
Weighted Gene Co-Expression Network Analysis Combined with Machine Learning Validation to Identify Key Modules and Hub Genes Associated with SARS-CoV-2 Infection

 , , , ,
, , , ,  , , , , , , and
, , , , , , and
Abstract
:1. Introduction
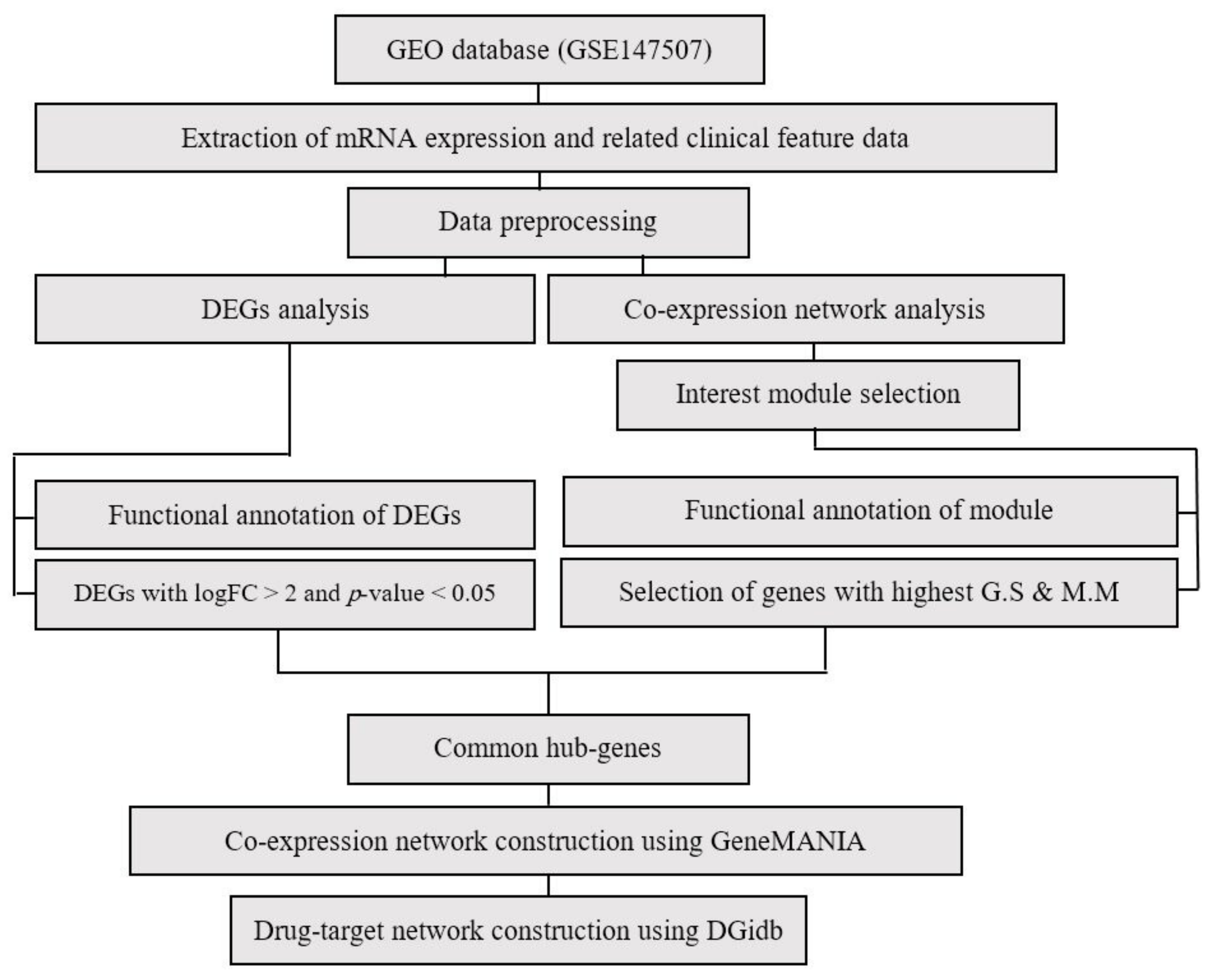
2. Materials and Methods
2.1. Gene Expression Dataset Acquisition and Preprocessing
2.2. Identification of Differentially Expressed Genes
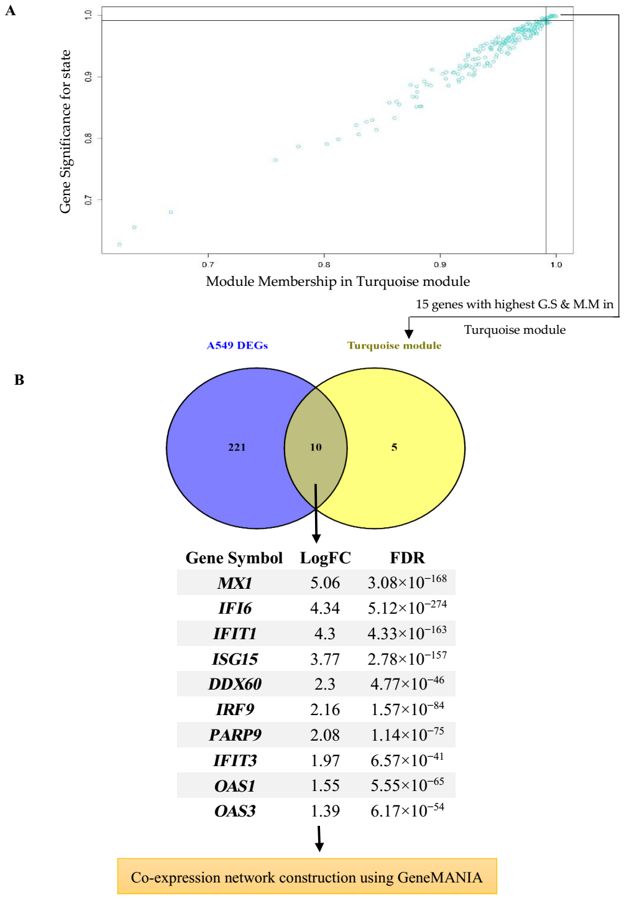
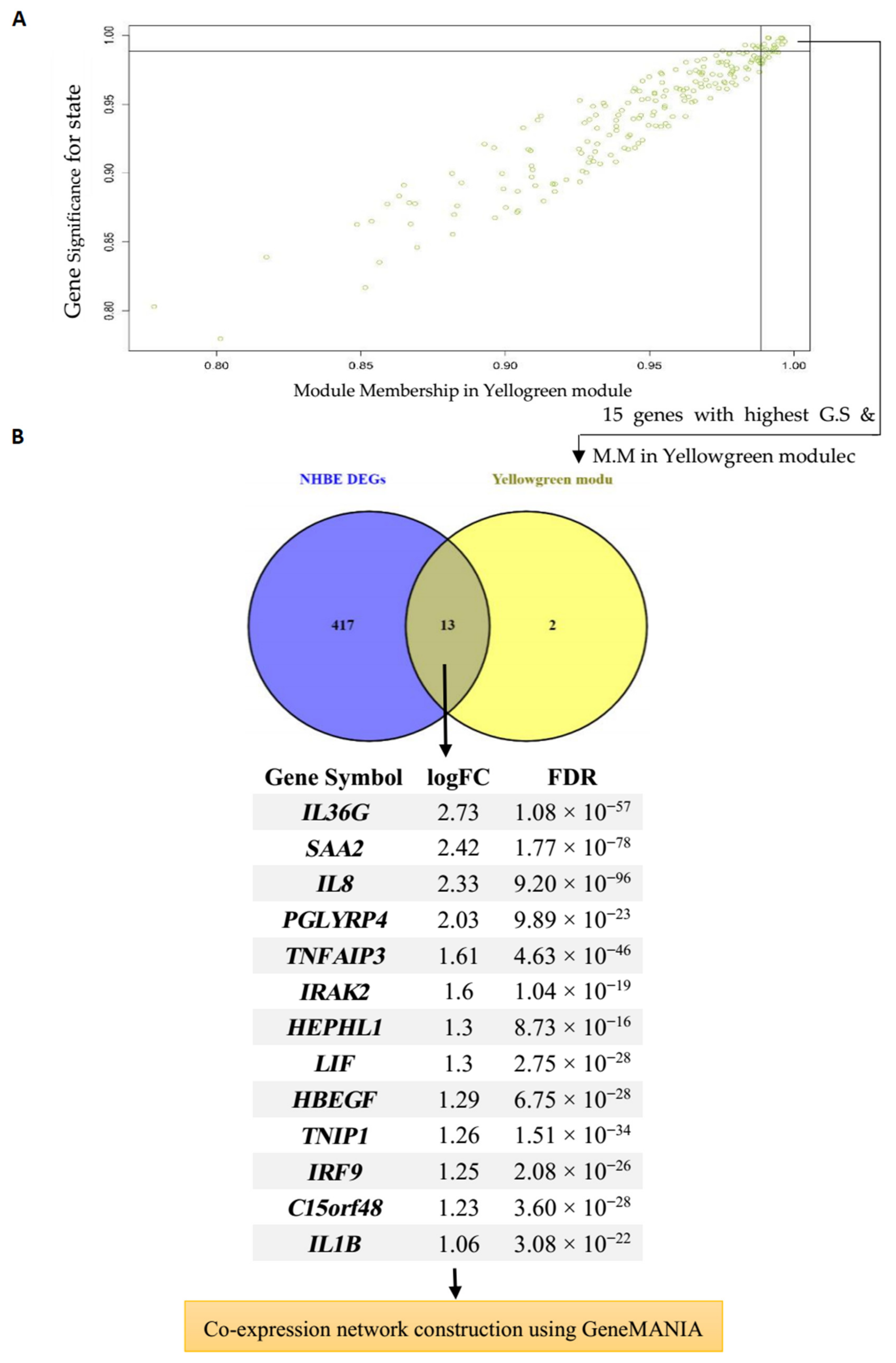
2.3. WGCNA Network Construction and Identification of Significant Modules
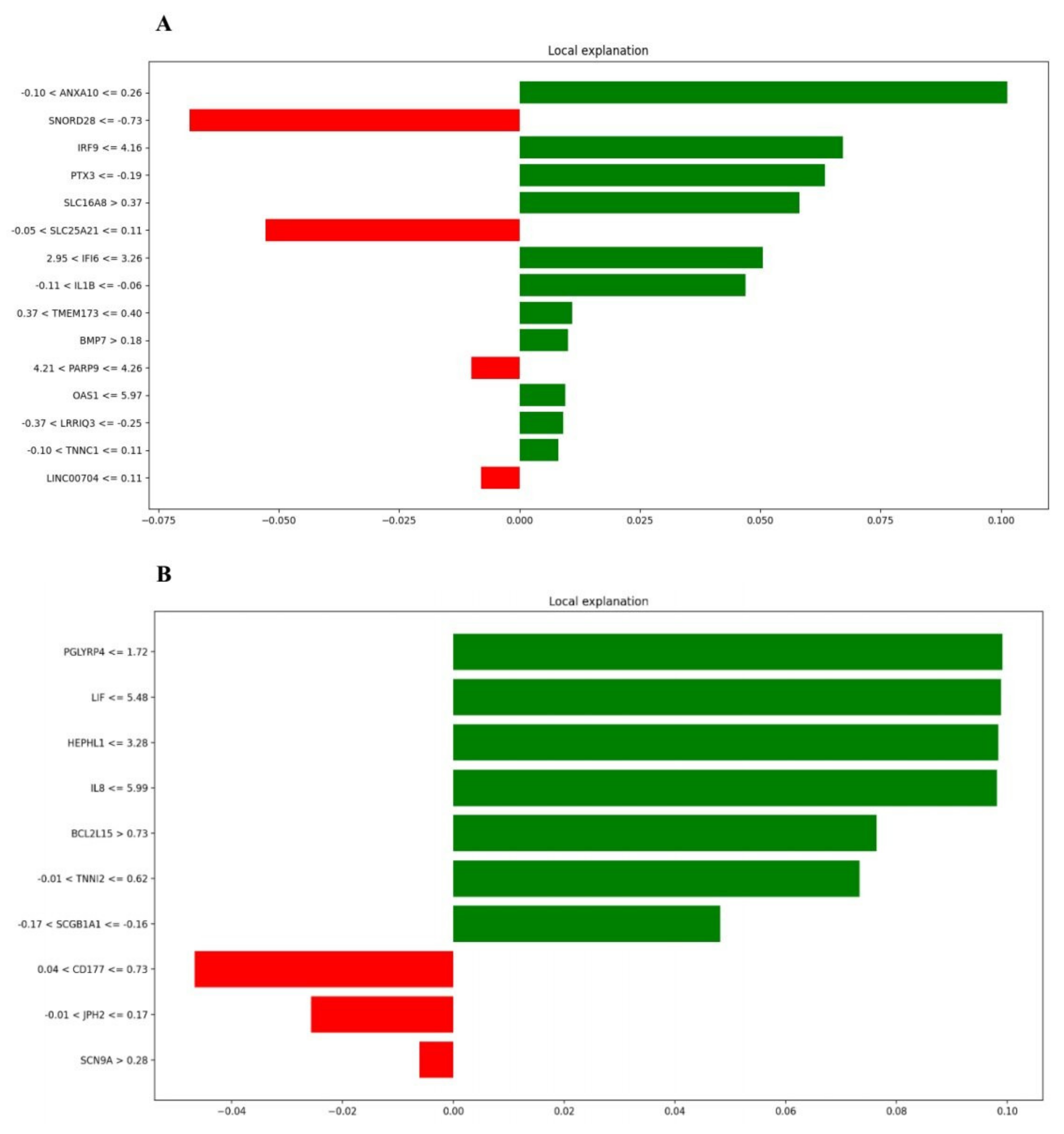
2.4. Explain the Gene Importance by SP-LIME
| Algorithm 1: SP-LIME algorithm. |
| Inputs: : The set of samples : The set of features : The set of instances for explanations
Feature importance Instances that cover the important features |
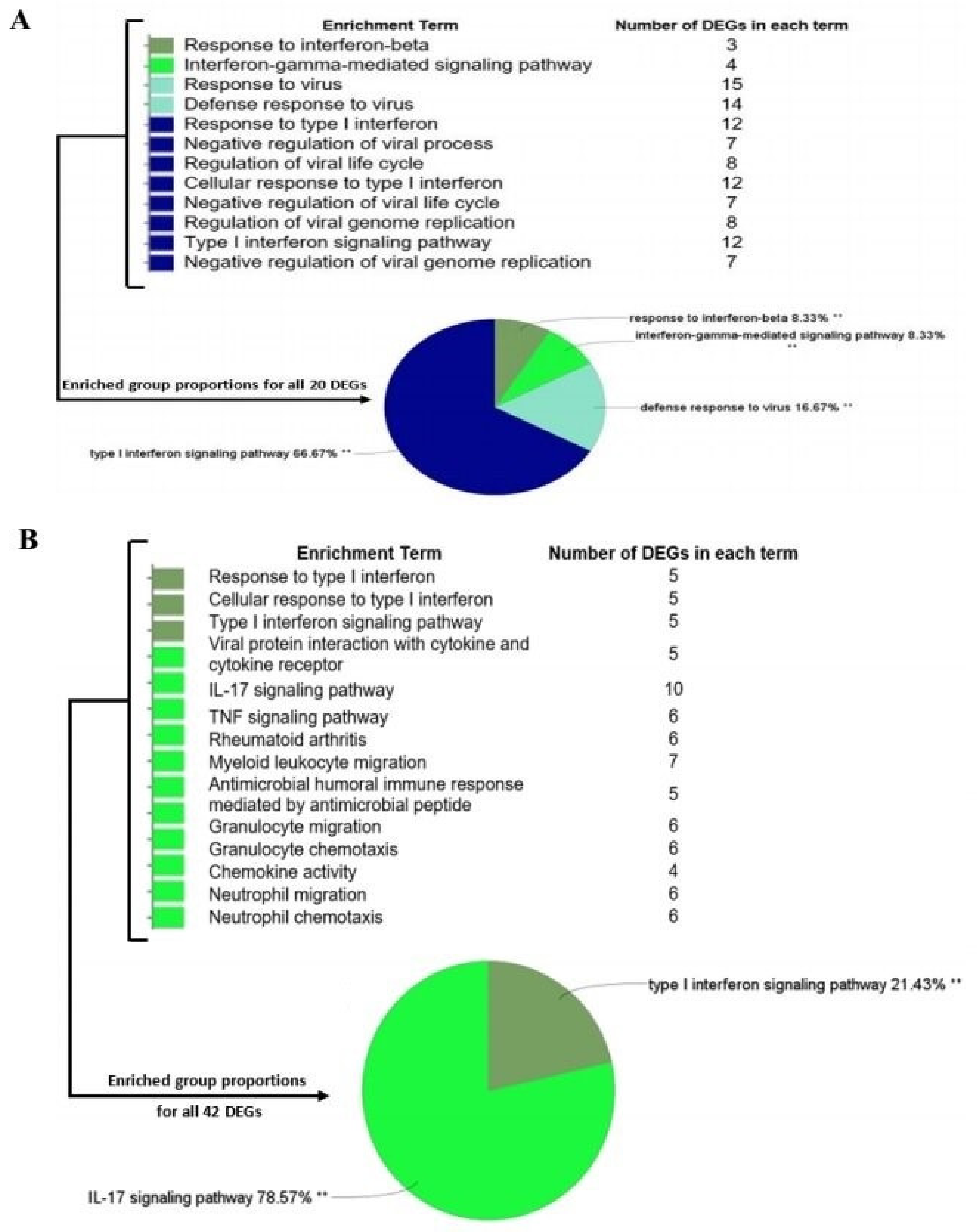
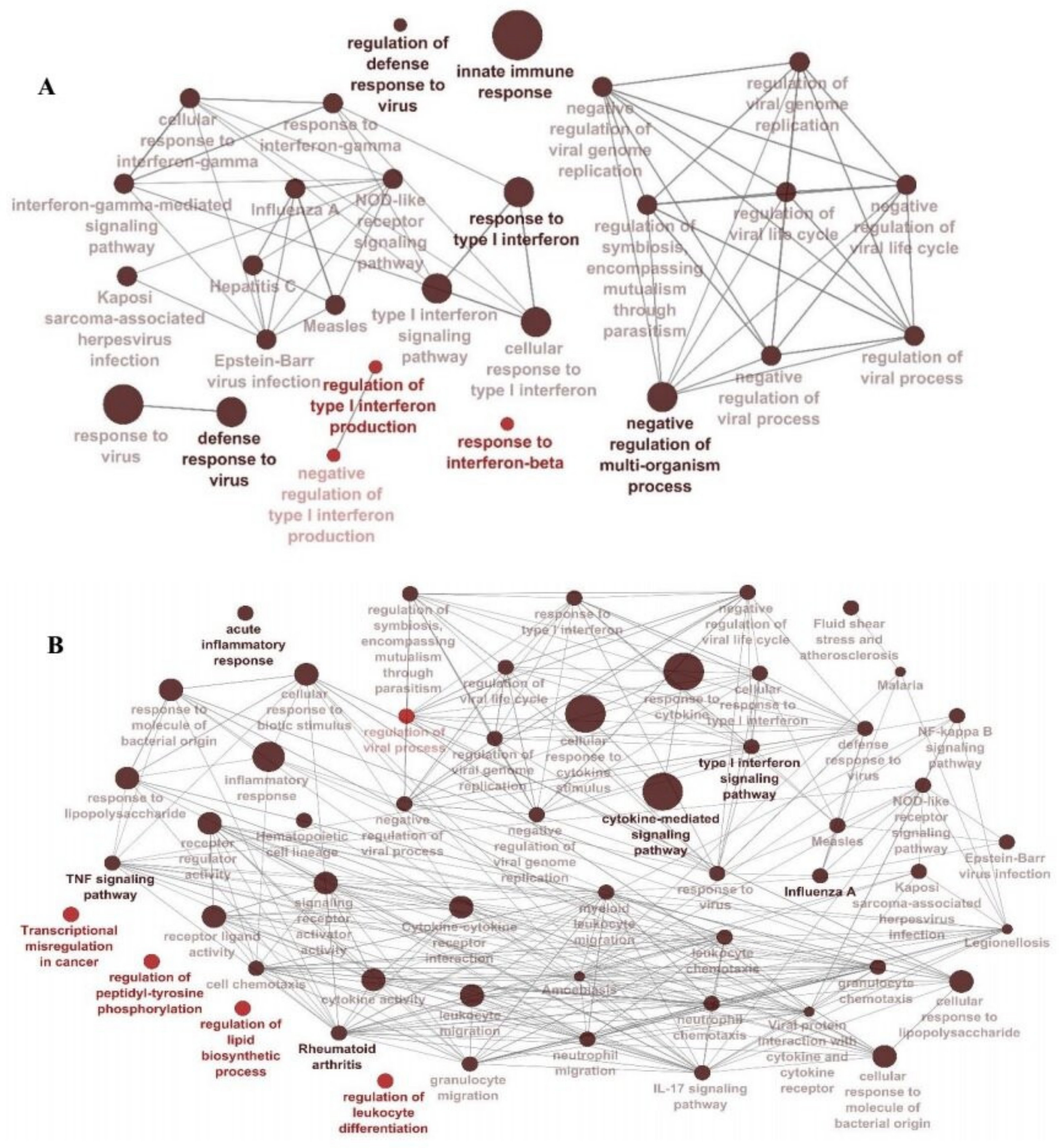
2.5. Functional Enrichment of Significant Modules
2.6. Hub Gene Detection and Co-Expression Network Reconstruction
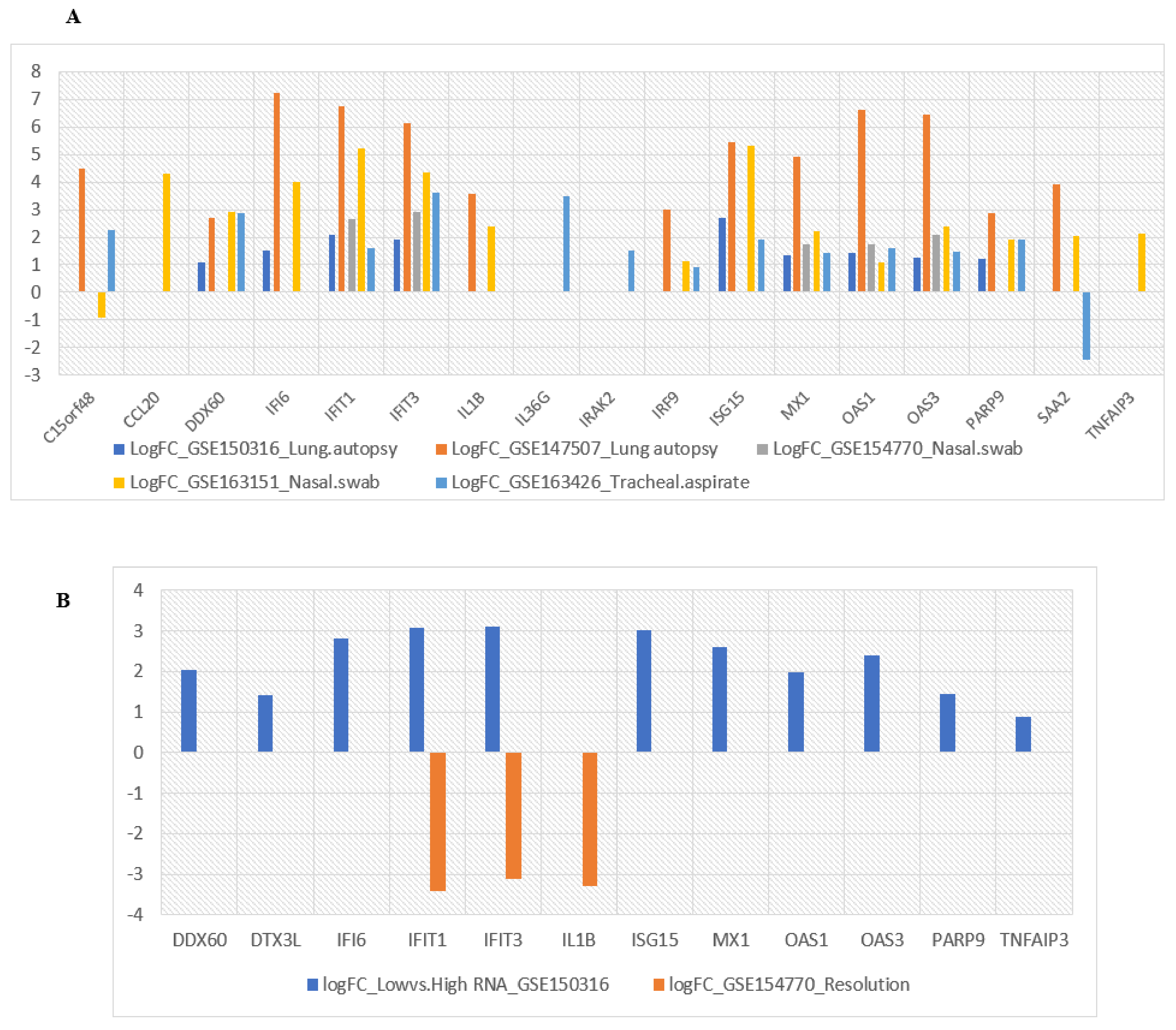
2.7. Evaluation of Selected Hub Genes’ Behavior in Other Virus-Based Infections
2.8. Evaluation of Selected Hub Genes’ Behavior in Lung Tissues and Secretions Isolated from COVID-19 Patients
2.9. Evaluation of Selected Hub Genes’ Behavior in COVID-19 Patients with Severe Pneumonia Varied by Viral Load
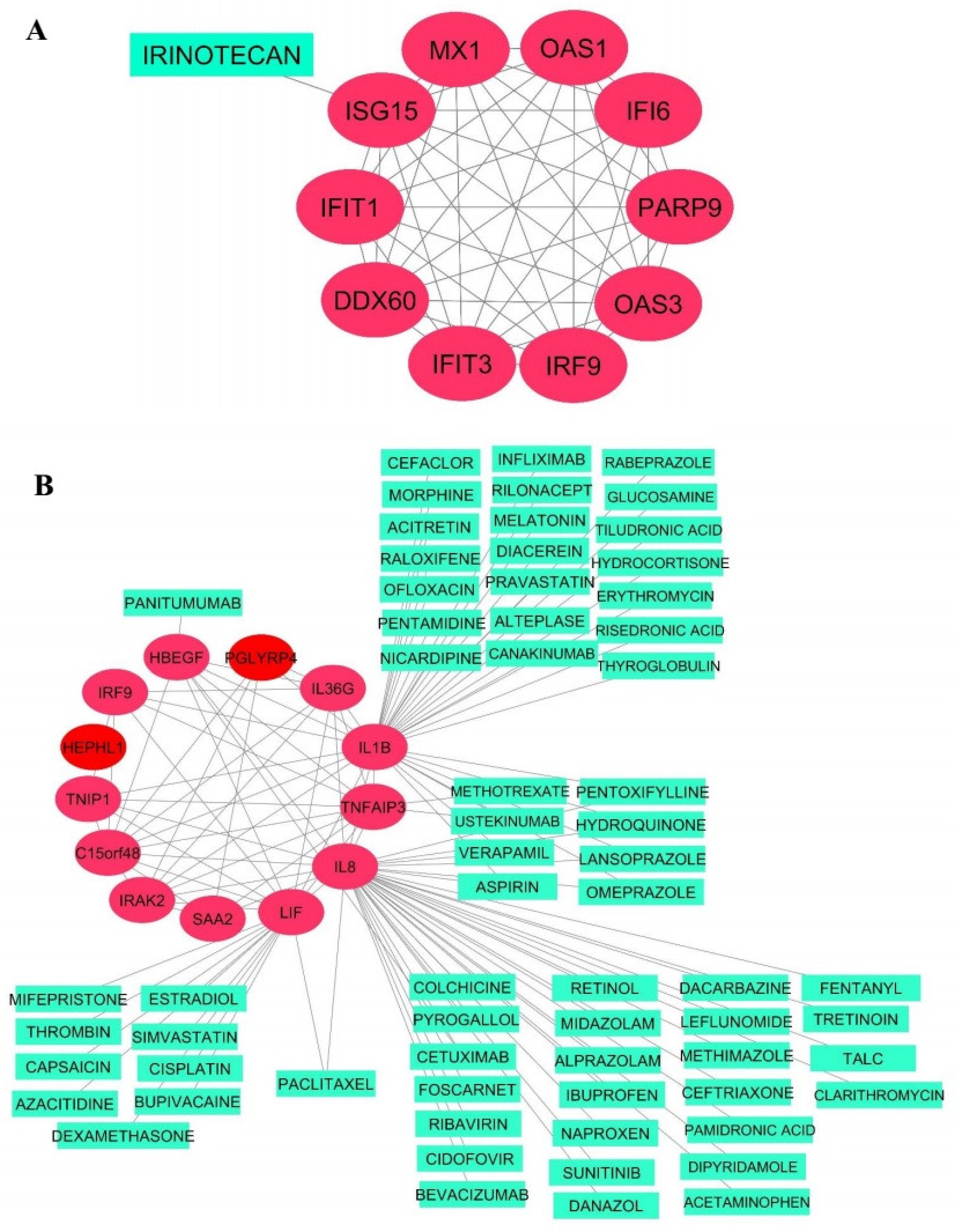
2.10. Identification of Candidate Regulatory Drugs
3. Results
3.1. Preprocessing and DEG Analysis
3.2. Identification of WGCNA Modules
3.3. Module–Trait Association Analysis and Functional Annotation Analysis of Interesting Modules
3.4. Hub Gene Identification and Network Analysis of Interesting Modules
3.5. SP-LIME as an Explainable AI Method for Identifying Important Genes
3.6. Evaluation of Selected Hub Genes’ Behavior in Other Virus-Based Infections
3.7. Evaluation of Selected Hub Genes’ Behavior in Respiratory Specimens of COVID-19 Patients
3.8. Evaluation of Selected Hub Genes’ Behavior in High and Low Viral Load of Samples from SARS-CoV-2-Infected Patients

3.9. Drug–Target Network Construction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yuan, S.; Kok, K.-H.; To, K.K.-W.; Chu, H.; Yang, J.; Xing, F.; Liu, J.; Yip, C.C.-Y.; Poon, R.W.-S.; et al. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 2020, 395, 514–523. [Google Scholar] [CrossRef]
- Young, B.E.; Ong, S.W.X.; Kalimuddin, S.; Low, J.G.; Tan, S.Y.; Loh, J.; Ng, O.-T.; Marimuthu, K.; Ang, L.W.; Mak, T.M.; et al. Epidemiologic features and clinical course of patients infected with SARS-CoV-2 in Singapore. JAMA 2020, 323, 1488–1494. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Fowler, R.; Hayden, F.G. Critical care management of adults with community-acquired severe respiratory viral infection. Intensiv. Care Med. 2020, 46, 315–328. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Fehr, A.; Channappanavar, R.; Perlman, S. Middle East Respiratory Syndrome: Emergence of a Pathogenic Human Coronavirus. Annu. Rev. Med. 2017, 68, 387–399. [Google Scholar] [CrossRef]
- Newton, A.H.; Cardani, A.; Braciale, T.J. The host immune response in respiratory virus infection: Balancing virus clearance and immunopathology. Semin. Immunopathol. 2016, 38, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Ren, X.; Wang, S.; Chen, X.; Wei, X.; Li, G.; Ren, S.; Zhang, T.; Zhang, X.; Lu, Z.; You, Z. Multiple expression assessments of ACE2 and TMPRSS2 SARS-CoV-2 entry molecules in the urinary tract and their associations with clinical manifestations of COVID-19. Infect. Drug Resist. 2020, 13, 3977. [Google Scholar] [CrossRef]
