
Exploring Toxins for Hunting SARS-CoV-2 Main Protease Inhibitors: Molecular Docking, Molecular Dynamics, Pharmacokinetic Properties, and Reactome Study

 , ,
, ,  , , ,
, , ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
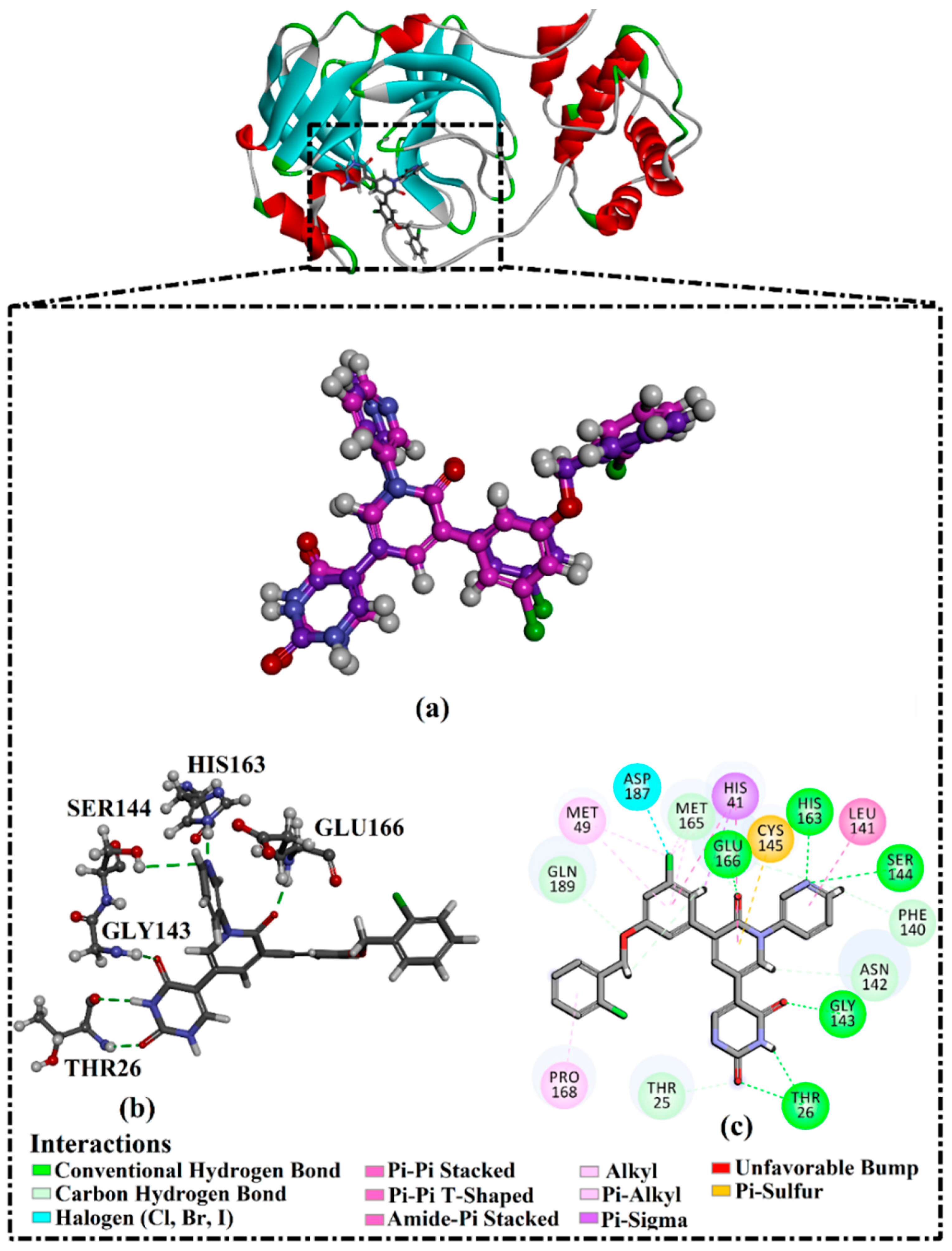
2.1. Validation of In Silico Protocol
2.2. T3DB Database Virtual Screening
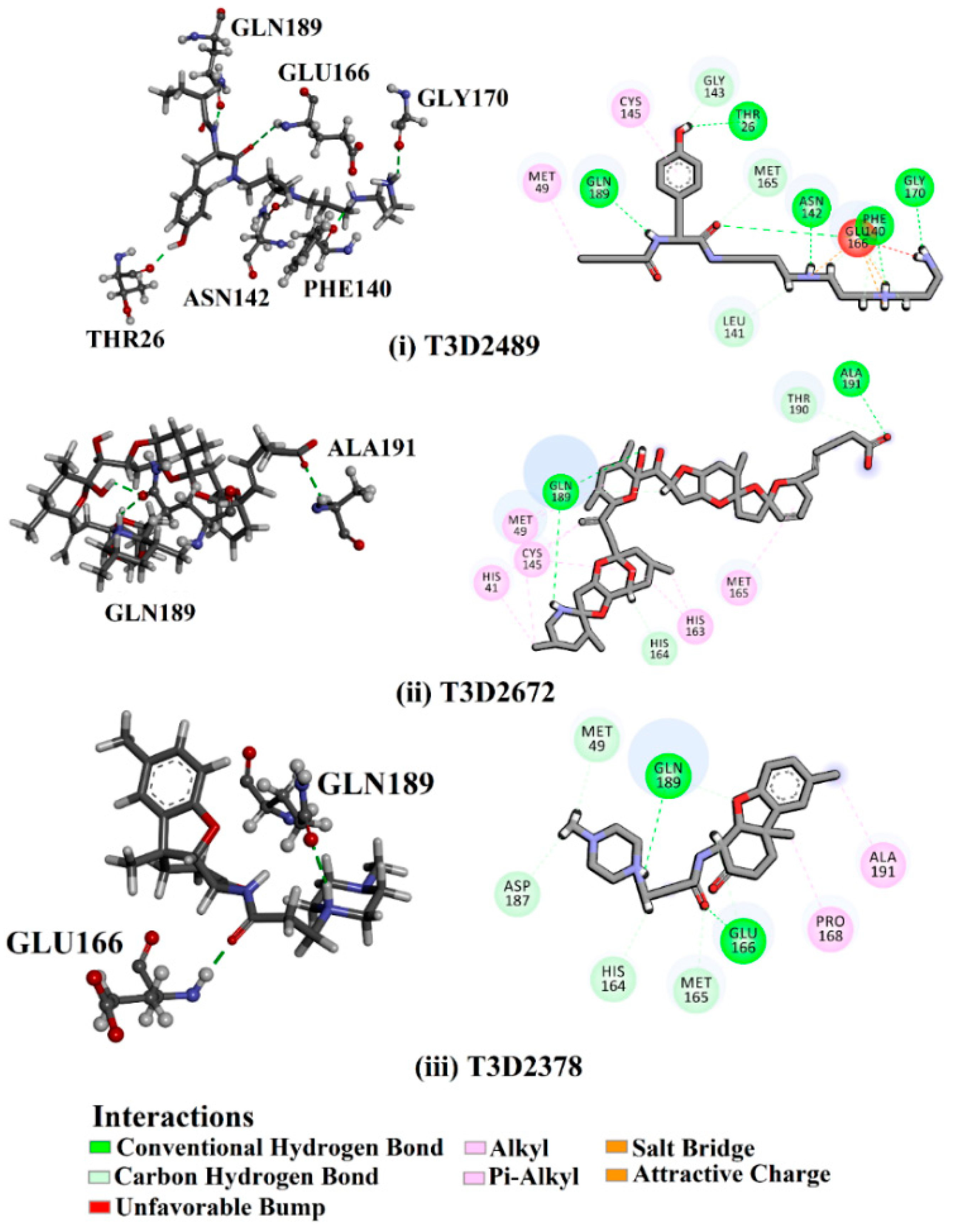
2.3. Molecular Dynamics (MD) Simulations
2.4. Post-MD Analyses
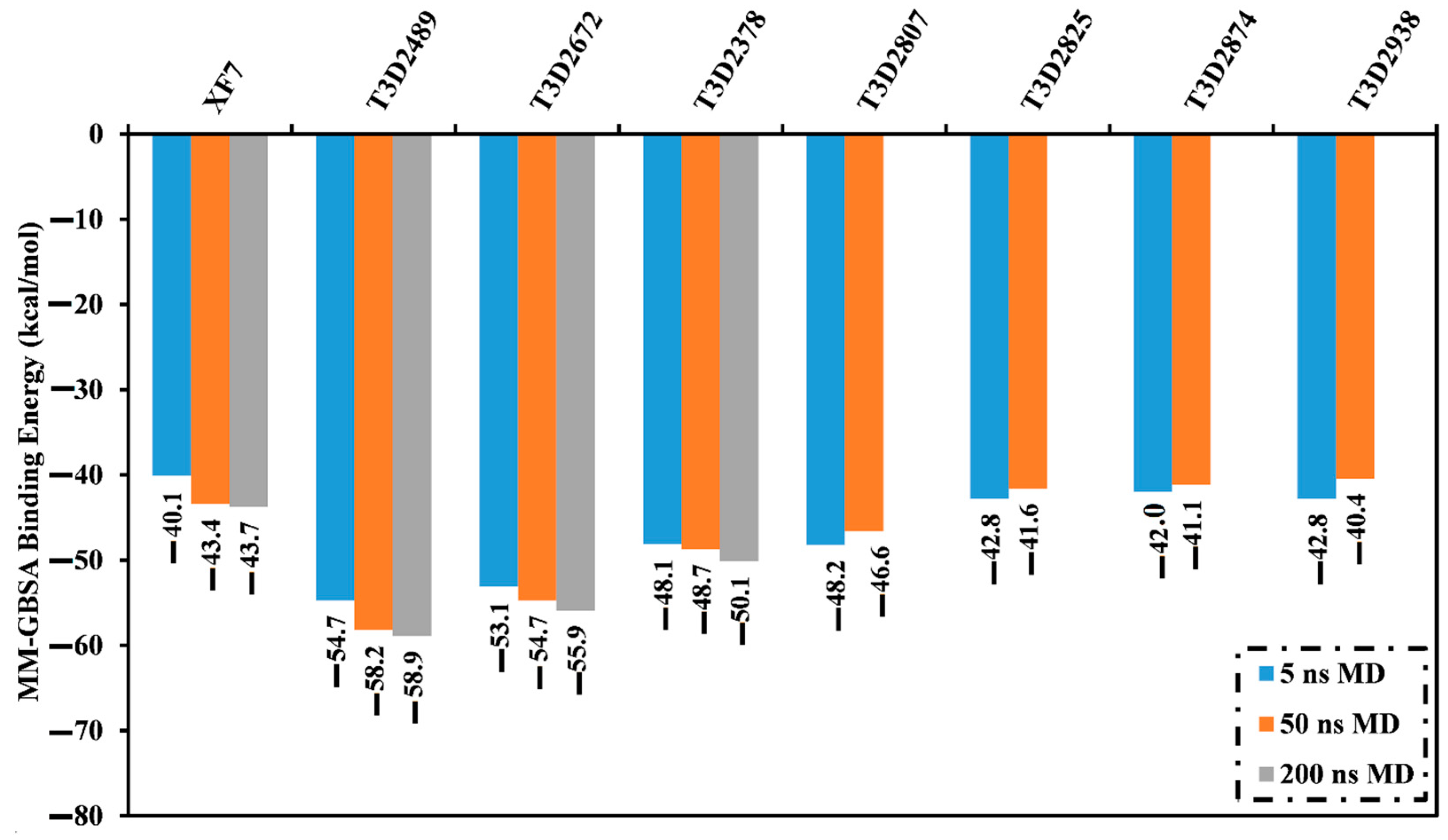
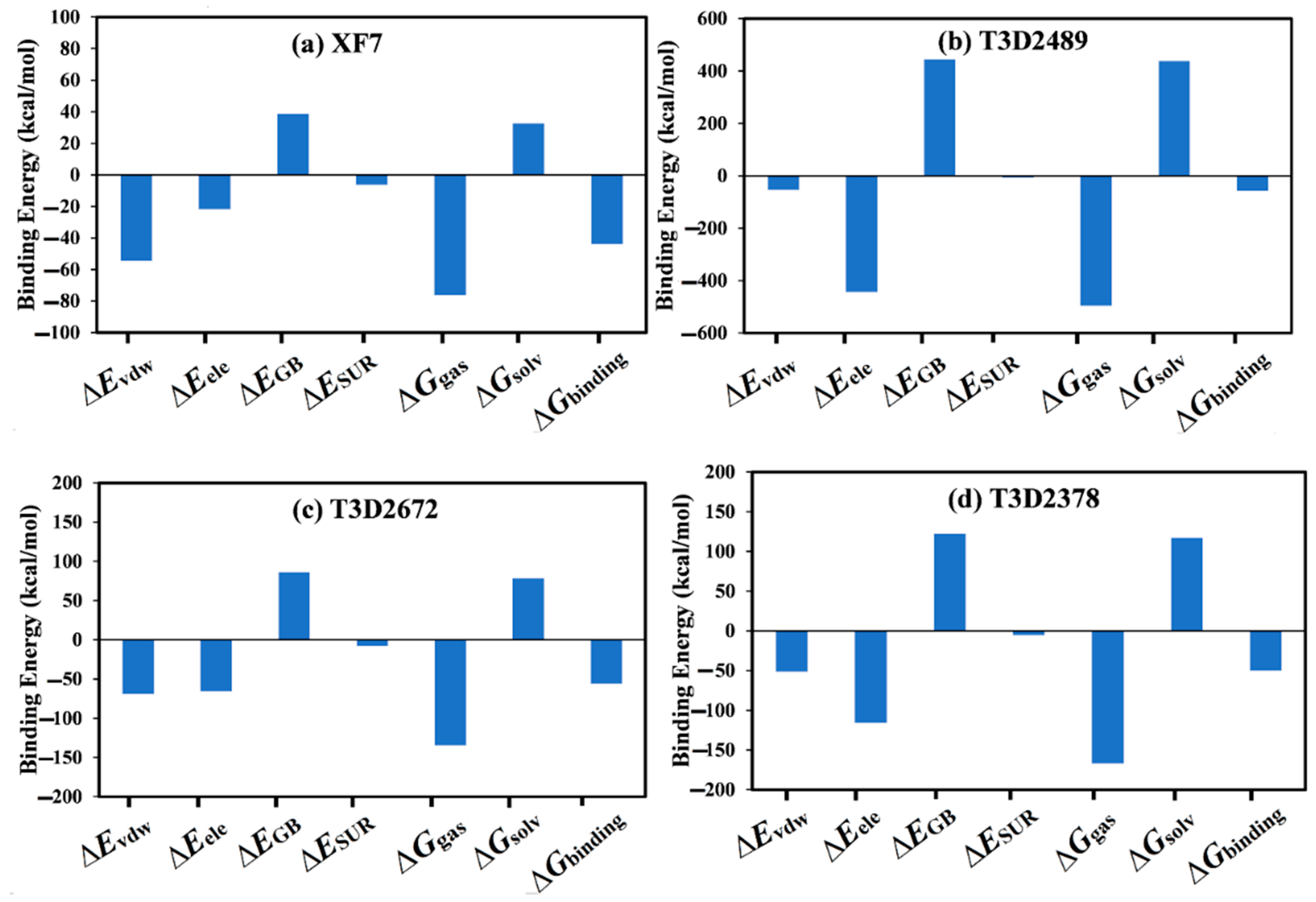
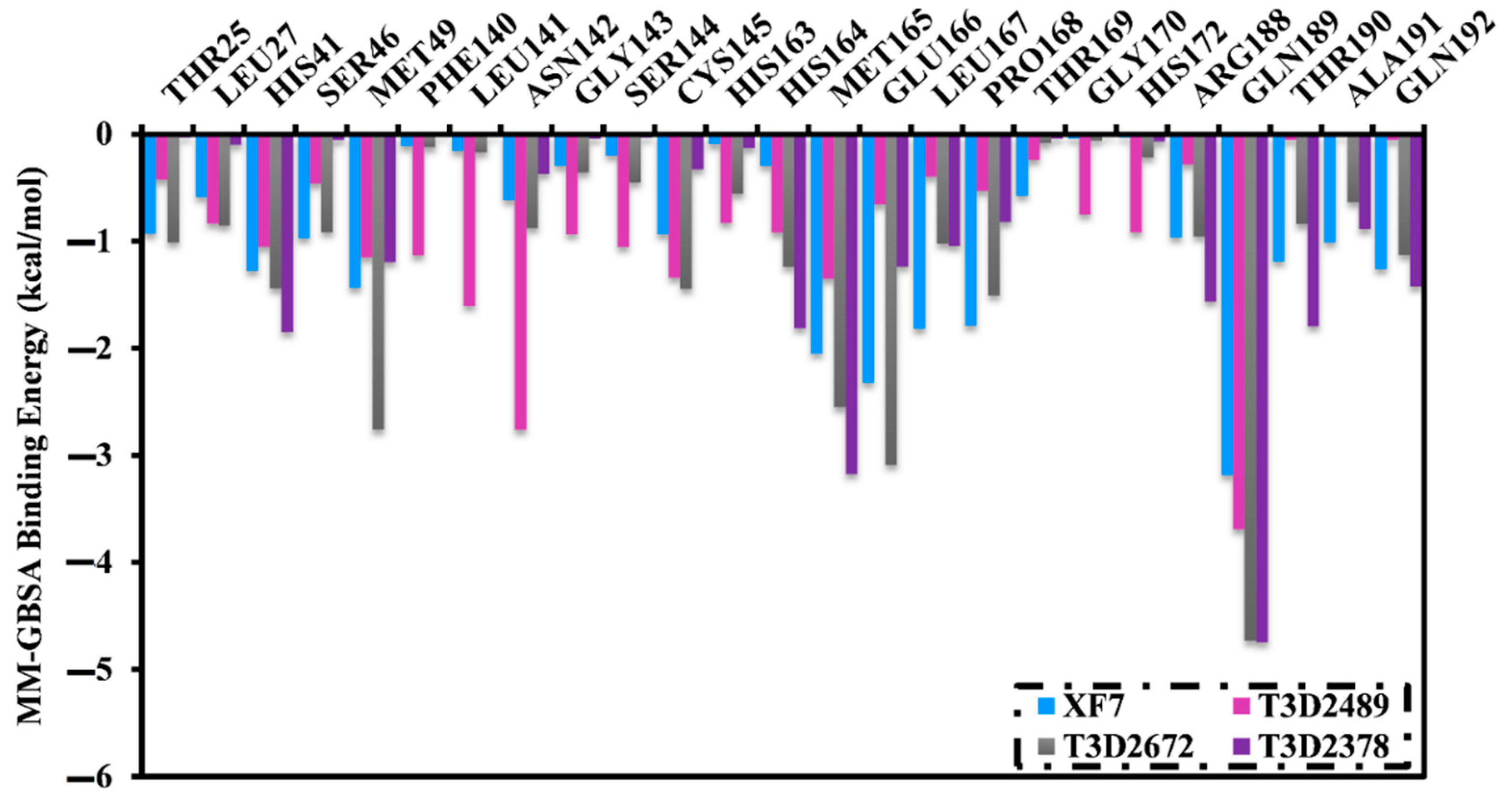
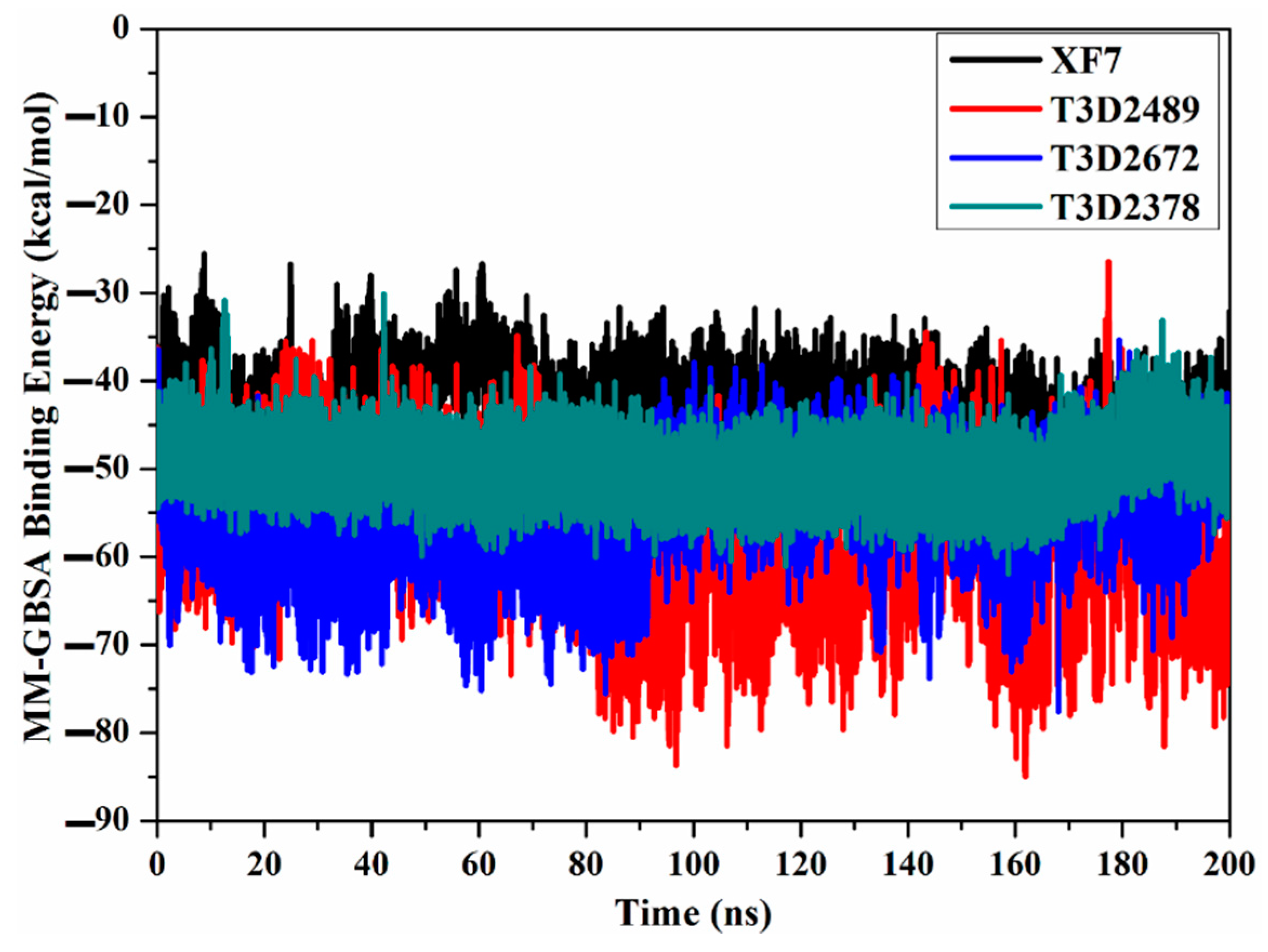
2.4.1. Binding Energy per Frame
2.4.2. Intermolecular Hydrogen Bonds
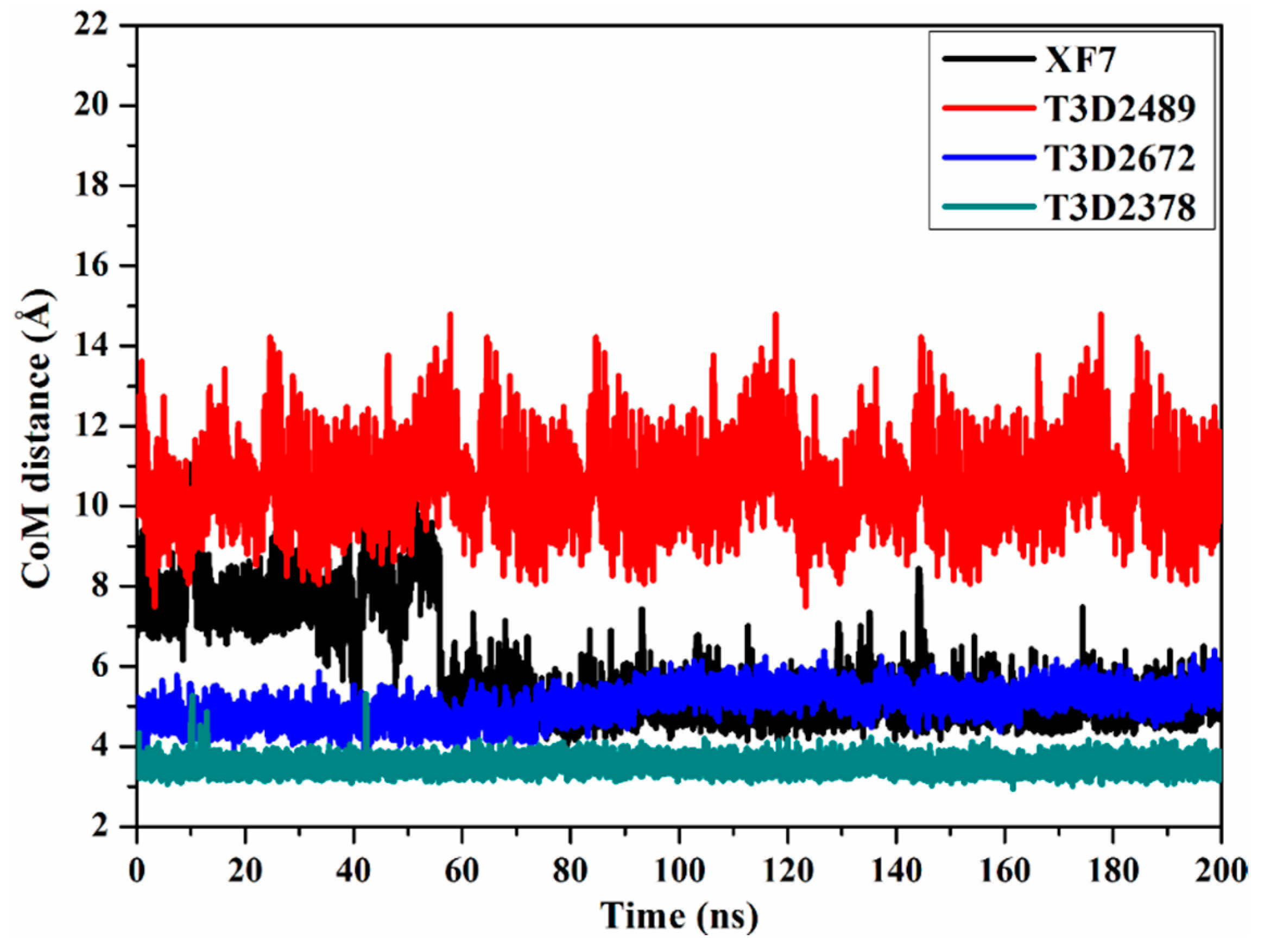
2.4.3. Center-of-Mass Distance
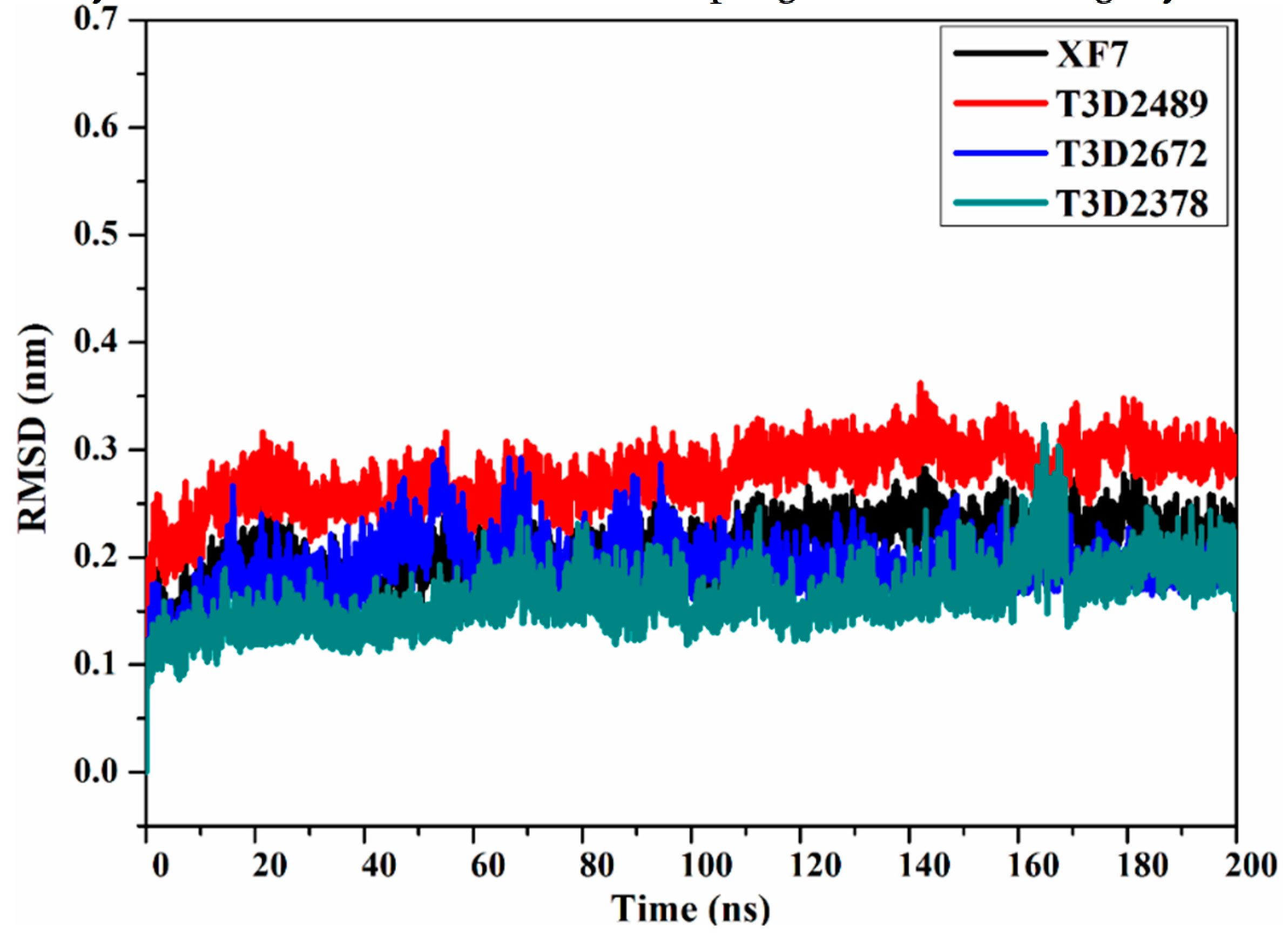
