 Xiaoxia Wang1,2†
Xiaoxia Wang1,2† Jing Yang
Jing Yang Chuan Wang
Chuan Wang Wentao Zhu
Wentao Zhu- 1West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, Sichuan, China
- 2Central and Clinical Laboratory of Sanya People's Hospital, Sanya, Hainan, China
- 3State Key Laboratory of Infectious Disease Prevention and Control, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
- 4Department of Infectious Diseases and Clinical Microbiology, Beijing Institute of Respiratory Medicine and Beijing Chao-Yang Hospital, Capital Medical University, Beijing, China
The SARS-CoV-2 is still undergoing rapid evolution, resulting in the emergence of several variants of concern, especially the Omicron variants (B.1.1.529), which are surging worldwide. In this study, we tracked Omicron subvariant BA.5.1.3 as the causative agent in the Hainan Province wave in China, which started on 1 August 2022. This was China’s first case of Omicron subvariant BA.5.1.3 and led to an indefinite total lockdown in Hainan with more than 8,500 confirmed cases. We obtained 391 whole genomes from positive nasopharyngeal swab samples in the city of Sanya in Hainan Province, which was the center of this outbreak. More than half of the infected cases were female (58%, 227/391) with a median age of 37.0 years (IQR 23.0–53.0). Median Ct values were 24.9 (IQR 22.6–27.3) and 25.2 (IQR 22.9–27.6) for ORF1ab and N genes, respectively. The total single-nucleotide polymorphism (SNP) numbers of Omicron BA.5.1.3 sampled in Sanya (median 69.0, IQR = 69.0–70.0) compared to those worldwide (median 63.0, IQR = 61.0–64.0) showed a significant difference (p < 0.05). Unique core mutations, including three non-synonymous mutations in ORF1ab (Y1064N, S2844G, and R3574K) and one synonymous mutation in ORF3a (S74S), were found. Phylogenetic analysis showed that virus from Sanya formed an independent sub-clade within the BA.5.1.3 subvariant, and could be divided into 15 haplotypes based on the S gene. The most recent common ancestor for the virus from Sanya was estimated as appearing on 5 July 2022, with 95% HPD ranging from 15 May to 20 September 2022. Thanks to our results, we were also able to delineate the mutational profile of this outbreak and highlight the importance of global genomic surveillance and data sharing.
Introduction
In December 2019, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first isolated from several workers who were suffering from pneumonia in China (Roychoudhury et al., 2020). It was declared as a global pandemic on 11 March 2020 by the World Health Organization (WHO), which is one of the greatest public health emergencies in human history (Roychoudhury et al., 2020). Globally, the COVID-19 pandemic has resulted in more than 628 million confirmed cases, with over 3.8 million deaths as of 2 November 2022.1 The spike (S) protein, this virus’s most important structural protein, plays a dominant role in the viral invasion (Lan et al., 2020; Jackson et al., 2022). The process of SARS-CoV-2 entry is initiated by the interaction between the receptor-binding domain (RBD) in the C-terminus of the S protein sub-segment S1 and angiotensin-converting enzyme 2 (ACE2) receptor on the host cell (Roychoudhury et al., 2020, 2021). This is followed by a conformational change of S protein initiated by type 2 transmembrane serine protease (TMPRSS2) on host cell surface, which allows the virus to enter the cell (Roychoudhury et al., 2021). Then, SARS-CoV-2 hijacks the cellular machinery for viral replication. As the virus continued to evolve, many mutations, especially those in the S gene, have gradually increased over time, leading to the emergence of several SARS-CoV-2 variants, and expanding our understanding of the impact of virus variants (WHO, 2022). Viral variants with mutations in the receptor-binding domain (RBD) within the S gene that can enhance viral transmissibility and virulence or affect vaccine effectiveness (immune escape potential) and diagnostic testing are designated as variants of concerns (VOCs) by the World Health Organization (WHO; Kumar et al., 2022). Thus far, SARS-CoV-2 has evolved into four previously circulating VOCs (Alpha, Beta, Delta, and Gamma) as well as one currently circulating VOC (Omicron) that has rapidly spread throughout the world (Shrestha et al., 2022; Wang et al., 2022).