- Ho, J.S.Y.; Mok, B.W.-Y.; Campisi, L.; Jordan, T.; Yildiz, S.; Parameswaran, S.; Wayman, J.A.; Gaudreault, N.N.; Meekins, D.A.; Indran, S.V.; et al. Topoisomerase 1 inhibition therapy protects against SARS-CoV-2-induced inflammation and death in animal models. bioRxiv 2020. [Google Scholar] [CrossRef]
- Weingarten-Gabbay, S.; Klaeger, S.; Sarkizova, S.; Pearlman, L.R.; Chen, D.-Y.; Bauer, M.R.; Taylor, H.B.; Conway, H.L.; Tomkins-Tinch, C.H.; Finkel, Y.; et al. SARS-CoV-2 infected cells present HLA-I peptides from canonical and out-of-frame ORFs. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hoagland, D.A.; Clarke, D.J.; Moeller, R.; Han, Y.; Yang, L.; Wojciechowicz, M.L.; Lachmann, A.; Oguntuyo, K.Y.; Stevens, C.; Lee, B.; et al. Modulating the transcriptional landscape of SARS-CoV-2 as an effective method for developing antiviral compounds. bioRxiv 2020. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.-H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 2021, 184, 92–105.e16. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, T.; Liu, Y.; Chen, J.; Xin, B.; Wu, M.; Cui, W. Coronary artery disease associated specific modules and feature genes revealed by integrative methods of WGCNA, MetaDE and machine learning. Gene 2019, 710, 122–130. [Google Scholar] [CrossRef]
- Derakhshani, A.; Hashemzadeh, S.; Asadzadeh, Z.; Shadbad, M.A.; Rasibonab, F.; Safarpour, H.; Jafarlou, V.; Solimando, A.G.; Racanelli, V.; Singh, P.K.; et al. Cytotoxic T-Lymphocyte Antigen-4 in Colorectal Cancer: Another Therapeutic Side of Capecitabine. Cancers 2021, 13, 2414. [Google Scholar] [CrossRef]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; Ladu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 1–12. [Google Scholar] [CrossRef]
- Guo, S.M.; Wang, J.X.; Li, J.; Xu, F.Y.; Wei, Q.; Wang, H.M.; Huang, H.Q.; Zheng, S.L.; Xie, Y.J.; Zhang, C. Identification of gene expression profiles and key genes in subchondral bone of osteoarthritis using weighted gene coexpression network analysis. J. Cell. Biochem. 2018, 119, 7687–7695. [Google Scholar] [CrossRef]
- Miao, L.; Yin, R.-X.; Pan, S.-L.; Yang, S.; Yang, D.-Z.; Lin, W.-X. Weighted Gene Co-Expression Network Analysis Identifies Specific Modules and Hub Genes Related to Hyperlipidemia. Cell. Physiol. Biochem. 2018, 48, 1151–1163. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Derakhshani, A.; Mollaei, H.; Parsamanesh, N.; Fereidouni, M.; Miri-Moghaddam, E.; Nasseri, S.; Luo, Y.; Safarpour, H.; Baradaran, B. Gene Co-expression Network Analysis for Identifying Modules and Functionally Enriched Pathways in Vitiligo Disease: A Systems Biology Study. Iran. J. Allergy Asthma Immunol. 2020, 19, 517–528. [Google Scholar] [CrossRef]
- Daamen, A.R.; Bachali, P.; Owen, K.A.; Kingsmore, K.M.; Hubbard, E.L.; Labonte, A.C.; Robl, R.; Shrotri, S.; Grammer, A.C.; Lipsky, P.E. Comprehensive transcriptomic analysis of COVID-19 blood, lung, and airway. Sci. Rep. 2021, 11, 1–19. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045.e9. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Burkart, N.; Huber, M.F. A survey on the explainability of supervised machine learning. J. Artif. Intell. Res. 2021, 70, 245–317. [Google Scholar] [CrossRef]
- Linardatos, P.; Papastefanopoulos, V.; Kotsiantis, S. Explainable AI: A Review of Machine Learning Interpretability Methods. Entropy 2020, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why should I trust you?” Explaining the predictions of any classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data mining, San Francisco, CA, USA, 13–17 August 2016; pp. 1135–1144. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Cao, W.; Wu, W.; Yan, M.; Tian, F.; Ma, C.; Zhang, Q.; Li, X.; Han, P.; Liu, Z.; Gu, J.; et al. Multiple region whole-exome sequencing reveals dramatically evolving intratumor genomic heterogeneity in esophageal squamous cell carcinoma. Oncogenesis 2015, 4, e175. [Google Scholar] [CrossRef]