2.4.4. Root-Mean-Square Deviation
2.5. Drug-like Properties
2.6. In Silico ADMET Analysis
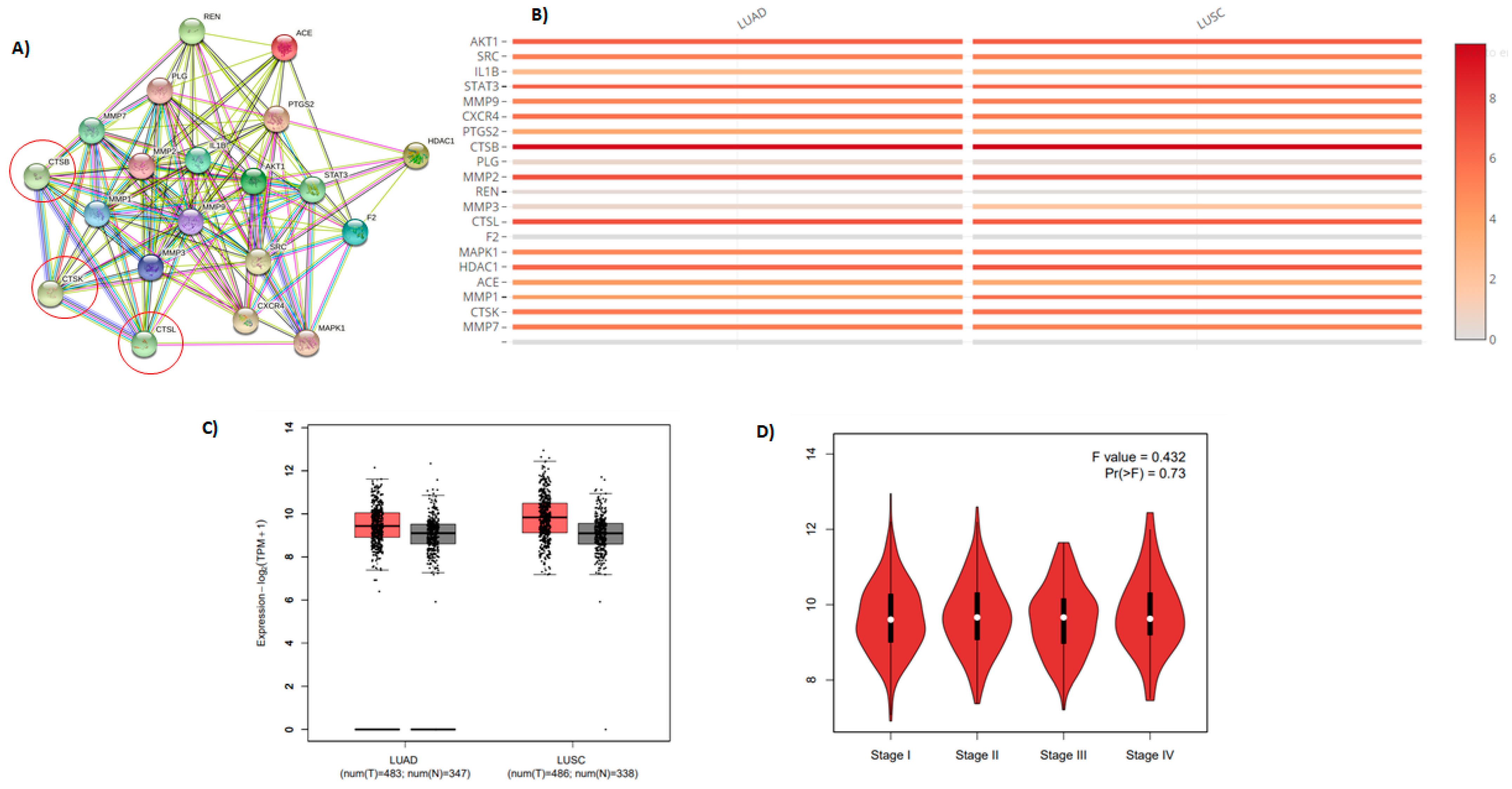
2.7. Molecular Target Prediction and Network Analysis
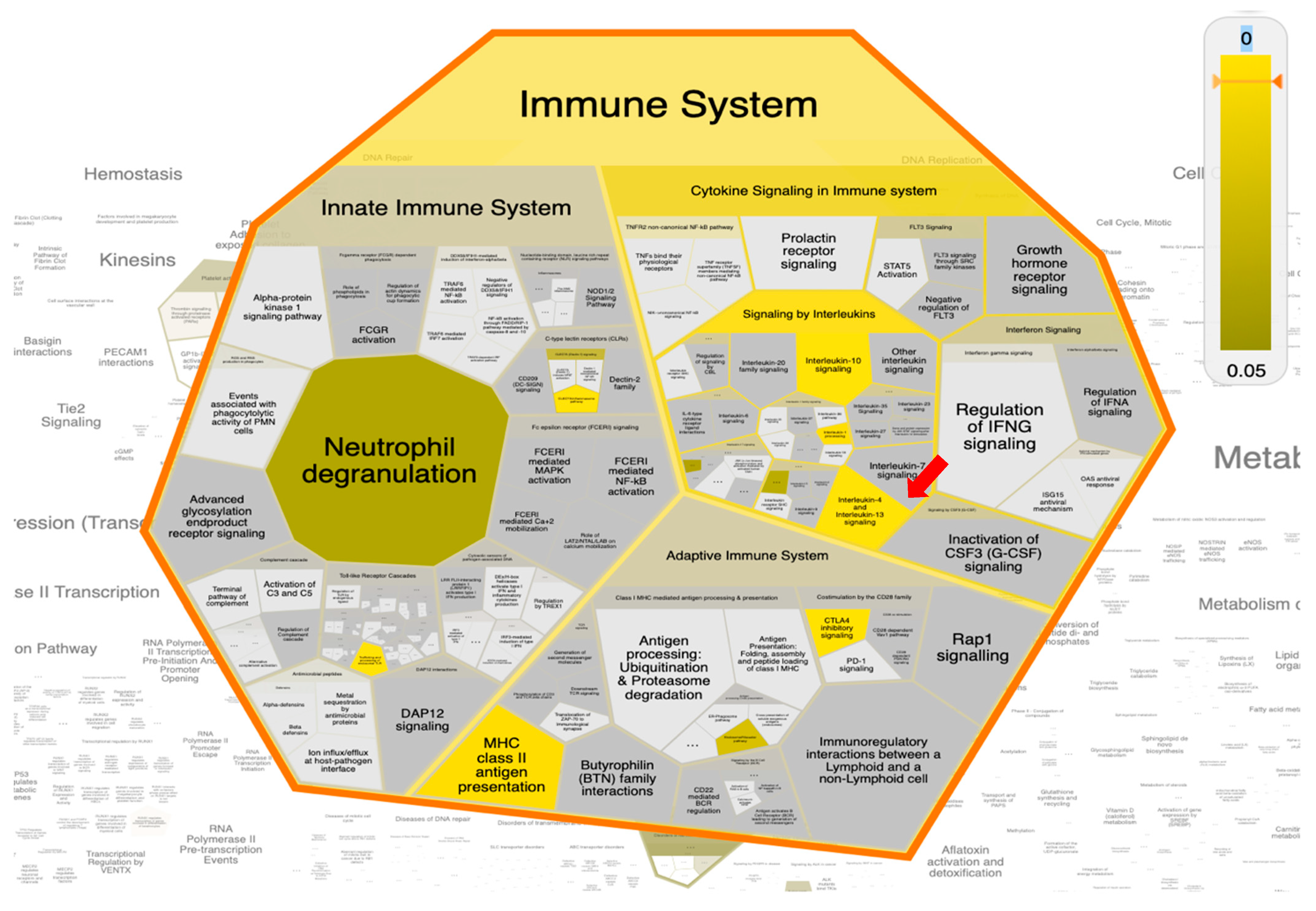
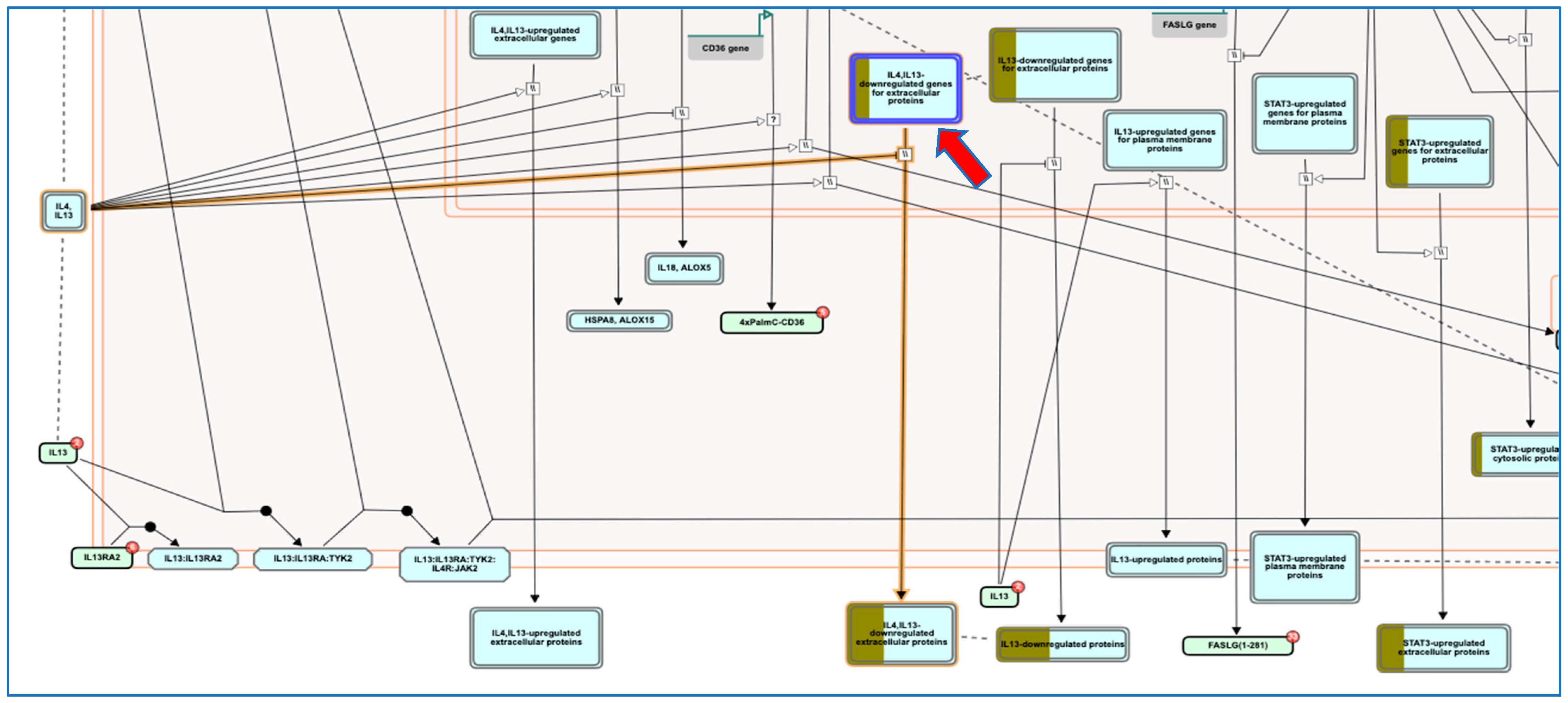
2.8. Pathway Enrichment Analysis (PEA) and Reactome Mining
3. Materials and Methods
3.1. Target Preparation
3.2. Database Preparation
3.3. Molecular Docking
3.4. Molecular Dynamics Simulations
3.5. MM-GBSA Binding Energy
3.6. Drug-Likeness Properties
3.7. In Silico ADMET Analysis
3.8. Protein Interaction Network Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 29 December 2021).
- WHO COVID-19 Vaccines. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/covid-19-vaccines (accessed on 29 December 2021).