The ongoing Omicron (B.1.1.529) variant was first detected in South Africa in mid-November 2021 (Araf et al., 2022; Saxena et al., 2022), and categorized as a VOC subsequently by WHO (Maxmen, 2022). It quickly spread to 80 countries within 3 weeks (Viana et al., 2022). Currently, SARS-CoV-2 Omicron is still the dominant variant circulating worldwide (in more than 170 countries and territories), accounting for >98% of SARS-CoV-2 genomes uploaded to the GISAID database since February 2022 (WHO, 2022). The Omicron variants have continued to evolve, resulting in Omicron subvariants that have been classified into several lineages, namely BA.1, BA.2, BA.3, BA.4, BA.5, and descendent lineages (Shrestha et al., 2022). The first three lineages (BA.1, BA.2, and BA.3) were identified almost simultaneously, and they are different not only from each other but also from previously circulating VOCs (Berkhout and Herrera-Carrillo, 2022; Shrestha et al., 2022). More recently, two additional lineages (BA.4 and BA.5) were first discovered in South Africa and have subsequently spread worldwide (Tegally et al., 2021; Maxmen, 2022). The most recent common ancestor of BA.4 and BA.5 has been estimated to appear in mid-November 2021 (Tegally et al., 2022b).
Omicron is the most divergent variant so far, due to its unprecedented set of non-synonymous mutation sites including 60 substitutions, deletions, and insertions (Kumar et al., 2022). Eight substitutions (K856R, L2084I, A2710T, T3255I, P3395H, and I3758V for ORF1a, and P314L and I1566V for ORF1b) and two deletions (four amino acids) have been identified in ORF1a and ORF1b of the Omicron variant contains (He et al., 2021; Kumar et al., 2022). More than 30 mutations have been identified on S protein of this variant, with half of these mutations (G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, and Y505H) accumulated in the RBD, enhancing binding affinity to ACE2 as well as strengthening viral transmission speed (Kumar et al., 2022; Viana et al., 2022). Furthermore, Omicron possesses D614G in the spike protein that shared by five VOCs, correlating with higher viral loads in the upper respiratory tract and a higher rate of infection in younger persons (Korber et al., 2020; Plante et al., 2021). For envelope (E) and membrane (M) proteins, one substitution (T9I) and three substitutions (D3G, Q19E, and A63T) were identified, respectively (He et al., 2021). In addition, three substitutions (P13L, R203K, and G204R) and a deletion of three amino acid residues (ERS31-33del) in the nucleocapsid (N) proteins were also found (He et al., 2021).
Omicron subvariant BA.5.1.3 was first detected in Romania on 24 February 2022, and has since become the world’s prevalent subvariant, identified in 59 countries and territories as of 2 October 2022. The densest concentration of cases has been mainly found in Germany, United States, Denmark, and United Kingdom. Compared with other subvariants, Omicron BA.5.1.3 is more contagious and spreads faster, thanks to the S:V289I mutation (Chen et al., 2021). The S protein of BA.5.1.3 has a common mutation pattern to the BA.4 and BA.5 lineages and is most similar to the BA.2 lineage, excluding 69–70 deletions, L452R, and F486V mutations, and the wild-type amino acid at Q493 (Tegally et al., 2022a).
From 1 August 2022 until early September 2022, a SARS-CoV-2 epidemic raged in Hainan Province in China. In this study, we performed whole-genome SARS-CoV-2 sequencing on RT-PCR positive nasopharyngeal swabs from the city of Sanya (which was the center of the outbreak), detecting the presence of the highly transmissible Omicron BA.5.1.3 subvariant. We then investigated the diversity of 384 genomes including their unique mutations, and compared the binding affinity of several mutations in the S protein. Finally, we conducted phylogenetic analysis and characterized the global distribution of BA.5.1.3.
