage COVID-19 Patients. Front. Immunol. 2020, 11, 598444. [Google Scholar] [CrossRef]
- Berretta, A.A.; Silveira, M.A.D.; Capcha, J.M.C.; De Jong, D. Propolis and its potential against SARS-CoV-2 infection mechanisms and COVID-19 disease: Running title: Propolis against SARS-CoV-2 infection and COVID-19. Biomed. Pharmacother. 2020, 131, 110622. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, J.; Bhadra, B.; Dattaroy, T.; Nagle, V.; Dasgupta, S. Potential of natural astaxanthin in alleviating the risk of cytokine storm in COVID-19. Biomed. Pharmacother. 2020, 132, 110886. [Google Scholar] [CrossRef] [PubMed]
- Peritore, A.; D’Amico, R.; Siracusa, R.; Cordaro, M.; Fusco, R.; Gugliandolo, E.; Genovese, T.; Crupi, R.; Di Paola, R.; Cuzzocrea, S.; et al. Management of Acute Lung Injury: Palmitoylethanolamide as a New Approach. Int. J. Mol. Sci. 2021, 22, 5533. [Google Scholar] [CrossRef]
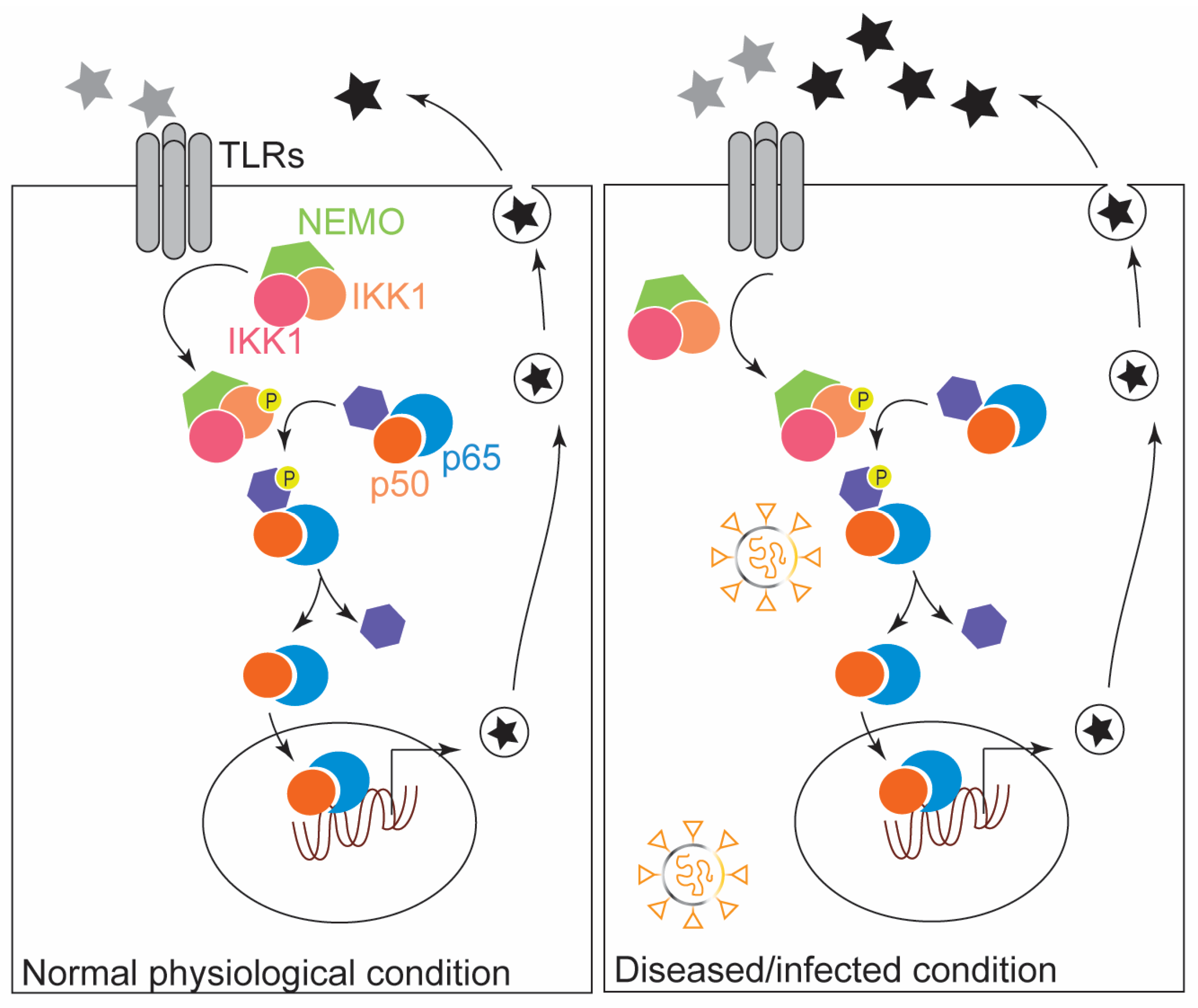
Figure 1.
NF-κB signaling during normal development and pathological infection is shown. In response to stimuli (grey stars), cytokines and chemokines are produced (black stars). The production of these cytokines and chemokines is elevated during the infection, often resulting in cytokine storm syndrome during sever infections such as COVID-19.
Figure 1.
NF-κB signaling during normal development and pathological infection is shown. In response to stimuli (grey stars), cytokines and chemokines are produced (black