
Computational Screening of Phenylamino-Phenoxy-Quinoline Derivatives against the Main Protease of SARS-CoV-2 Using Molecular Docking and the ONIOM Method

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Pharmacokinetics Study
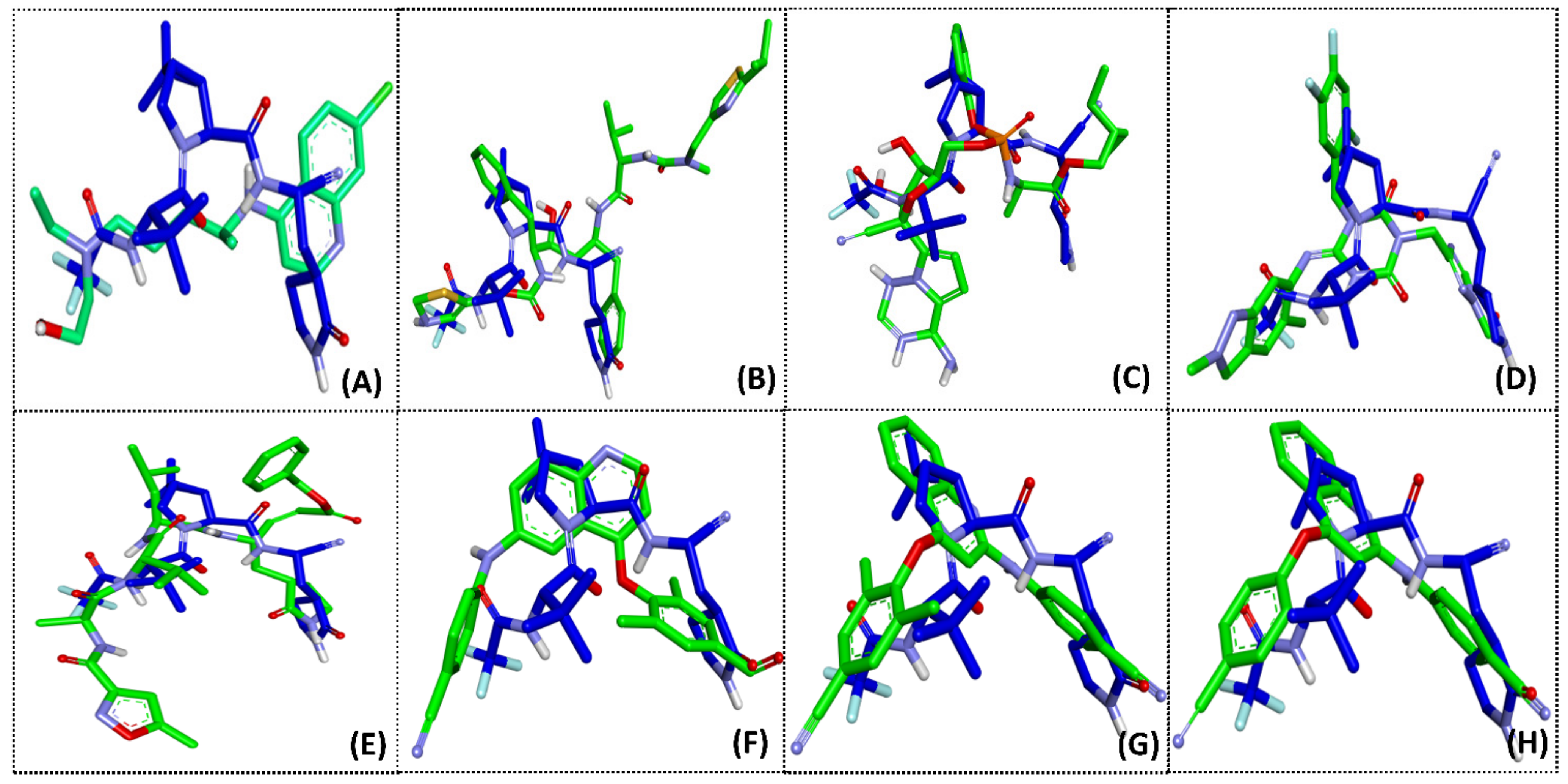
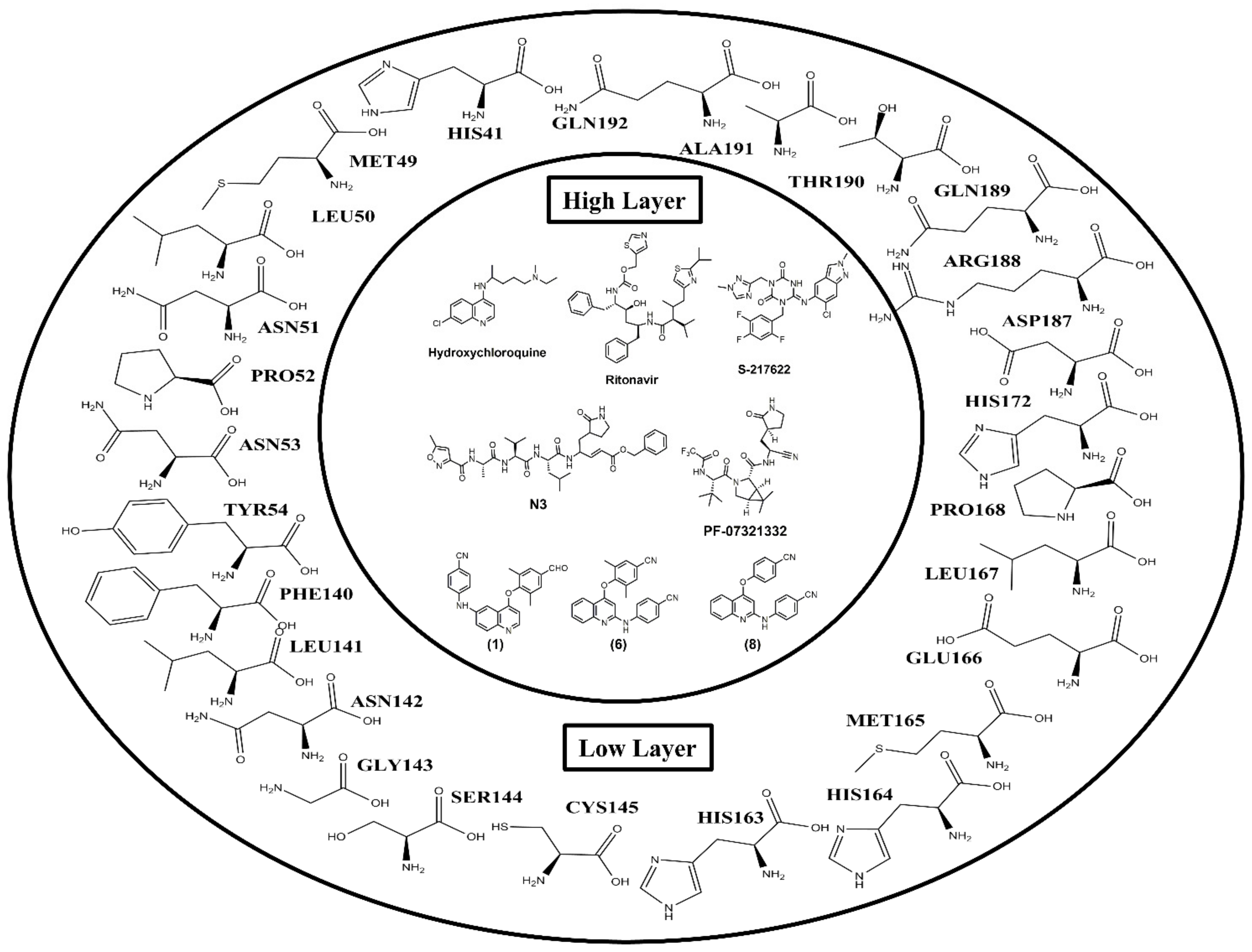
2.2. Molecular Docking
2.3. ONIOM Study
3. Materials and Methods
3.1. Pharmacokinetics Study
3.2. Ligand and Protein Structure Preparation
3.3. Molecular Docking
3.4. ONIOM Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Liu, Y.; Liang, C.; Xin, L.; Xie, X.; Zhang, D.; Wan, M.; Li, H.; Fu, X.; Liu, H.; et al. An update review of emerging small-molecule therapeutic options for COVID-19. Biomed. Pharmacother. 2021, 137, 111313–111329. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yu, X.; Kuo, C.; Min, J.; Chen, S.; Ma, L.; Liu, K.; Guo, R. Overview of antiviral drug candidates targeting coronaviral 3C-Like main proteases. FEBS J. 2021, 288, 5089–5121. [Google Scholar] [CrossRef]
- Cheng, V.; Lau, S.; Woo, P.; Yuen, K. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007, 20, 660–694. [Google Scholar] [CrossRef] [Green Version]
- Zumla, A.; Chan, J.; Azhar, E.; Hui, D.; Yuen, K. Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, B.; Batool, M.; Ain, Q.; Kim, M.; Choi, S. Exploring the binding mechanism of PF-07321332 SARS-Cov-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int. J. Mol. Sci. 2021, 22, 9124–9136. [Google Scholar] [CrossRef]