Materials and methods
Sampling
From August 1st to 18th August 2022, as mentioned previously, SARS-CoV-2 positive (RT-PCR detection) oropharyngeal swab samples were preserved in 3 mL viral sample preservation solution (Inactivated) and collected as part of national COVID-19 routine surveillance from the city of Sanya in Hainan Province in China and preserved in viral transport medium (Zhu et al., 2021, 2022). The total RNA of was extracted from 200 μL sample using the QIAamp viral RNA mini kit (Qiagen, Germany) and eluted using 40 μL RNase-free water. The 5 μL eluted water of each sample was conducted to clinical one-step RT-PCR using detection kit for 2019-nCoV (PCR-Fluorescence) according to the manufacturer’s instructions (DaAnGene, China) targeting the ORF1ab and N genes, respectively. All sampling work was done in collaboration with the national network for COVID-19 surveillance and approved by the Ethics Committee of Sanya People’s Hospital (NO: SYPH-2022-043).
Whole-genome sequencing of SARS-CoV-2
The DNA of the positive oropharyngeal samples was removed using DNase I, and the concentration of total RNA was determined using Qubit 2.0 (Invitrogen, United States). The sequencing libraries were constructed using a gene-specific multiplexed amplicon-based sequencing strategy (ATOPlex SARS-CoV-2) and sequenced on the MGISEQ-2000 (MGI, China) platform (paired reads 100). The raw reads were qualified using FastQC v0.11.92 (Andrews, 2010), and the clean reads were then aligned to the genome of the Wuhan-Hu-1 virus (MN908947.3) using Bowtie 2 (Langmead and Salzberg, 2012) and assembled by SPAdes v3.15.4 (Bankevich et al., 2012).
Lineage classification and variant calling
The lineages of whole SARS-CoV-2 genomes were classified using pangoLEARN v4.03 (O’Toole et al., 2021). The global Omicron BA.5.1.3 genomes (sampling location designated as non-China), along with their complete genomes and collection dates, were downloaded from the GISAID database (date of access 2 October 2022).4 Those genomes with low coverage and > 0.01% of N were parsed. The mutations of genomes were called using SARS-CoV-2-freebayes with Wuhan-Hu-1 as the reference (Farkas et al., 2021), and the obtained VCF files were filtered and annotated using the SnpEff package (Cingolani et al., 2012). The frequency of each variant was calculated, which was then divided into two groups based on sampling location (non-China or China). The single nucleotide polymorphisms (SNPs) of the amino acids and genomes were summarized and visualized using ESPript v3.0 (Robert and Gouet, 2014) and snipit,5 respectively.
Structural modeling
The native protein structure of hACE2 (1R42) was downloaded from the Protein Data Bank (PDB).6 The SARS-CoV-2 RBD structures were modeled using SWISS-MODEL with PDB: 7a91.1 as the template (Waterhouse et al., 2018). Receptor (hACE2)-ligand (RBD) docking was conducted via the Hex protein docking algorithm, using the default parameter settings (shape dimension: 0.6, receptor range: 180, ligand range: 180, distance range: 40, and box size: 10; Macindoe et al., 2010).
Phylogenetic analysis
Whole genome sequences from this study were aligned using MAFFT v7.471 set to the default parameters (Katoh and Standley, 2013). The alignment was submitted to Nextstrain v12 using SARS-CoV-2 workflow and visualized using auspice (Hadfield et al., 2018). Additionally, the BA.5.1.3 globally-sourced and China-sourced genomes were aligned using MAFFT set to the fast option (FFT-NS-2) and trimmed using Gblocks v0.91b (Castresana, 2000), which were then used to build phylogenetic trees using FastTree v2.1 (Price et al., 2010). The haplotype phylogenetic network of S gene was clustered by sampling date and built using PopART v1.7 (Leigh and Bryant, 2015).
Molecular clock estimation
The genomes from this study and representatives of Nextstrain clades belonging to Omicron were aligned and used to infer its preliminary maximum-likelihood tree, which was also inspected for the presence of a temporal signal using TempEst v.1.5.3 (Rambaut et al., 2016). Bayesian time-calibrated tree was build using BEAST v1.10.4 (Suchard et al., 2018), with a strict molecular clock model, an exponential growth coalescent model, and an HKY + I substitution model (Tegally et al., 2021). The length of the Markov chain of the Monte Carlo (MCMC) tree was 1 × 108, with sampling every 1,000 steps. The result was evaluated using Tracer v1.7.1 (effective sample size >200) and summarized in TreeAnnotator v1.10.4, discarding 10% as burn-in (Rambaut et al., 2018; Suchard et al., 2018).