- Li, Q.; Kang, C. Progress in Developing Inhibitors of SARS-CoV-2 3C-Like Protease. Microorganisms 2020, 8, 1250. [Google Scholar] [CrossRef]
- Suarez, D.; Diaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model. 2020, 60, 5815–5831. [Google Scholar] [CrossRef]
- Verma, S.; Twilley, D.; Esmear, T.; Oosthuizen, C.B.; Reid, A.M.; Nel, M.; Lall, N. Anti-SARS-CoV Natural Products With the Potential to Inhibit SARS-CoV-2 (COVID-19). Front. Pharmacol. 2020, 11, 561334. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Allemailem, K.S.; Almatroudi, A.; Moustafa, M.F.; Hegazy, M.F. In Silico Evaluation of Prospective Anti-COVID-19 Drug Candidates as Potential SARS-CoV-2 Main Protease Inhibitors. Protein J. 2021, 40, 296–309. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Hegazy, M.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 5756–5767. [Google Scholar] [CrossRef]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.Y.; Xu, M.Y.; Wang, X.; Zhang, H.Y.; Hu, H.R.; Li, Y.F.; Hu, Z.H.; Zhong, W.; Wang, M.L. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Hussien, T.A.; Badr, E.A.A.; Mohamed, T.A.; El-Seedi, H.R.; Pare, P.W.; Efferth, T.; Hegazy, M.F. In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors. Comput. Biol. Med. 2020, 126, 104046. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Mohamed, E.A.R.; Abdelrahman, A.H.M.; Allemailem, K.S.; Moustafa, M.F.; Shawky, A.M.; Mahzari, A.; Hakami, A.R.; Abdeljawaad, K.A.A.; Atia, M.A.M. Rutin and flavone analogs as prospective SARS-CoV-2 main protease inhibitors: In silico drug discovery study. J. Mol. Graph. Model. 