- Ji, W.; Wang, W.; Zhao, X.; Zai, J.; Li, X. Cross-species transmission of the newly identified coronavirus 2019-nCoV. J. Med. Virol. 2020, 92, 433–440. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Singh, A.-K.; Singh, A.; Singh, R.; Misra, A. Hydroxychloroquine in patients with COVID-19: A Systematic Review and meta-analysis. Diabetes Metab. Syndr. 2020, 14, 589–596. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schafer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef] [Green Version]
- Naik, V.R.; Munikumar, M.; Ramakrishna, U.; Srujana, M.; Gouda, G.; Naresh, P.; Kumar, B.N.; Hemalatha, R. Remdesivir (GS-5734) as a therapeutic option of 2019-nCOV main protease—In silico approach. J. Biomol. Struct. Dyn. 2020, 39, 4701–4714. [Google Scholar] [CrossRef]
- Eastman, R.T.; Jacob, S.R.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef]
- Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.; Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.; et al. Discovery of S-217622, a non-covalent oral SARS-CoV-2 3cl protease inhibitor clinical candidate for treating COVID-19. bioRxiv 2022. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Arafet, K.; Serrano-Aparicio, N.; Lodola, A.; Mulholland, A.; González, F.; Świderek, K.; Moliner, V. mechanism of inhibition of sars-cov-2 mpro by N3 peptidyl michael acceptor explained by qm/mm simulations and design of new derivatives with tun-able chemical reactivity. Chem. Sci. 2021, 12, 1433–1444. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Evaluation of green tea polyphenols as novel corona virus (SARS CoV-2) main protease (Mpro) inhibitors an in-silico docking and molecular dynamics simulation study. J. Biomol. Struct. Dyn. 2020, 39, 4362–4374. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M pro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 24, 1586–1593. [Google Scholar] [CrossRef]
- Pavan, M.; Bolcato, G.; Bassani, D.; Sturlese, M.; Moro, S. Supervised molecular dynamics (sumd) insights into the mechanism of action of SARS-CoV-2 main protease inhibitor PF-07321332. J. Enzym. Inhib. Med. Chem. 2021, 36, 1646–1650. [Google Scholar] [CrossRef]
- Abdelnabi, R.; Foo, C.S.; Jochmans, D.; Vangeel, L.; De Jonghe, S.; Augustijns, P.; Mols, R.; Weynand, B.; Wattanakul, T.; Hoglund, R.M.; et al. The oral protease inhibitor (PF-07321332) protects Syrian hamsters against infection with SARS-CoV-2 variants of concern. Nat Commun. 2022, 13, 719. [Google Scholar] [CrossRef]
- COVID-19: EMA Recommends Conditional Marketing Authorisation for Paxlovid. Available online: www.ema.europa.eu/en/news/covid-19-ema-recommends-conditional-marketing-authorisation-paxlovid (accessed on 16 February 2022).