Phylogeographic analysis
To infer the phylogenetic diffusion, the genomic alignment and corresponding location traits were jointly inferred at 1 × 108 generations and sampled every 10,000 steps using BEAST v1.10.4 (Suchard et al., 2018). The geographic transition routes (country level) and posterior probability (PP) were summarized and visualized using SPREAD3 v0.9.7.1 with the Bayesian stochastic search variable selection (BSSVS) method (Bielejec et al., 2016).
Statistical analysis
Statistical plots were conducted using Origin Pro 2021 version. The significant difference was estimated by GraphPad Prism v8.3.0, using either the Mann–Whitney test or Kruskal–Wallis test with p < 0.05 indicating statistical significance.
Results
Outbreak investigation
On 1 August 2022, a SARS-CoV-2 outbreak erupted in Sanya, the southernmost city on the island province of Hainan in China. This epidemic lasted until early September 2022, resulting in 8,630 confirmed cases with 6,595 cases from the city of Sanya (Supplementary Figure S1). The nasopharyngeal swab specimen of the first case was analyzed using next-generation sequencing, indicating that the causative agent was SARS-CoV-2 Omicron BA.5.1.3 (Nextstrain clade 22B), which had spread to more than 60 countries and territories worldwide at the time this sample was collected (Figure 1A). The outbreak in Sanya