- Asokananthan, N.; Graham, P.T.; Fink, J.; Knight, D.A.; Bakker, A.J.; McWilliam, A.S.; Thompson, P.J.; Stewart, G.A. Activation of Protease-Activated Receptor (PAR)-1, PAR-2, and PAR-4 Stimulates IL-6, IL-8, and Prostaglandin E2Release from Human Respiratory Epithelial Cells. J. Immunol. 2002, 168, 3577–3585. [Google Scholar] [CrossRef]
- Zaas, A.K.; Chen, M.; Varkey, J.; Veldman, T.; Hero, A.O., III; Lucas, J.; Huang, Y.; Turner, R.; Gilbert, A.; Lambkin-Williams, R.; et al. Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe 2009, 6, 207–217. [Google Scholar] [CrossRef]
- Fink, K.; Martin, L.; Mukawera, E.; Chartier, S.; De Deken, X.; Brochiero, E.; Miot, F.; Grandvaux, N. IFNβ/TNFα synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH Oxidase-mediated airway antiviral response. Cell Res. 2013, 23, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Wein, A.N.; Dunbar, P.R.; McMaster, S.R.; Li, Z.-R.T.; Denning, T.; Kohlmeier, J.E. IL-36γ Protects against Severe Influenza Infection by Promoting Lung Alveolar Macrophage Survival and Limiting Viral Replication. J. Immunol. 2018, 201, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Leong, W.; Tan, H.; Ooi, E.E.; Koh, D.; Chow, V.T. Microarray and real-time RT-PCR analyses of differential human gene expression patterns induced by severe acute respiratory syndrome (SARS) coronavirus infection of Vero cells. Microbes Infect. 2005, 7, 248–259. [Google Scholar] [CrossRef]
- Pillai, P.S.; Molony, R.D.; Martinod, K.; Dong, H.; Pang, I.K.; Tal, M.; Solis, A.G.; Bielecki, P.; Mohanty, S.; Trentalange, M.; et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science 2016, 352, 463–466. [Google Scholar] [CrossRef]
- Kroeker, A. A Proteomic Approach to Discovering Novel Anti-Influenza Mechanisms in Primary Human Airway Epithelial Cells. Ph.D. Thesis, University of Manitoba, Winnipeg, MB, Canada, 2012. [Google Scholar]
- Barik, S. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. In Challenges and Opportunities for Respiratory Syncytial Virus Vaccines; Springer: Berlin/Heidelberg, Germany, 2013; pp. 173–191. [Google Scholar]
- Imajoh, M.; Hashida, Y.; Murakami, M.; Maeda, A.; Sato, T.; Fujieda, M.; Wakiguchi, H.; Daibata, M. Characterization of Epstein–Barr virus (EBV) BZLF1 gene promoter variants and comparison of cellular gene expression profiles in Japanese patients with infectious mononucleosis, chronic active EBV infection, and EBV-associated hemophagocytic lymphohistiocytosis. J. Med. Virol. 2012, 84, 940–946. [Google Scholar]
- Zhang, Y.; Mao, D.; Roswit, W.T.; Jin, X.; Patel, A.C.; Patel, D.A.; Agapov, E.; Wang, Z.; Tidwell, R.M.; Atkinson, J.J.; et al. PARP9-DTX3L ubiquitin ligase targets host histone H2BJ and viral 3C protease to enhance interferon signaling and control viral infection. Nat. Immunol. 2015, 16, 1215–1227. [Google Scholar] [CrossRef]
- Tatebe, K.; Zeytun, A.; Ribeiro, R.M.; Hoffmann, R.; Harrod, K.S.; Forst, C.V. Response network analysis of differential gene expression in human epithelial lung cells during avian influenza infections. BMC Bioinform. 2010, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.; Liu, W.-C.; Møller, R.; Panis, M.; Sachs, D.; Albrecht, R.A.; tenOever, B.R. SARS-CoV-2 launches a unique transcriptional signature from in vitro, ex vivo, and in vivo systems. bioRxiv 2020. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Jumeau, C.; Awad, F.; Assrawi, E.; Cobret, L.; Duquesnoy, P.; Giurgea, I.; Valeyre, D.; Grateau, G.; Amselem, S.; Bernaudin, J.-F.; et al. Expression of SAA1, SAA2 and SAA4 genes in human primary monocytes and monocyte-derived macrophages. PLoS ONE 2019, 14, e0217005. [Google Scholar] [CrossRef] [PubMed]