stars). The production of these cytokines and chemokines is elevated during the infection, often resulting in cytokine storm syndrome during sever infections such as COVID-19.

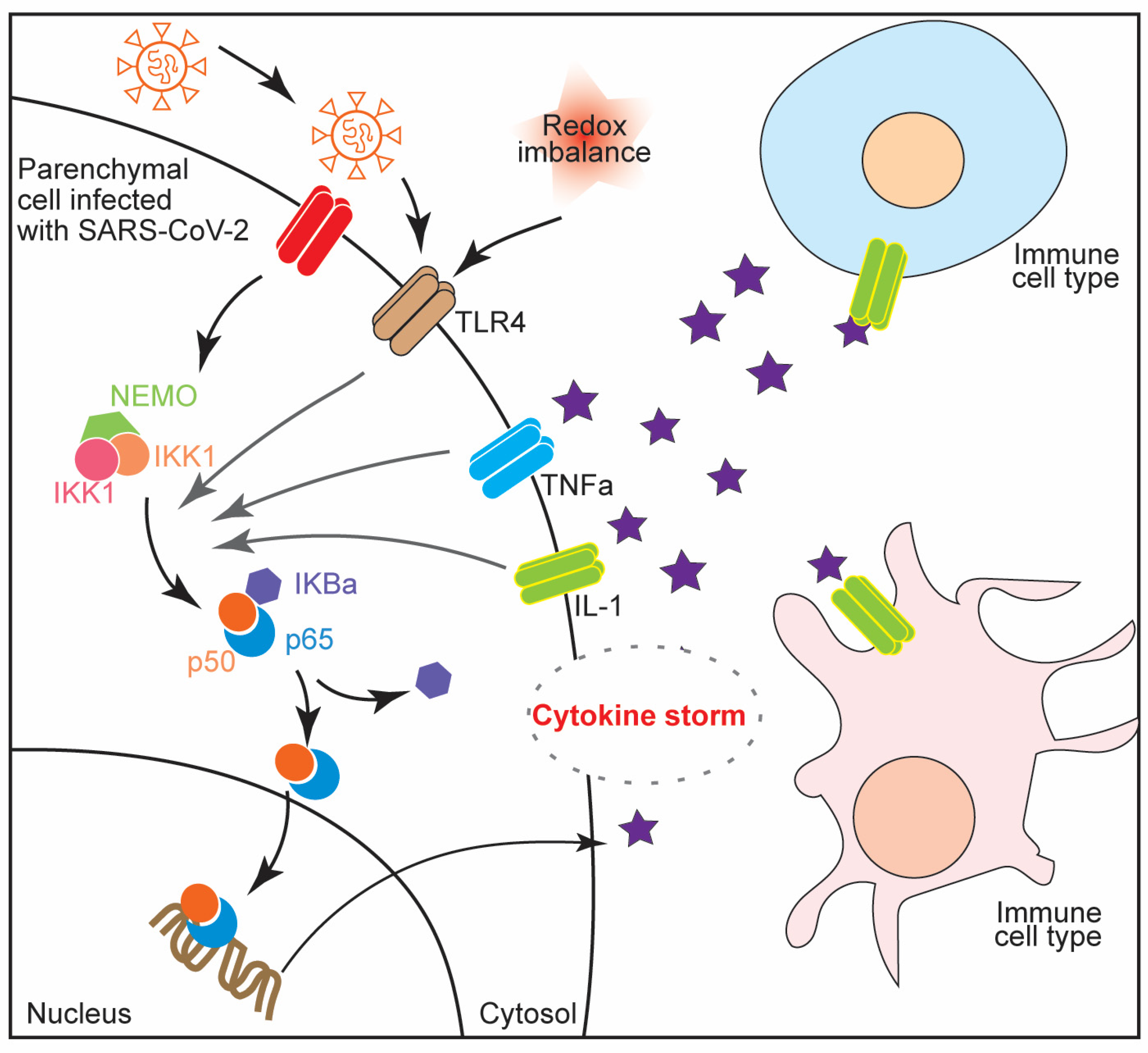
Figure 2.
NF-κB signaling in COVID-19 infection is illustrated. In response to viral infections, the downstream signaling results in amplified cytokine response, activating different immune cell types such as T cells and dendritic cells. This hyperactivation of immune response causes cytokine storm syndrome.
Figure 2.
NF-κB signaling in COVID-19 infection is illustrated. In response to viral infections, the downstream signaling results in amplified cytokine response, activating different immune cell types such as T cells and dendritic cells. This hyperactivation of immune response causes cytokine storm syndrome.