2021, 105, 107904. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Atia, M.A.M.; Mohamed, T.A.; Moustafa, M.F.; Hakami, A.R.; Khalifa, S.A.M.; Alhumaydhi, F.A.; Alrumaihi, F.; Abidi, S.H.; et al. Blue Biotechnology: Computational Screening of Sarcophyton Cembranoid Diterpenes for SARS-CoV-2 Main Protease Inhibition. Mar Drugs 2021, 19, 391. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Mohamed, T.A.; Atia, M.A.M.; Al-Hammady, M.A.M.; Abdeljawaad, K.A.A.; Elkady, E.M.; Moustafa, M.F.; Alrumaihi, F.; Allemailem, K.S.; et al. In Silico Mining of Terpenes from Red-Sea Invertebrates for SARS-CoV-2 Main Protease (M(pro)) Inhibitors. Molecules 2021, 26, 2082. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Hegazy, M.F. Natural-like products as potential SARS-CoV-2 M(pro) inhibitors: In-silico drug discovery. J. Biomol. Struct. Dyn. 2021, 39, 5722–5734. [Google Scholar] [CrossRef]
- Zakaryan, H.; Arabyan, E.; Oo, A.; Zandi, K. Flavonoids: Promising natural compounds against viral infections. Arch. Virol. 2017, 162, 2539–2551. [Google Scholar] [CrossRef]
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS ONE 2020, 15, e0240653–e0240666. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.-S.; Shin, D.H. Flavonoids with inhibitory activity against SARS-CoV-2 3CLpro. J. Enzym. Inhib. Med. Chem. 2020, 35, 1539–1544. [Google Scholar] [CrossRef]
- Fact Sheet for Healthcare Providers: Emergency Use Authorization for PAXLOVID. Available online: https://www.fda.gov/media/155050/download (accessed on 20 December 2021).