- Makarasen, A.; Kuno, M.; Patnin, S.; Reukngam, N.; Khlaychan, P.; Deeyohe, S.; Intachote, P.; Saimanee, B.; Sengsai, S.; Boonsri, P.; et al. Molecular Docking Studies and Synthesis of Amino-oxydiarylquinoline Derivatives as Potent Non-nucleoside HIV-1 Reverse Transcriptase Inhibitors. Drug Res. 2019, 69, 671–682. [Google Scholar]
- Makarasen, A.; Patnin, S.; Vijitphan, P.; Reukngam, N.; Khlaychan, P.; Kuno, M.; Intachote, P.; Saimanee, B.; Sengsai, S.; Techasakul, S. Structural basis of 2-phenylamino-4-phenoxyquinoline derivatives as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Molecules 2022, 27, 461–481. [Google Scholar] [CrossRef]
- Patnin, S.; Makarasen, A.; Kuno, M.; Deeyohe, S.; Techasakul, S.; Chaivisuthangkura, A. Binding interaction of potent HIV-1 NNRTIs, amino-oxy-diarylquinoline with the transport protein using spectroscopic and molecular docking. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2020, 233, 118159. [Google Scholar] [CrossRef]
- Boonsri, P.; Kuno, M.; Hannongbua, S. Key interactions of the mutant HIV-1 reverse transcriptase/efavirenz: An evidence obtained from ONIOM method. Med. Chem. Commun. 2011, 2, 1181–1187. [Google Scholar] [CrossRef]
- Samanta, P.N.; Das, K.K. Inhibition activities of catechol diether based non-nucleoside inhibitors against the HIV reverse transcriptase variants: Insights from molecular docking and ONIOM calculations. J. Mol. Graph. Model. 2017, 75, 294–305. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharm. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond: In Structural Chemistry and Biology. In International Union of Crystallography; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Ghosh, S.; Chopra, P.; Wategaonkar, S. C–H⋯S interaction exhibits all the characteristics of conventional hydrogen bonds. Phys. Chem. Chem. Phys. 2020, 22, 17482–17493. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef]
- Jin, Z.; Zhao, Y.; Sun, Y.; Zhang, B.; Wang, H.; Wu, Y.; Zhu, Y.; Zhu, C.; Hu, T.; Du, X.; et al. Structural basis for the inhibition of SARS-CoV-2 main protease by antineoplastic drug carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. [Google Scholar] [CrossRef]