has been attributed to China’s first cases of the Omicron BA. 5.1.3. From 1 August 1 to 18 August, a total of 391 SARS-CoV-2 positive specimens with corresponding metadata were randomly collected to perform further analysis (Figure 1B). More than half of the confirmed cases were female (58%, 227/391; Figure 1B). The age of individuals ranged from 2 months to 83.0 years old, with a median of 37.0 (IQR 23.0–53.0) years old (Figure 1C). The cycle threshold (Ct) value of ORF1ab gene ranged from 14.8 to 35.2 with median of 24.9 (IQR 22.6–27.3; Figures 2A,B). The Ct value of N gene ranged from 13.7 to 35.0, with a median of 25.2 (IQR 22.9–27.6; Figures 2A,C). The difference in Ct values between female and male cases was insignificant (p > 0.05) for both ORF1ab and N gene (Figures 2B,C).
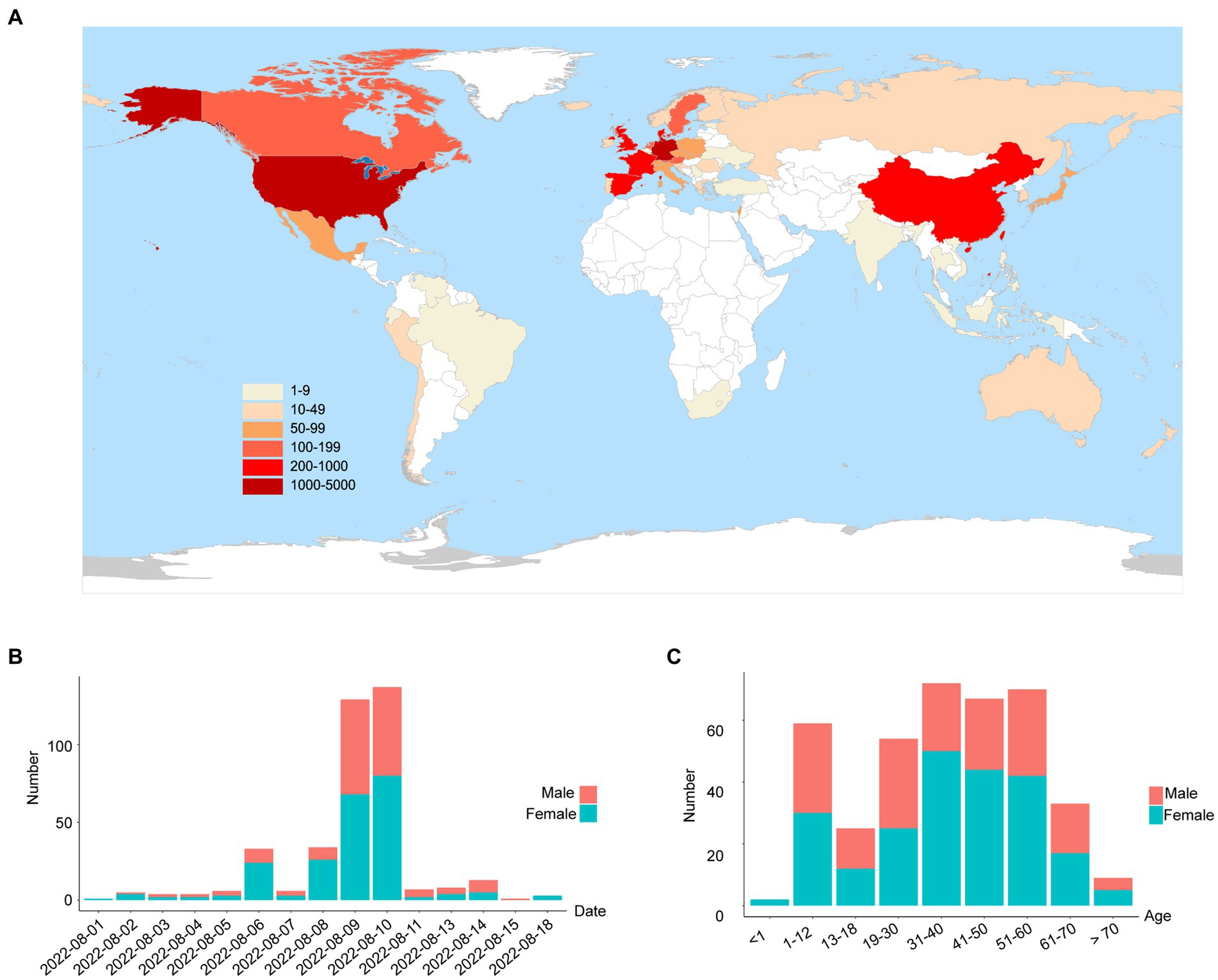
Figure 1. The global emergence of Omicron BA.5.1.3 and cases identified in this study. (A) Map showing the global distribution of Omicron BA.5.1.3. The locations of genomes from this study and GISAID (date of access 2 October 2022) were grouped by countries and territories. The shade of each color represents the corresponding confirmed cases per country. (B) The sampling date of confirmed cases in the city of Sanya. (C) The age range of confirmed cases in Sanya. Orange represents male cases and blue represents female cases.