Figure 3.
Activation of NF-κB signaling in response to stimuli such as infections, antigen receptors, cytokines, or oxidative stress and their downstream cellular responses are highlighted.
Figure 3.
Activation of NF-κB signaling in response to stimuli such as infections, antigen receptors, cytokines, or oxidative stress and their downstream cellular responses are highlighted.

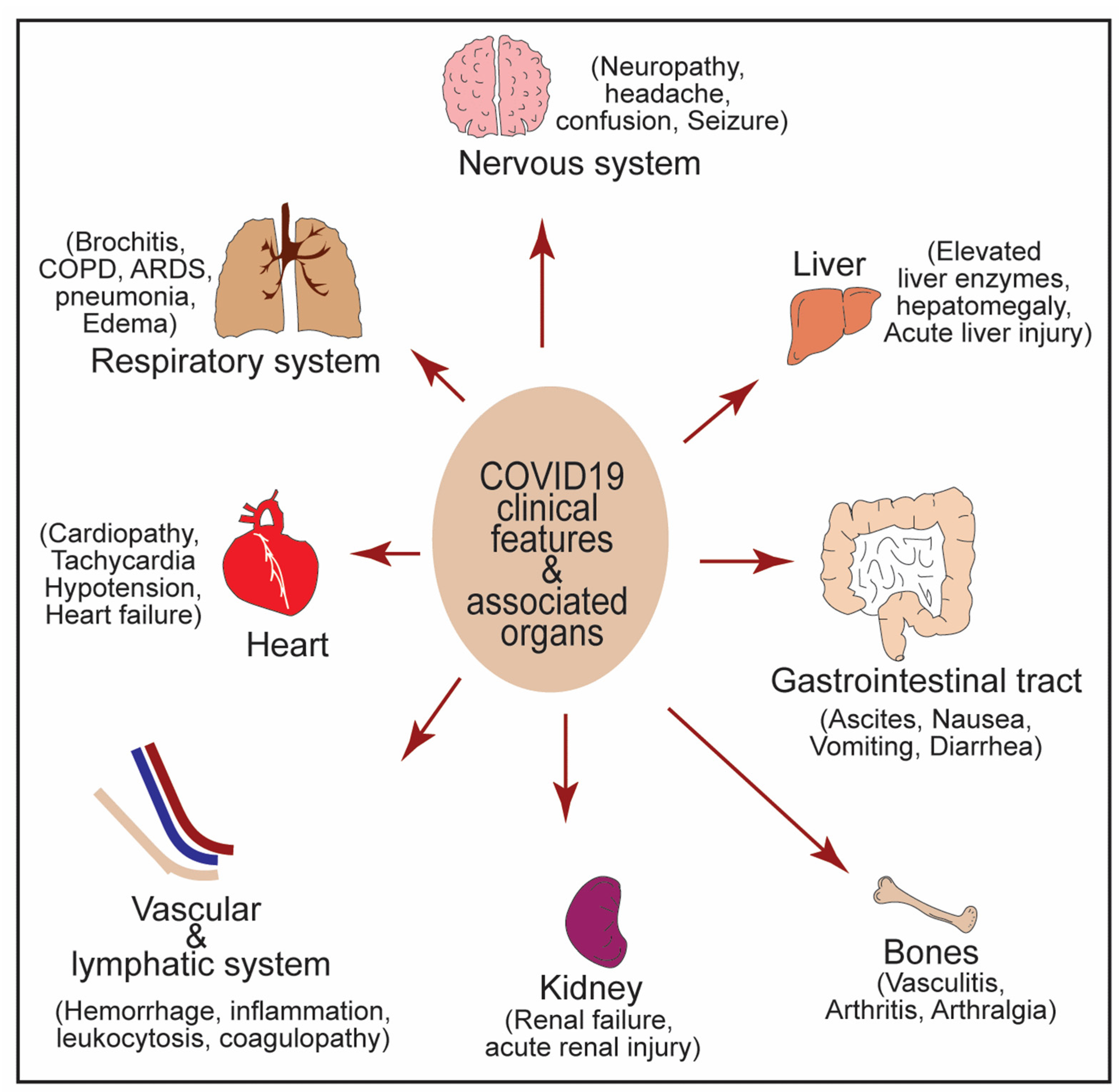
Figure 4.
Clinical manifestations associated with COVID-19 infection in multiple organs are highlighted.
Figure 4.
Clinical manifestations associated with COVID-19 infection in multiple organs are highlighted.