- Zhou, J.; Fang, L.; Yang, Z.; Xu, S.; Lv, M.; Sun, Z.; Chen, J.; Wang, D.; Gao, J.; Xiao, S. Identification of novel proteolytically inactive mutations in coronavirus 3C-like protease using a combined approach. FASEB J. 2019, 33, 14575–14587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of Two Coronavirus Main Proteases: Implications for Substrate Binding and Antiviral Drug Design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, B.; Goyal, D. Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy. ACS Comb. Sci. 2020, 22, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-C.; Chang, G.-G.; Chou, C.-Y. Mutation of Glu-166 Blocks the Substrate-Induced Dimerization of SARS Coronavirus Main Protease. Biophys. J. 2010, 98, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDockTools4. AutoDock4 Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]





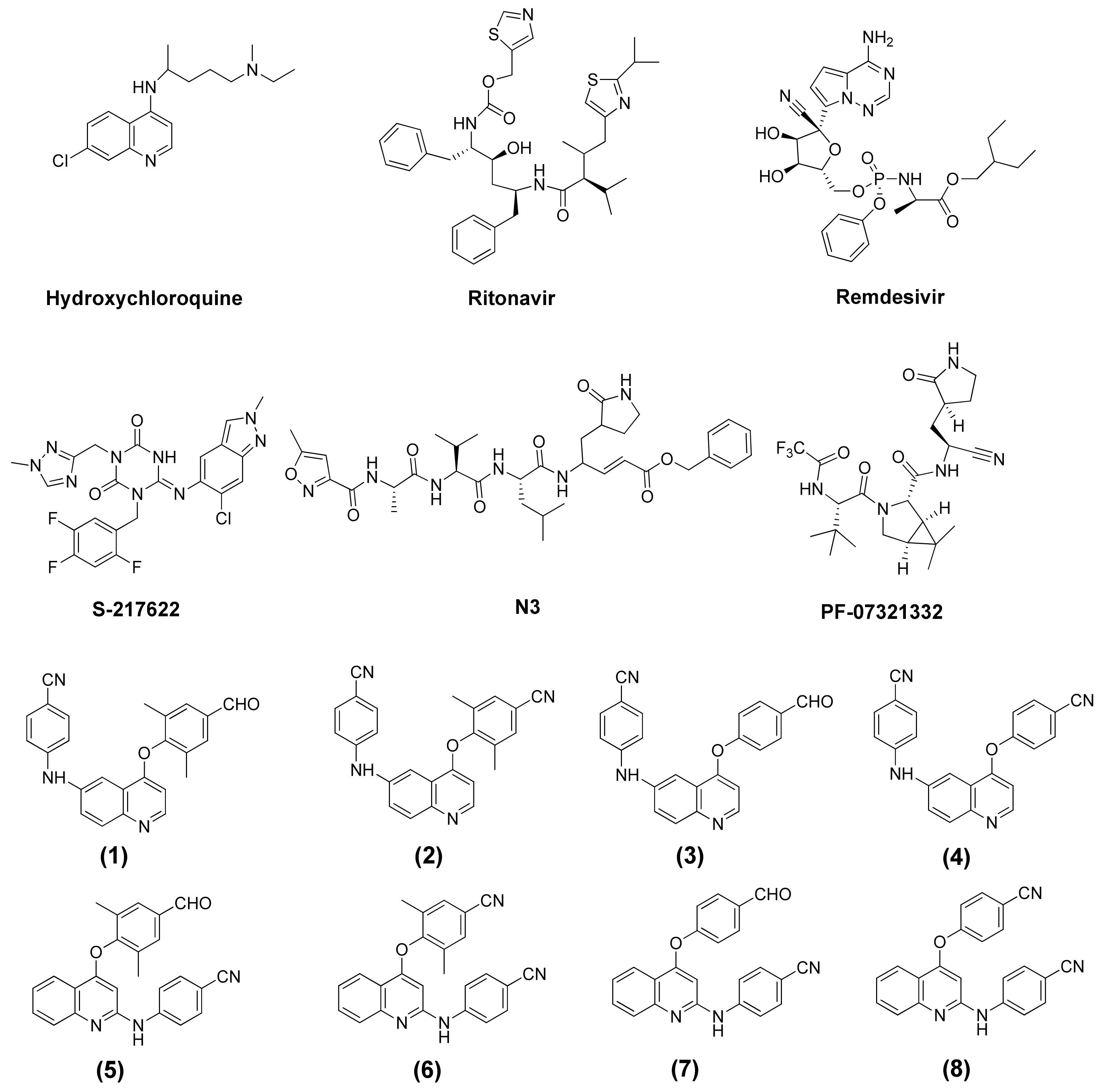{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Number of H-Bond Acceptors | Number of H-Bond Donors | LogP | Number of Rotatable Bonds | Molecular Weight (g/mol) | TPSA (Å2) |
|---|---|---|---|---|---|---|
| Hydroxychloroquine | 3 | 2 | 2.35 | 9 | 335.87 | 48.39 |
| Ritonavir | 7 | 4 | 1.80 | 22 | 720.94 | 202.26 |
| Remdesivir | 12 | 4 | 0.18 | 14 | 602.58 | 213.36 |
| S-217622 | 9 | 1 | 3.70 | 5 | 531.88 | 120.68 |
| N3 | 9 | 5 | 0.38 | 22 | 680.79 | 197.83 |
| PF-07321332 | 8 | 3 | 0.41 | 11 | 499.53 | 131.40 |
| (1) | 4 | 1 | 2.77 | 5 | 393.44 | 75.01 |
| (2) | 4 | 1 | 2.77 | 5 | 390.52 | 81.73 |
| (3) | 4 | 1 | 2.35 | 5 | 365.14 | 75.01 |
| (4) | 4 | 1 | 2.35 | 4 | 362.65 | 81.73 |
| (5) | 4 | 1 | 3.17 | 5 | 393.42 | 75.01 |
| (6) | 4 | 1 | 3.17 | 4 | 390.44 | 81.73 |
| (7) | 4 | 1 | 2.75 | 5 | 365.29 | 75.01 |
| (8) | 4 | 1 | 2.75 | 4 | 362.38 | 81.73 |
| Ligand | Binding Energy (kcal/mol) | H-Bond | Pi-Pi Stacking | ||
|---|---|---|---|---|---|
| a | b | c | |||
| Hydroxychloroquine | −7.06 ± 0.11 | LEU141 GLY143 CYS145 HIS164 | ASN142 | - | HIS41 |
| Ritonavir | −8.56 ± 0.25 | ASN142 GLY143 CYS145 GLN189 | GLU166 MET165 | HIS163 | - |
| Remdesivir | −8.63 ± 0.25 | GLY143 CYS145 LEU167 | ASN142 GLU166 | - | HIS41 |
| S-217622 | −9.62 ± 0.08 | TYR54 CYS145 GLU166 GLN189 | PHE140 | MET165 | HIS41 |
| N3 | −9.44 ± 0.16 | PHE140 GLY143 GLU166 | HIS41 SER144 | - | - |
| PF-07321332 | −10.61 ± 0.12 | PHE140 SER144 CYS145 HIS163 HIS164 GLU166 THR190 | GLN189 | - | - |
| (1) | −10.12 ± 0.01 | HIS164 GLN192 | MET49 PRO52 PRO168 | GLU166 | HIS41 |
| (2) | −9.75 ± 0.08 | HIS164 GLN192 | MET49 PRO52 | GLU166 | HIS41 |
| (3) | −9.67 ± 0.04 | HIS164 GLN192 | MET49 PRO52 THR190 | GLU166 | HIS41 |