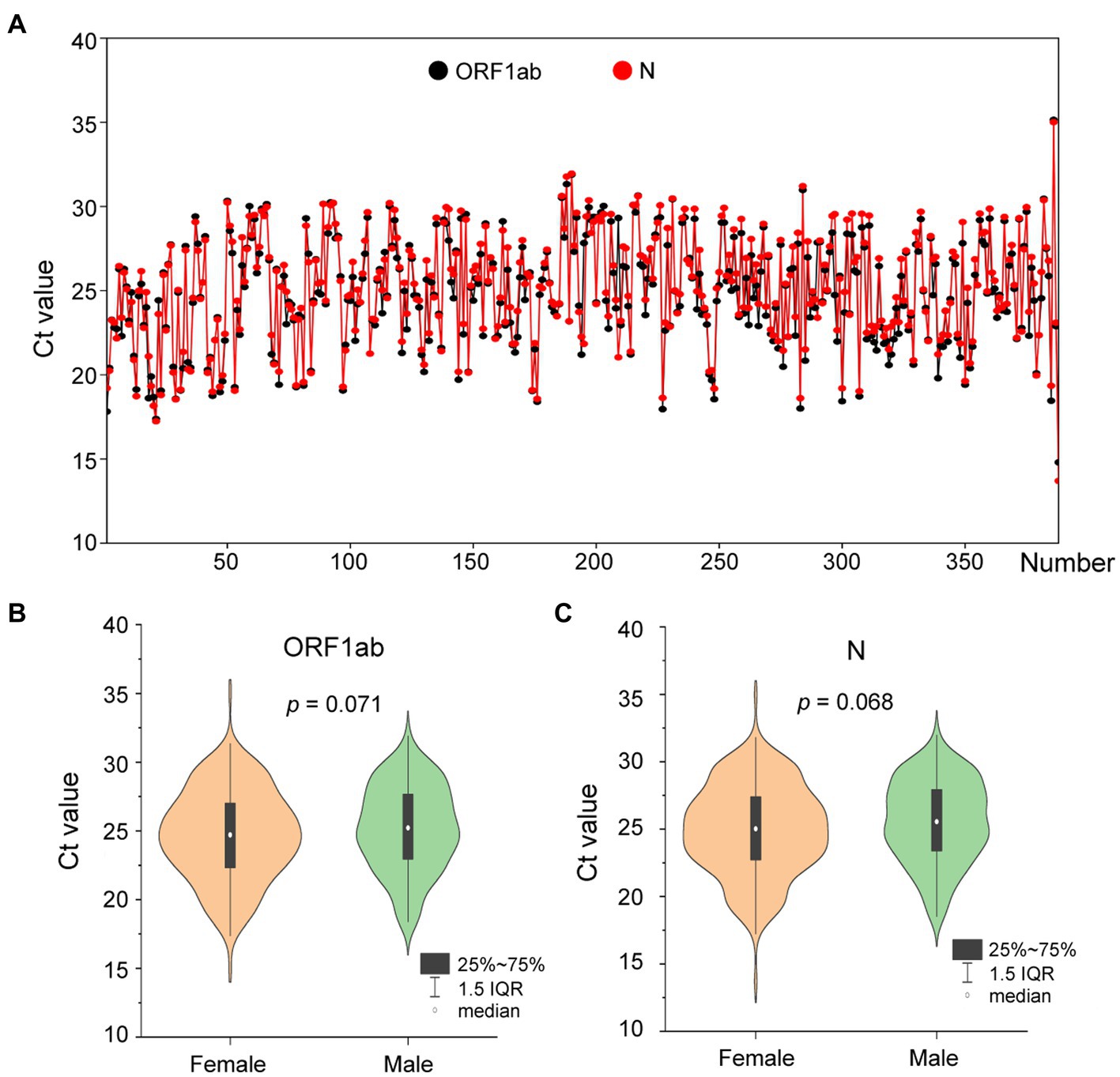
Figure 2. The Ct values of confirmed cases from the city of Sanya. (A) The Ct value of each case. The values of ORF1ab and N genes are labeled with black- and red-filled circles, respectively. (B) The comparison between female and male cases based on Ct value of ORF1ab. (C) The comparison between female and male cases based on Ct value of N.
Evolutionary analysis
After removing the low-quality genomes from the total genomes collected, a total of 384 SARS-CoV-2 genomes were retained for phylogenetic analysis. The results from Nextstrain and Nextclade analyses showed that all these SARS-CoV-2 genomes clustered together and belonged to Omicron BA.5.1.3 (Nextstrain clade 22B; Figure 3A). To assess their detailed phylogenetic relationship, all available BA.5.1.3 Omicron genomes (4,123 genomes retained after parsing) were retrieved from the GISAID database (2 October 2022) and used to build a maximum-likelihood tree. Results revealed that SARS-CoV-2 genomes from the city of Sanya formed a monophyletic cluster within all BA.5.1.3 genomes (Figure 3B). The S gene of Omicron BA.5.1.3 genomes from Sanya could be divided into 15 representative haplotypes, which indicated that the vast majority of genomes (83.3%, 320/384) belonged to haplotype 1 (H1), followed by H2 (8.6%, 33/384) and H3 (3.6%, 14/384; Figure 3C).