- Sarma, A.; Christenson, S.; Mick, E.; Deiss, T.; DeVoe, C.; Pisco, A.; Ghale, R.; Jauregui, A.; Byrne, A.; Moazed, F.; et al. COVID-19 ARDS is characterized by a dysregulated host response that differs from cytokine storm and is modified by dexamethasone. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Desai, N.; Neyaz, A.; Szabolcs, A.; Shih, A.R.; Chen, J.H.; Thapar, V.; Nieman, L.T.; Solovyov, A.; Mehta, A.; Lieb, D.J.; et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Hemmat, N.; Asadzadeh, Z.; Ahangar, N.K.; Alemohammad, H.; Najafzadeh, B.; Derakhshani, A.; Baghbanzadeh, A.; Baghi, H.B.; Javadrashid, D.; Najafi, S.; et al. The roles of signaling pathways in SARS-CoV-2 infection; lessons learned from SARS-CoV and MERS-CoV. Arch. Virol. 2021, 166, 675–696. [Google Scholar] [CrossRef]
- Frohman, E.M.; Villemarette-Pittman, N.R.; Cruz, R.A.; Longmuir, R.; Rowe, V.; Rowe, E.S.; Varkey, T.C.; Steinman, L.; Zamvil, S.Z.; Frohman, T.C. Part II. High-dose methotrexate with leucovorin rescue for severe COVID-19: An immune stabilization strategy for SARS-CoV-2 induced ‘PANIC’ attack. J. Neurol. Sci. 2020, 415, 116935. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, K.M.; Dickmanns, A.; Gerber, S.; Nikolova, V.; Klemke, L.; Manzini, V.; Schloesser, D.; Bierwirth, C.; Freund, J.; Sitte, M.; et al. The folate antagonist methotrexate diminishes replication of the coronavirus SARS-CoV-2 and enhances the antiviral efficacy of remdesivir in cell culture models. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lee, V.S.; Chong, W.L.; Sukumaran, S.D.; Nimmanpipug, P.; Letchumanan, V.; Goh, B.H.; Lee, L.-H.; Zain, S.M.; Abd Rahman, N. Computational screening and identifying binding interaction of anti-viral and anti-malarial drugs: Toward the potential cure for SARS-CoV-2. Prog. Drug Discov. Biomed. Sci. 2020, 3. [Google Scholar] [CrossRef]
- Galvez, J.; Zanni, R.; Galvez-Llompart, M. Drugs Repurposing for Coronavirus Treatment: Computational Study Based On Molecular Topology. Nereis 2020, 15–18. [Google Scholar] [CrossRef]
- Chang, Y.; Tung, Y.; Lee, K.; Chen, T.; Hsiao, Y.; Chang, H.; Hsieh, T.; Su, C.; Wang, S.; Yu, J.; et al. Potential Therapeutic Agents for COVID-19 Based on the Analysis of Protease and RNA Polymerase Docking. Preprints 2020. [Google Scholar] [CrossRef]
- Contini, A. Virtual screening of an FDA approved drugs database on two COVID-19 coronavirus proteins. Am. Chem. S. 2020. [Google Scholar] [CrossRef]
- Cava, C.; Bertoli, G.; Castiglioni, I. In Silico Discovery of Candidate Drugs against COVID-19. Viruses 2020, 12, 404. [Google Scholar] [CrossRef]
- Jeon, S.; Ko, M.; Lee, J.; Choi, I.; Byun, S.Y.; Park, S.; Shum, D.; Kim, S. Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs. Antimicrob. Agents Chemother. 2020, 64. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, X.; Ni, L.; Di, X.; Ma, B.; Niu, S.; Liu, C.; Reiter, R.J. COVID-19: Melatonin as a potential adjuvant treatment. Life Sci. 2020, 250, 117583. [Google Scholar] [CrossRef]
- Liu, X.; Li, Z.; Liu, S.; Chen, Z.; Zhao, Z.; Huang, Y.-Y.; Zhang, Q.; Wang, J.; Shi, Y.; Xu, Y.; et al. Therapeutic effects of dipyridamole on COVID-19 patients with coagulation dysfunction. medRxiv 2020. [Google Scholar] [CrossRef]
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef]
- Zhang, R.; Li, Y.; Pan, B.; Li, Y.; Liu, A.; Li, X. Increased expression of hub gene CXCL10 in peripheral blood mononuclear cells of patients with systemic lupus erythematosus. Exp. Ther. Med. 2019, 18, 4067–4075. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef]
- Thaker, S.K.; Ch’Ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef]