| (4) | −9.65 ± 0.03 | HIS164 GLN192 | MET49 PRO52 | - | HIS41 |
| (5) | −9.72 ± 0.04 | GLN189 GLN192 | ASP187 | GLU166 | HIS41 |
| (6) | −10.02 ± 0.01 | GLN189 GLN192 | MET49 ALA191 | GLU166 | HIS41 |
| (7) | −9.78 ± 0.05 | HIS164 GLN192 | MET49 PRO52 PRO168 | GLU166 | HIS41 |
| (8) | −9.97 ± 0.02 | CYS145 HIS164 GLN192 | MET49 PRO52 | GLU166 | HIS41 |
| Amino Acid Residue | Hydroxychloroquine | Ritonavir | Remdesivir | S-217622 | N3 | PF-07321332 | (1) | (6) | (8) |
|---|---|---|---|---|---|---|---|---|---|
| HIS41 | 4.208 | 3.954 | 4.158 | 4.457 | 3.412 | 2.295 | 4.178 | 3.897 | 3.758 |
| MET49 | - | - | - | - | - | - | 2.256 | 2.225 | 2.185 |
| PRO52 | - | - | - | - | - | - | 2.694 | - | 2.806 |
| PHE140 | - | - | - | 3.445 | 2.815 | 2.489 | - | - | - |
| LEU141 | 2.015 | - | - | - | - | - | - | - | |
| ASN142 | 3.421 | 1.899 | 2.892 | - | - | - | - | - | |
| GLY143 | 2.915 | 2.454 | 2.914 | - | 2.203 | - | - | - | - |
| SER144 | 2.036 | - | - | - | - | 2.725 | - | - | - |
| CYS145 | 2.614 | 2.701 | 3.725 | 3.170 | - | 2.245 | - | - | 3.697 |
| HIS163 | - | - | - | - | - | 1.877 | - | - | - |
| HIS164 | 2.299 | - | - | - | - | 2.167 | 2.104 | - | 2.054 |
| MET165 | - | 3.039 | 3.156 | 3.053 | - | - | - | - | - |
| GLU166 | - | 2.964 | 2.937 | 2.943 | 2.089/2.263 | 2.012/2.192/2.954 | 2.962 | 2.432 | 2.937 |
| PRO168 | - | - | - | - | - | - | 2.321 | - | - |
| GLN189 | - | 1.984 | - | 2.546 | - | 2.352 | - | 3.141 | - |
| THR190 | - | - | - | - | - | 2.585 | - | - | - |
| ALA191 | - | - | - | - | - | - | - | 2.910 | - |
| GLN192 | - | - | - | - | - | 2.838 | 1.954 | 1.872 | 2.251 |
| Amino Acid | Interaction Energy (kcal/mol) | |||||||
|---|---|---|---|---|---|---|---|---|
| Hydroxychloroquine | Ritonavir | S-217622 | N3 | PF-07321332 | (1) | (6) | (8) | |
| HIS41 | −1.06 | −0.16 | −2.32 | −3.42 | −0.36 | −2.06 | −2.18 | −2.2 |
| MET49 | 0.64 | −0.08 | −0.41 | −0.23 | −0.87 | −1.04 | −1.08 | −1.47 |
| LEU50 | 0.06 | −0.21 | 0.10 | −0.05 | 0.01 | −0.67 | 0.19 | −0.43 |
| ASN51 | −0.11 | −0.14 | 0.08 | 0.03 | 0.06 | −0.42 | −0.06 | −0.26 |
| PRO52 | 0.87 | −0.14 | 0.74 | 0.01 | −0.03 | −0.96 | −1.01 | −1.86 |
| ASN53 | −0.09 | 0.07 | 0.05 | 0.04 | 0.12 | −0.28 | −0.07 | −0.26 |
| TYR54 | −0.09 | −0.27 | −3.45 | 0.01 | 0.07 | 1.09 | 1.01 | 0.9 |
| PHE140 | 1.16 | −0.21 | −2.32 | −4.32 | −3.61 | 0.22 | −0.16 | −0.47 |
| LEU141 | −2.71 | −0.32 | 1.04 | 2.3 | −0.77 | −0.62 | −0.88 | 0.02 |