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Bordon, K.C.F.; Cologna, C.T.; Fornari-Baldo, E.C.; Pinheiro-Junior, E.L.; Cerni, F.A.; Amorim, F.G.; Anjolette, F.A.P.; Cordeiro, F.A.; Wiezel, G.A.; Cardoso, I.A.; et al. From Animal Poisons and Venoms to Medicines: Achievements, Challenges and Perspectives in Drug Discovery. Front. Pharmacol. 2020, 11, 1132–1160. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.; Pon, A.; Djoumbou, Y.; Knox, C.; Shrivastava, S.; Guo, A.C.; Neveu, V.; Wishart, D.S. T3DB: A comprehensively annotated database of common toxins and their targets. Nucleic Acids Res. 2010, 38, D781–D786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, T.A.; Elshamy, A.I.; Ibrahim, M.A.A.; Atia, M.A.M.; Ahmed, R.F.; Ali, S.K.; Mahdy, K.A.; Alshammari, S.O.; Al-Abd, A.M.; Moustafa, M.F.; et al. Gastroprotection against Rat Ulcers by Nephthea Sterol Derivative. Biomolecules 2021, 11, 1247. [Google Scholar] [CrossRef]
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475. [Google Scholar] [CrossRef]
- Wellendorph, P.; Jaroszewski, J.W.; Hansen, S.H.; Franzyk, H. A sequential high-yielding large-scale solution-method for synthesis of philanthotoxin analogues. Eur. J. Med. Chem. 2003, 38, 117–122. [Google Scholar] [CrossRef]
- Satake, M.; Ofuji, K.; Naoki, H.; James, K.J.; Furey, A.; McMahon, T.; Silke, J.; Yasumoto, T. Azaspiracid, a new marine toxin having unique spiro ring assemblies, isolated from Irish mussels, Mytilus edulis. J. Am. Chem. Soc. 1998, 120, 9967–9968. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Shen, M.; Tian, S.; Li, Y.; Li, Q.; Xu, X.; Wang, J.; Hou, T. Drug-likeness analysis of traditional Chinese medicines: 1. property distributions of drug-like compounds, non-drug-like compounds and natural compounds from traditional Chinese medicines. J. Cheminform. 2012, 4, 31. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Zhang, J.; Hu, C.Q.; Zhang, X.; Ma, B.; Zhang, P. In silico ADME and Toxicity Prediction of Ceftazidime and Its Impurities. Front. Pharmacol. 2019, 10, 434. [Google Scholar] [CrossRef]
- Bakht, M.A.; Yar, M.S.; Abdel-Hamid, S.G.; Al Qasoumi, S.I.; Samad, A. Molecular properties prediction, synthesis and antimicrobial activity of some newer oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5862–5869. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Lennernas, H. Intestinal Permeability and Drug Absorption: Predictive Experimental, Computational and In Vivo Approaches. Pharmaceutics 2019, 11, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pohlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral. Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.M.; Yang, W.L.; Yang, F.Y.; Zhang, L.; Huang, W.J.; Hou, W.; Fan, C.F.; Jin, R.H.; Feng, Y.M.; Wang, Y.C.; et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target. Ther. 2021, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Luo, S.; Libby, P.; Shi, G.P. Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef]
- Smieszek, S.P.; Przychodzen, B.P.; Polymeropoulos, M.H. Amantadine disrupts lysosomal gene expression: A hypothesis for COVID19 treatment. Int. J. Antimicrob. Agents 2020, 55, 106004. [Google Scholar] [CrossRef]
- Gomes, C.P.; Fernandes, D.E.; Casimiro, F.; da Mata, G.F.; Passos, M.T.; Varela, P.; Mastroianni-Kirsztajn, G.; Pesquero, J.B. Cathepsin L in COVID-19: From Pharmacological Evidences to Genetics. Front. Cell. Infect. Microbiol. 2020, 10, 589505. [Google Scholar] [CrossRef]
- Vaz de Paula, C.B.; de Azevedo, M.L.V.; Nagashima, S.; Martins, A.P.C.; Malaquias, M.A.S.; Miggiolaro, A.; da Silva Motta Junior, J.; Avelino, G.; do Carmo, L.A.P.; Carstens, L.B.; et al. IL-4/IL-13 remodeling pathway of COVID-19 lung injury. Sci. Rep. 2020, 10, 18689. [Google Scholar] [CrossRef]
- Guizani, I.; Fourti, N.; Zidi, W.; Feki, M.; Allal-Elasmi, M. SARS-CoV-2 and pathological matrix remodeling mediators. Inflamm. Res. 2021, 70, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef] [PubMed]
- OMEGA, 2.5.1.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2013.
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- SZYBKI 1.9.0.3, 1.9.0.3; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Halgren, T.A. MMFF VI. MMFF94s option for energy minimization studies. J. Comput. Chem. 1999, 20, 720–729. [Google Scholar] [CrossRef]
- QUACPAC, 1.7.0.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminform. 2015, 7, 23. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [Green Version]
- Mansourian, M.; Fassihi, A.; Saghaie, L.; Madadkar-Sobhani, A.; Mahnam, K.; Abbasi, M. QSAR and docking analysis of A2B adenosine receptor antagonists based on non-xanthine scaffold. Med. Chem. Res. 2014, 24, 394–407. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges—The Resp Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E01, Gaussian Inc.: Wallingford, CT, USA, 2009.