- Pacha, O.; Sallman, M.A.; Evans, S.E. COVID-19: A case for inhibiting IL-17? Nat. Rev. Immunol. 2020, 20, 345–346. [Google Scholar] [CrossRef]
- Lu, L.; Zhang, H.; Zhan, M.; Jiang, J.; Yin, H.; Dauphars, D.J.; Li, S.-Y.; Li, Y.; He, Y.-W. Preventing Mortality in COVID-19 Patients: Which Cytokine to Target in a Raging Storm? Front. Cell Dev. Biol. 2020, 8, 677. [Google Scholar] [CrossRef]
- Okabayashi, T.; Kariwa, H.; Yokota, S.-I.; Iki, S.; Indoh, T.; Yokosawa, N.; Takashima, I.; Tsutsumi, H.; Fujii, N. Cytokine regulation in SARS coronavirus infection compared to other respiratory virus infections. J. Med. Virol. 2006, 78, 417–424. [Google Scholar] [CrossRef]
- Hemmat, N.; Derakhshani, A.; Baghi, H.B.; Silvestris, N.; Baradaran, B.; De Summa, S. Neutrophils, Crucial, or Harmful Immune Cells Involved in Coronavirus Infection: A Bioinformatics Study. Front. Genet. 2020, 11, 641. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, J.; Zhan, Y.; Wu, L.; Yu, X.; Zhang, W.; Ye, L.; Xu, S.; Sun, R.; Wang, Y.; et al. Analysis of Serum Cytokines in Patients with Severe Acute Respiratory Syndrome. Infect. Immun. 2004, 72, 4410–4415. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Han, M.; Su, X.; Li, H.; Chen, J.; Guo, X. Identification of Hub genes associated with infection of three lung cell lines by SARS-CoV-2 with integrated bioinformatics analysis. J. Cell. Mol. Med. 2020, 24, 12225–12230. [Google Scholar] [CrossRef]
- Cheng, L.-C.; Kao, T.-J.; Phan, N.N.; Chiao, C.-C.; Yen, M.-C.; Chen, C.-F.; Hung, J.-H.; Jiang, J.-Z.; Sun, Z.; Wang, C.-Y. Novel signaling pathways regulate SARS-CoV and SARS-CoV-2 infectious disease. Medicine 2021, 100, e24321. [Google Scholar] [CrossRef]
- Fang, C.; Mei, J.; Tian, H.; Liou, Y.-L.; Rong, D.; Zhang, W.; Liao, Q.; Wu, N. CSF3 Is a Potential Drug Target for the Treatment of COVID-19. Front. Physiol. 2021, 11, 605792. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, E.; Petralia, M.C.; Basile, M.S.; Bramanti, A.; Bramanti, P.; Nicoletti, F.; Spandidos, D.A.; Shoenfeld, Y.; Fagone, P. Transcriptomic analysis of COVID-19 lungs and bronchoalveolar lavage fluid samples reveals predominant B cell activation responses to infection. Int. J. Mol. Med. 2020, 46, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Sevimoglu, T.; Arga, K.Y. The role of protein interaction networks in systems biomedicine. Comput. Struct. Biotechnol. J. 2014, 11, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Ciurleo, R.; Lombardo, S.D.; Iacobello, C.; Palermo, C.I.; Shoenfeld, Y.; Bendtzen, K.; Bramanti, P.; Nicoletti, F. Transcriptional landscape of SARS-CoV-2 infection dismantles pathogenic pathways activated by the virus, proposes unique sex-specific differences and predicts tailored therapeutic strategies. Autoimmun. Rev. 2020, 19, 102571. [Google Scholar] [CrossRef]
- Li, G.; Wang, J.; He, X.; Zhang, L.; Ran, Q.; Xiong, A.; Wu, D.; Hu, L.; Song, Q.; Zhu, D. An integrative analysis identifying transcriptional features and key genes involved in COVID-19. Epigenomics 2020, 12, 1969–1981. [Google Scholar] [CrossRef]
- El-Hachem, N.; Eid, E.; Nemer, G.; Dbaibo, G.; Abbas, O.; Rubeiz, N.; Zeineldine, S.; Matar, G.M.; Bikorimana, J.-P.; Shammaa, R.; et al. Integrative Transcriptome Analyses Empower the Anti-COVID-19 Drug Arsenal. iScience 2020, 23, 101697. [Google Scholar] [CrossRef]
- O’Donovan, S.M.; Imami, A.; Eby, H.; Henkel, N.D.; Creeden, J.F.; Asah, S.; Zhang, X.; Wu, X.; Alnafisah, R.; Taylor, R.T.; et al. Identification of candidate repurposable drugs to combat COVID-19 using a signature-based approach. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Marrero, M.C.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 2020, 182, 685–712.e19. [Google Scholar] [CrossRef]