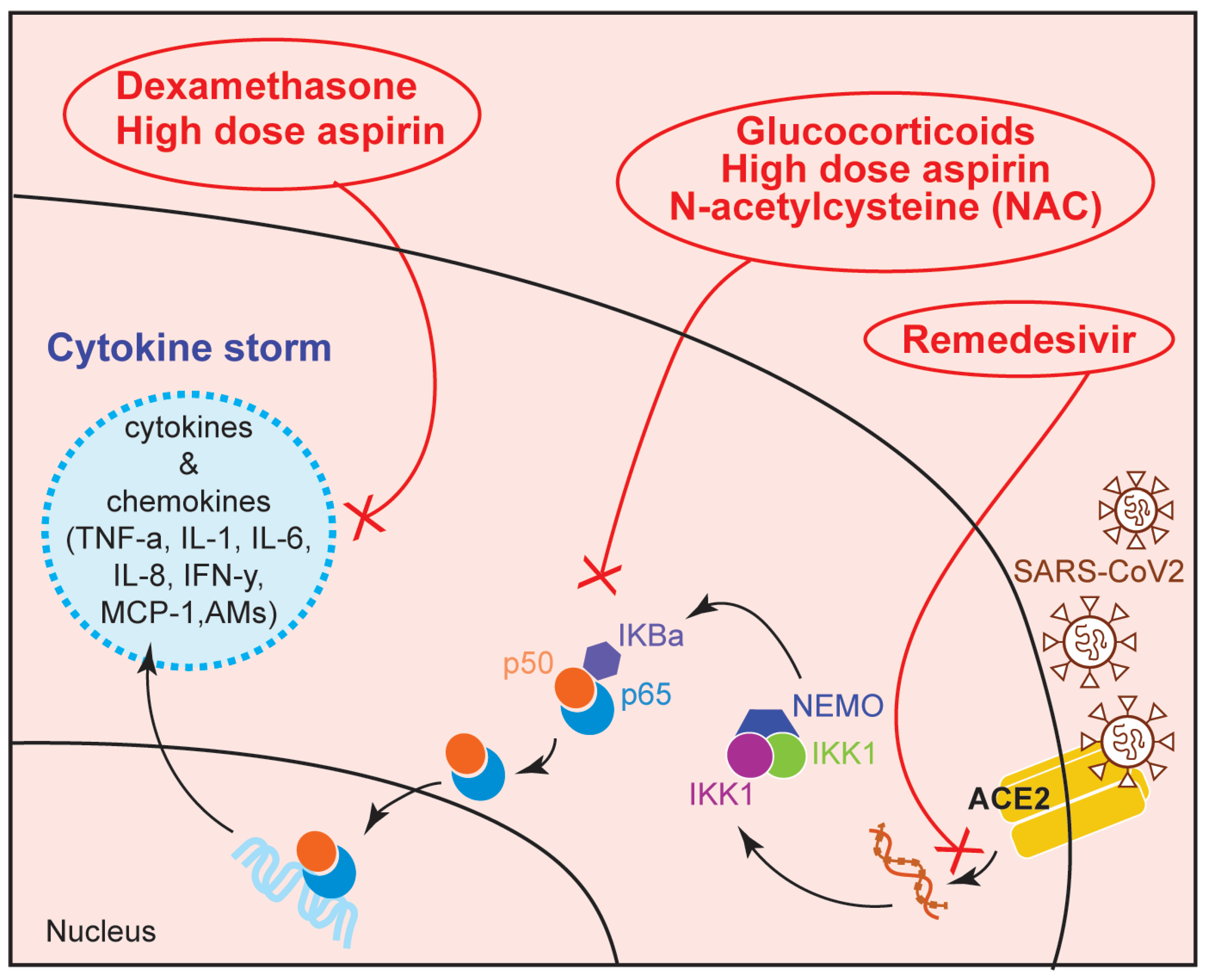
Figure 5.
Schematic diagram showing the NF-κB-associated targets of potential COVID-19 drugs. AMs (Adhesion molecules).
Figure 5.
Schematic diagram showing the NF-κB-associated targets of potential COVID-19 drugs. AMs (Adhesion molecules).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pandey, A.; Mishra, A.K. From Innate Immunity to Inflammation: A Primer on Multiple Facets of NF-κB Signaling in COVID-19. Physiologia 2022, 2, 34-45. https://doi.org/10.3390/physiologia2020004
AMA Style
Pandey A, Mishra AK. From Innate Immunity to Inflammation: A Primer on Multiple Facets of NF-κB Signaling in COVID-19. Physiologia. 2022; 2(2):34-45. https://doi.org/10.3390/physiologia2020004
Chicago/Turabian StylePandey, Ashutosh, and Abhinava K. Mishra. 2022. "From Innate Immunity to Inflammation: A Primer on Multiple Facets of NF-κB Signaling in COVID-19" Physiologia 2, no. 2: 34-45. https://doi.org/10.3390/physiologia2020004