| ASN142 | −1.36 | −1.01 | −1.01 | −0.36 | −1.21 | 0.46 | 0.77 | 0.21 |
| GLY143 | −3.14 | −2.45 | −1.25 | −5.02 | −0.59 | 0.2 | 0.25 | 0.05 |
| SER144 | 0.43 | −0.18 | −0.24 | −3.37 | −3.96 | −0.02 | 0.41 | −0.07 |
| CYS145 | −2.47 | −2.4 | −2.89 | −2.5 | −3.44 | −0.67 | −0.98 | −2.87 |
| HIS163 | −0.78 | −1.69 | −1.14 | −2.08 | −3.05 | −0.57 | −0.49 | −0.66 |
| HIS164 | −3.2 | −1.07 | −1.33 | 0.15 | −2.75 | −2.98 | −0.55 | −3.16 |
| MET165 | 2.29 | 0.94 | −1.89 | 0.03 | 0.13 | 0.48 | 1.53 | 1.5 |
| GLU166 | −9.51 | −18.44 | −22.49 | −23.14 | −24.99 | −14.47 | −21.33 | −21.75 |
| LEU167 | −0.94 | −0.82 | −0.22 | −0.64 | −0.32 | 0.09 | 0.17 | 0.11 |
| PRO168 | −0.42 | 0.25 | 0.10 | −0.08 | −0.05 | −1.09 | −0.03 | −0.83 |
| HIS172 | −0.19 | −0.19 | −0.67 | −2.28 | −0.21 | 0.26 | −0.18 | −0.76 |
| ASP187 | 2.57 | −0.26 | 0.22 | 0.49 | −1.03 | −0.44 | −0.43 | 0.02 |
| ARG188 | −3.2 | 0.33 | 0.98 | −0.45 | 0.49 | −0.31 | −1.17 | −0.57 |
| GLN189 | −0.44 | −3.03 | −3.17 | −0.08 | −3.54 | −1.36 | −2.15 | −1.07 |
| THR190 | −0.22 | 1.48 | 1.01 | 0.07 | 0.21 | −0.85 | −0.31 | 0.42 |
| ALA191 | −0.18 | −0.06 | 0.02 | 0.09 | 0.05 | −0.36 | −1.19 | −0.78 |
| GLN192 | −0.88 | −0.41 | 0.21 | 0.07 | 0.13 | −5.31 | −3.38 | −3.66 |
| Total | −22.97 | −30.47 | −40.25 | −44.73 | −49.51 | −31.68 | −33.30 | −39.90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patnin, S.; Makarasen, A.; Vijitphan, P.; Baicharoen, A.; Chaivisuthangkura, A.; Kuno, M.; Techasakul, S. Computational Screening of Phenylamino-Phenoxy-Quinoline Derivatives against the Main Protease of SARS-CoV-2 Using Molecular Docking and the ONIOM Method. Molecules 2022, 27, 1793. https://doi.org/10.3390/molecules27061793
Patnin S, Makarasen A, Vijitphan P, Baicharoen A, Chaivisuthangkura A, Kuno M, Techasakul S. Computational Screening of Phenylamino-Phenoxy-Quinoline Derivatives against the Main Protease of SARS-CoV-2 Using Molecular Docking and the ONIOM Method. Molecules. 2022; 27(6):1793. https://doi.org/10.3390/molecules27061793
Chicago/Turabian StylePatnin, Suwicha, Arthit Makarasen, Pongsit Vijitphan, Apisara Baicharoen, Apinya Chaivisuthangkura, Mayuso Kuno, and Supanna Techasakul. 2022. "Computational Screening of Phenylamino-Phenoxy-Quinoline Derivatives against the Main Protease of SARS-CoV-2 Using Molecular Docking and the ONIOM Method" Molecules 27, no. 6: 1793. https://doi.org/10.3390/molecules27061793