- Pinto, B.; Oliveira, A.E.R.; Singh, Y.; Jimenez, L.; Gonçalves, A.N.A.; Ogava, R.L.T.; Creighton, R.; Peron, J.P.S.; I Nakaya, H. ACE2 Expression Is Increased in the Lungs of Patients With Comorbidities Associated With Severe COVID-19. J. Infect. Dis. 2020, 222, 556–563. [Google Scholar] [CrossRef]
- Gong, J.; Dong, H.; Xia, S.Q.; Huang, Y.Z.; Wang, D.; Zhao, Y.; Liu, W.; Tu, S.; Zhang, M.; Wang, Q.; et al. Correlation Analysis Between Disease Severity and Inflammation-related Parameters in Patients with COVID-19 Pneumonia. medRxiv 2020. [Google Scholar] [CrossRef]
- Belinky, F.; Nativ, N.; Stelzer, G.; Zimmerman, S.; Stein, T.I.; Safran, M.; Lancet, R. PathCards: Multi-source consolidation of human biological pathways. Database 2015, 2015, bav006. [Google Scholar] [CrossRef]
- Skerry, C.; Goldman, W.E.; Carbonetti, N.H. Peptidoglycan Recognition Protein 4 Suppresses Early Inflammatory Responses to Bordetella pertussis and Contributes to Sphingosine-1-Phosphate Receptor Agonist-Mediated Disease Attenuation. Infect. Immun. 2019, 87, 87. [Google Scholar] [CrossRef]
- Dabrowski, A.N.; Shrivastav, A.; Conrad, C.; Komma, K.; Weigel, M.; Dietert, K.; Gruber, A.D.; Bertrams, W.; Wilhelm, J.; Schmeck, B.; et al. Peptidoglycan Recognition Protein 4 Limits Bacterial Clearance and Inflammation in Lungs by Control of the Gut Microbiota. Front. Immunol. 2019, 10, 2106. [Google Scholar] [CrossRef]
- Ma, P.; Wang, Z.; Pflugfelder, S.C.; Li, D.-Q. Toll-like receptors mediate induction of peptidoglycan recognition proteins in human corneal epithelial cells. Exp. Eye Res. 2010, 90, 130–136. [Google Scholar] [CrossRef]
- Moni, M.A.; Quinn, J.M.; Sinmaz, N.; Summers, M.A. Gene expression profiling of SARS-CoV-2 infections reveal distinct primary lung cell and systemic immune infection responses that identify pathways relevant in COVID-19 disease. Brief. Bioinform. 2021, 22, 1324–1337. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Manne, U.; Varambally, S. Comparative transcriptome analyses reveal genes associated with SARS-CoV-2 infection of human lung epithelial cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Al Heialy, S.; Hachim, M.Y.; Senok, A.; Gaudet, M.; Tayoun, A.A.; Hamoudi, R.; Alsheikh-Ali, A.; Hamid, Q. Regulation of Angiotensin- Converting Enzyme 2 in Obesity: Implications for COVID-19. Front. Physiol. 2020, 11, 11. [Google Scholar] [CrossRef]
- Karakurt, H.U.; Pınar, P. Integration of transcriptomic profile of SARS-CoV-2 infected normal human bronchial epi-thelial cells with metabolic and protein-protein interaction networks. Turk. J. Biol. 2020, 44, 168. [Google Scholar] [CrossRef]
- Kang, K.; Kim, H.H.; Choi, Y. Tiotropium Is Predicted to Be a Promising Drug for COVID-19 Through Transcriptome-Based Comprehensive Molecular Pathway Analysis. Viruses 2020, 12, 776. [Google Scholar] [CrossRef]
- Goswami, R.; Russell, V.S.; Tu, J.J.; Hughes, P.F.; Kelly, F.; Langel, S.N.; Steppe, J.; Palmer, S.M.; Haystead, T.; Blasi, M.; et al. Oral Hsp90 inhibitor, SNX-5422, attenuates SARS-CoV-2 replication and dampens inflammation in airway cells. SSRN 2021. [Google Scholar] [CrossRef]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a phar-macological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 1–10. [Google Scholar] [CrossRef]
- Meacci, E.; Garcia-Gil, M.; Pierucci, F. SARS-CoV-2 infection: A role for S1P/S1P receptor signaling in the nervous system? Int. J. Mol. Sci. 2020, 21, 6773. [Google Scholar] [CrossRef] [PubMed]
- Zrzavy, T.; Wimmer, I.; Rommer, P.S.; Berger, T. Immunology of COVID-19 and disease-modifying therapies: The good, the bad and the unknown. Eur. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Cronin, S.J.F.; Woolf, C.J.; Weiss, G.; Penninger, J.M. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front. Mol. Biosci. 2019, 6, 116. [Google Scholar] [CrossRef]