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle—An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Fish, R.H.; Jaouen, G. Bioorganometallic Chemistry: Structural Diversity of Organometallic Complexes with Bioligands and Molecular Recognition Studies of Several Supramolecular Hosts with Biomolecules, Alkali-Metal Ions, and Organometallic Pharmaceuticals. Organometallics 2003, 22, 2166–2177. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Abraham, M.H.; Le, J.; Hersey, A.; Luscombe, C.N.; Beck, G.; Sherborne, B.; Cooper, I. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Blucher, A.S.; McWeeney, S.K.; Stein, L.; Wu, G. Visualization of drug target interactions in the contexts of pathways and networks with ReactomeFIViz. F1000Research 2019, 8, 908. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name/Code | Origin/Usage b | Chemical Structure | Docking Score (kcal/mol) | Compound Name/Code | Origin/Usage b | Chemical Structure | Docking Score kcal/mol) | ||
|---|---|---|---|---|---|---|---|---|---|
| Conv. c | Exp. d | Conv. c | Exp. d | ||||||
| XF7 |  | −9.1 | −9.5 | T3D2324 | Industrial/workplace toxin |  | −9.2 | −9.9 | |
| T3D2489 | Insect toxin (Egyptian solitary wasp) |  | −11.7 | −11.7 | T3D2680 | Synthetic compound (anticholesteremic agent) |  | −9.8 | −9.9 |
| T3D2672 | Marine toxin (Mytilus edulis) |  | −11.3 | −11.6 | T3D2884 | Synthetic compound (antineoplastic agent) |  | −9.8 | −9.9 |
| T3D2378 | Industrial/workplace toxin |  | −11.2 | −11.2 | T3D4082 | Plant toxin (Veratrum californicum) |  | −9.8 | −9.8 |
| T3D2807 | Synthetic compound (anti-anxiety agent) |  | −10.9 | −10.9 | T3D2694 | Food toxin (antihypoparathyroid agent) |  | −9.7 | −9.8 |
| T3D2825 | Synthetic compound (antihypertensive agent) |  | −10.9 | −10.9 | T3D2871 | Food toxin (antipsychotic agent) |  | −9.7 | −9.8 |
| T3D2874 | Synthetic compound (anti-allergic agent) |  | −10.8 | −10.9 | T3D4050 | Plant toxin (Solanum chacoense) |  | −9.6 | −9.8 |
| T3D2938 | Synthetic compound (anti-allergic agent) |  | −10.5 | −10.8 | T3D2536 | Animal toxin (B. rubescens, B. marinus) |  | −9.6 | −9.8 |
| T3D2913 | Synthetic compound (antipsychotic agent) |  | −10.5 | −10.8 | T3D2933 | Synthetic compound (antipsychotic agent) |  | −9.6 | −9.7 |
| T3D4084 | Plant toxin (genus Veratrum) |  | −10.2 | −10.6 | T3D4051 | Marine toxin (Tiostrea chilensis) |  | −9.5 | −9.7 |
| T3D2727 | Synthetic compound (antineoplastic agent) |  | −10.1 | −10.5 | T3D2527 | Animal toxin (genus Dendrobates and genus Phyllobates) |  | −9.5 | −9.7 |
| T3D2460 | Synthetic compound (vasoconstrictor agent) |  | −10.1 | −10.2 | T3D0233 | Synthetic compound (pesticide) |  | −9.5 | −9.7 |
| T3D2750 | Synthetic compound (vasoconstrictor agent) |  | −10.0 | −10.2 | T3D2910 | Synthetic compound (cholinesterase inhibitor) |  | −9.5 | −9.6 |
| T3D2801 | Synthetic compound (psychotropic drug) |  | −10.0 | −10.1 | T3D2863 | Synthetic compound (anti-HIV agent) |  | −9.5 | −9.6 |
| T3D4083 | Plant toxin (genus Veratrum) |  | −9.9 | −10.1 | T3D2535 | Animal toxin (B. gargarizans) |  | −9.4 | −9.6 |
| T3D2939 | Synthetic compound (vasoconstrictor agent) |  | −9.8 | −10.0 | T3D4232 | Bacterial toxin (cyanobacteria) |  | −9.3 | −9. 6 |
| T3D2143 | Industrial/workplace toxin |  | −9.8 | −10.0 | |||||
| Compound Name/Code | milogP | TPSA | nON | nOHNH | Nrotb | MWt | %ABS |
|---|---|---|---|---|---|---|---|
| T3D2489 | 0.03 | 128.5 | 8 | 7 | 18 | 435.6 | 64.7% |
| T3D2672 | 6.7 | 163.7 | 13 | 4 | 9 | 842.1 | 52.5% |
| T3D2378 | 1.7 | 61.9 | 6 | 1 | 4 | 385.5 | 87.6% |
| XF7 | 4.3 | 109.9 | 8 | 2 | 6 | 492.5 | 71.1% |
| ADME Parameters | XF7 | T3D2489 | T3D2672 | T3D2378 |
|---|---|---|---|---|
| Absorption | ||||
| Water solubility | −3.5 | −3.0 | −3.4 | −2.5 |
| Caco2 permeability | 0.5 | −0.2 | −0.1 | 0.3 |
| Intestinal absorption (human) | 93.5 | 47.6 | 56.7 | 95.0 |
| Skin Permeability | −2.7 | −2.7 | −2.7 | −3.2 |
| P-glycoprotein substrate | Yes | Yes | Yes | Yes |
| P-glycoprotein I inhibitor | Yes | No | Yes | No |
| P-glycoprotein II inhibitor | Yes | No | Yes | No |
| Distribution | ||||
| VDss (human) | 0.3 | 1.6 | 0.6 | 1.3 |
| BBB permeability | −1.1 | −0.7 | −1.7 | 0.2 |
| CNS permeability | −2.5 | −4.1 | −3.4 | −2.6 |
| Metabolism | ||||
| CYP2D6 substrate | No | No | No | No |
| CYP3A4 substrate | Yes | No | Yes | Yes |
| CYP1A2 inhibitior | No | No | No | No |
| CYP2C19 inhibitior | Yes | No | No | No |
| CYP2C9 inhibitior | Yes | No | No | No |
| CYP2D6 inhibitior | No | No | No | No |
| CYP3A4 inhibitior | Yes | No | No | No |
| Excretion | ||||
| Total Clearance | 0.8 | 1.4 | −0.1 | 0.8 |
| Toxicity | ||||
| AMES toxicity | No | No | No | No |
| Max. tolerated dose (human) | 0.4 | 0.3 | −0.3 | −0.4 |
| hERG I inhibitor | No | No | No | No |
| hERG II inhibitor | Yes | Yes | No | Yes |
| Oral Rat Acute Toxicity (LD50) | 3.3 | 2.7 | 2.8 | 2.4 |
| Oral Rat Chronic Toxicity (LOAEL) | 1.1 | 3.1 | 2.7 | 0.7 |
| Hepatotoxicity | Yes | Yes | Yes | Yes |
| Skin Sensitisation | No | No | No | No |
| T. Pyriformis toxicity | 0.3 | 0.3 | 0.3 | 0.6 |
| Minnow toxicity | 2.4 | 2.2 | 0.9 | 4.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Jaragh-Alhadad, L.A.; Atia, M.A.M.; Alzahrani, O.R.; Ahmed, M.N.; Moustafa, M.S.; Soliman, M.E.S.; Shawky, A.M.; Paré, P.W.; et al. Exploring Toxins for Hunting SARS-CoV-2 Main Protease Inhibitors: Molecular Docking, Molecular Dynamics, Pharmacokinetic Properties, and Reactome Study. Pharmaceuticals 2022, 15, 153. https://doi.org/10.3390/ph15020153
Ibrahim MAA, Abdelrahman AHM, Jaragh-Alhadad LA, Atia MAM, Alzahrani OR, Ahmed MN, Moustafa MS, Soliman MES, Shawky AM, Paré PW, et al. Exploring Toxins for Hunting SARS-CoV-2 Main Protease Inhibitors: Molecular Docking, Molecular Dynamics, Pharmacokinetic Properties, and Reactome Study. Pharmaceuticals. 2022; 15(2):153. https://doi.org/10.3390/ph15020153
Chicago/Turabian StyleIbrahim, Mahmoud A. A., Alaa H. M. Abdelrahman, Laila A. Jaragh-Alhadad, Mohamed A. M. Atia, Othman R. Alzahrani, Muhammad Naeem Ahmed, Moustafa Sherief Moustafa, Mahmoud E. S. Soliman, Ahmed M. Shawky, Paul W. Paré, and et al. 2022. "Exploring Toxins for Hunting SARS-CoV-2 Main Protease Inhibitors: Molecular Docking, Molecular Dynamics, Pharmacokinetic Properties, and Reactome Study" Pharmaceuticals 15, no. 2: 153. https://doi.org/10.3390/ph15020153