- Sharma, P.; Reichert, M.; Lu, Y.; Markello, T.C.; Adams, D.R.; Steinbach, P.J.; Fuqua, B.K.; Parisi, X.; Kaler, S.G.; Vulpe, C.D.; et al. Biallelic HEPHL1 variants impair ferroxidase activity and cause an abnormal hair phenotype. PLoS Genet. 2019, 15, e1008143. [Google Scholar] [CrossRef]
- Dalamaga, M.; Karampela, I.; Mantzoros, C.S. Commentary: Could iron chelators prove to be useful as an adjunct to COVID-19 Treatment Regimens? Metabolism 2020, 108, 154260. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Prentice, A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008, 6, 541–552. [Google Scholar] [CrossRef]
- Horie, S.; Harada, T.; Mitsunari, M.; Taniguchi, F.; Iwabe, T.; Terakawa, N. Progesterone and progestational compounds attenuate tumor necrosis factor alpha–induced interleukin-8 production via nuclear factor kappaB inactivation in endometriotic stromal cells. Fertil. Steril. 2005, 83, 1530–1535. [Google Scholar] [CrossRef] [PubMed]
- Barh, D.; Tiwari, S.; Weener, M.E.; Azevedo, V.; Góes-Neto, A.; Gromiha, M.M.; Ghosh, P. Multi-omics-based identification of SARS-CoV-2 infection biology and candidate drugs against COVID-19. Comput. Biol. Med. 2020, 126, 104051. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, S.; Zhang, Y.; Zhi, Y. Does hereditary angioedema make COVID-19 worse? World Allergy Organ. J. 2020, 13, 100454. [Google Scholar] [CrossRef]
- Soy, M.; Keser, G.; Atagündüz, P.; Tabak, F.; Atagündüz, I.; Kayhan, S. Cytokine storm in COVID-19: Pathogenesis and overview of anti-inflammatory agents used in treatment. Clin. Rheumatol. 2020, 39, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Cremers, S.; Papapoulos, S. Pharmacology of bisphosphonates. Bone 2011, 49, 42–49. [Google Scholar] [CrossRef]
- Yazdanifar, M.; Mashkour, N.; Bertaina, A. Making a case for using γδ T cells against SARS-CoV-2. Crit. Rev. Microbiol. 2020, 46, 689–702. [Google Scholar] [CrossRef]
- Fujimura, T.; Kambayashi, Y.; Furudate, S.; Kakizaki, A.; Aiba, S. Immunomodulatory Effect of Bisphosphonate Risedronate Sodium on CD163+ Arginase 1+ M2 Macrophages: The Development of a Possible Supportive Therapy for Angiosarcoma. Clin. Dev. Immunol. 2013, 2013, 1–7. [Google Scholar] [CrossRef]
- Brufsky, A.; Marti, J.L.G.; Nasrazadani, A.; Lotze, M.T. Boning up: Amino-bisphophonates as immunostimulants and endosomal disruptors of dendritic cell in SARS-CoV-2 infection. J. Transl. Med. 2020, 18, 1–6. [Google Scholar] [CrossRef]
- Karami, H.; Derakhshani, A.; Fereidouni, M.; Miri-Moghaddam, E.; Baradaran, B.; Silvestris, N.; Paradiso, A.V.; Safarpour, H.; De Summa, S. Transcriptional analysis of lung epithelial cells using WGCNA revealed the role of IRF9 and IFI6 genes in SARS-CoV-2 pathogenicity. Res. Sq. 2020. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Selected Module | Hub Genes | SARS-CoV-2 | RSV | IAV | IAV-ΔNS1 | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| logFC | p-Value | logFC | p-Value | logFC | p-Value | logFC | p-Value | |||
| A549 | Turquoise | MX1 | 5.06 | 3.08 × 10−168 | 5.54 | 1.76 × 10−110 | −4.46 | ns | - | - |
| IFI6 | 4.29 | 5.12 × 10−274 | 2.92 | 5.97 × 10−43 | −0.57 | ns | - | - | ||
| IFIT1 | 3.76 | 4.33 × 10−163 | 5.09 | 7.98 × 10−105 | 0.03 | ns | - | - | ||
| IFIT3 | 1.97 | 5.28 × 10−44 | 4.99 | 4.88 × 10−101 | 0.34 | ns | - | - | ||
| OAS1 | 1.55 | 2.74 × 10−68 | 3.10 | 8.42 × 10−51 | 0.75 | 0.006989 | - | - | ||
| OAS3 | 1.39 | 3.43 × 10−57 | 2.98 | 1.27 × 10−47 | −0.02 | ns | - | - | ||
| ISG15 | 4.33 | 2.78 × 10−157 | 4.52 | 5.05 × 10−88 | −0.80 | ns | - | - | ||
| DDX60 | 2.30 | 4.77 × 10−46 | 3.10 | 1.61 × 10−38 | −0.06 | ns | - | - | ||
| IRF9 | 2.16 | 1.57 × 10−84 | 1.33 | 1.61 × 10−9 | 0.46 | ns | - | - | ||
| PARP9 | 2.07 | 1.14 × 10−75 | 3.02 | 6.60 × 10−43 | 0.42 | ns | - | - | ||
| NHBE | Yellow-green | IL36G | 2.72 | 1.08 × 10−57 | - | 0.88 | 0.004286 | 4.12 | 7.63 × 10−20 | |
| SAA2 | 2.41 | 1.77 × 10−78 | - | - | 0.74 | 4.02 × 10−7 | 2.69 | 4.02 × 10−07 | ||
| TNIP1 | 1.26 | 1.63 × 10−37 | - | - | 0.01 | ns | 1.46 | 9.28 × 10−8 | ||
| TNFAIP3 | 1.61 | 2.64 × 10−49 | - | - | 1.41 | 4.55 × 10−16 | 4.49 | 1.47 × 10−22 | ||
| HEPHL1 | 1.30 | 2.82 × 10−18 | - | - | 1.45 | 5.47 × 10−8 | 1.56 | 1.86 × 10−5 | ||
| IRAK2 | 1.60 | 2.89 × 10−22 | - | - | 0.97 | 1.25 × 10−5 | 2.86 | 3.05 × 10−23 | ||
| HBEGF | 1.29 | 1.07 × 10−30 | - | - | 1.09 | 0.005477 | 2.07 | 8.60 × 10−24 | ||
| LIF | 1.30 | 4.01 × 10−31 | - | - | 0.17 | ns | 2.89 | 4.10 × 10−9 | ||
| IRF9 | 1.25 | 3.69 × 10−29 | - | - | 1.39 | 9.52 × 10−10 | 2.75 | 3.45 × 10−43 | ||
| IL1B | 1.06 | 7.22 × 10−25 | - | - | −0.04 | ns | 1.24 | 5.98 × 10−9 | ||
| IL8 | 2.32 | 9.20 × 10−96 | - | - | - | - | - | - | ||
| PGLYRP4 | 2.02 | 9.89 × 10−23 | - | - | 1.08 | 0.088695 | 1.98 | 0.004146 | ||
| Modules | Genes | Drugs | Drug Class | PMIDs | DGIdb Score | Anti-Viral Properties |
|---|---|---|---|---|---|---|
| Yellow-green module | IL8 | Danazol | Androgen | - | 2 | - |
| IL8 | Cetuximab | Antineoplastic, Anti EGFR | - | 4 | - | |
| IL8 | Aluminum hydroxide | Antacid | - | 2 | - | |
| IL8 | Talc | Sclerosing agents | - | 2 | - | |
| IL8 | Alprazolam | Benzodiazepine | 12218154 | 2 | IAV | |
| IL8 | Pamidronic acid | Bisphosphonate | 21708931 | 2 | IAV | |
| 24154548 | EBV | |||||
| 25220446 | ||||||
| IL1B | Risedronic acid | Bisphosphonate | - | - | ||
| IL1B | Tiludronic acid | Bisphosphonate | - | - | ||
| IL1B | Rilonacept | Interleukin 1 inhibitor | - | - | ||
| IL1B | Pentamidine | Antiprotozoal | - | - | ||
| IL1B | Glucosamine | Non-steroidal anti-inflammatory and antirheumatic | - | - | ||
| IL1B | Ustekinumab | Anti-IL-12 and IL-23 | - | - | ||
| LIF | Capsaicin | Non-narcotic analgesic | 32528827 | - | ||
| 12537981 | - | |||||
| 27574502 | - | |||||
| LIF | Azacitidine | Antineoplastic, cytosine analogue | 32176725 | - | ||
| LIF | Paclitaxel | Antineoplastic, Antimicrotubular | 31797089 | 2 | - | |
| HBEGF | Cetuximab | Antineoplastic, Anti EGFR | - | 4 | - | |
| HBEGF | Panitumumab | Antineoplastic, Anti EGFR | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karami, H.; Derakhshani, A.; Ghasemigol, M.; Fereidouni, M.; Miri-Moghaddam, E.; Baradaran, B.; Tabrizi, N.J.; Najafi, S.; Solimando, A.G.; Marsh, L.M.; et al. Weighted Gene Co-Expression Network Analysis Combined with Machine Learning Validation to Identify Key Modules and Hub Genes Associated with SARS-CoV-2 Infection. J. Clin. Med. 2021, 10, 3567. https://doi.org/10.3390/jcm10163567
Karami H, Derakhshani A, Ghasemigol M, Fereidouni M, Miri-Moghaddam E, Baradaran B, Tabrizi NJ, Najafi S, Solimando AG, Marsh LM, et al. Weighted Gene Co-Expression Network Analysis Combined with Machine Learning Validation to Identify Key Modules and Hub Genes Associated with SARS-CoV-2 Infection. Journal of Clinical Medicine. 2021; 10(16):3567. https://doi.org/10.3390/jcm10163567
Chicago/Turabian StyleKarami, Hassan, Afshin Derakhshani, Mohammad Ghasemigol, Mohammad Fereidouni, Ebrahim Miri-Moghaddam, Behzad Baradaran, Neda Jalili Tabrizi, Souzan Najafi, Antonio Giovanni Solimando, Leigh M. Marsh, and et al. 2021. "Weighted Gene Co-Expression Network Analysis Combined with Machine Learning Validation to Identify Key Modules and Hub Genes Associated with SARS-CoV-2 Infection" Journal of Clinical Medicine 10, no. 16: 3567. https://doi.org/10.3390/jcm10163567