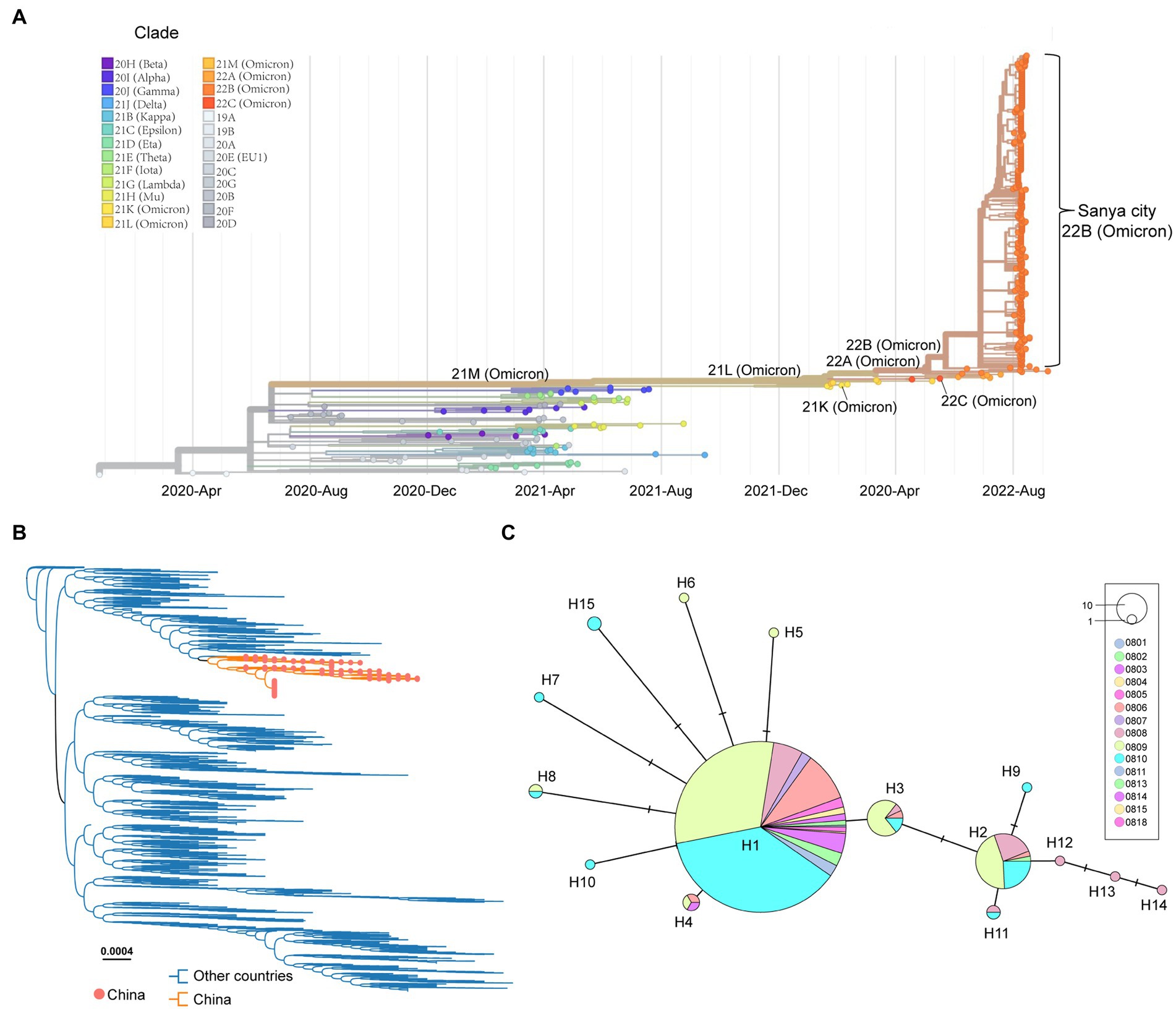
Figure 3. The overview of phylogenetic distribution of genomes identified from the city of Sanya. (A) Nextstrain’s phylodynamic analysis of genomes from Sanya City. (B) The phylogenetic analysis, including BA.5.1.3genomes from the GISAID database and the city of Sanya. The sequences from this study are labeled with orange-filled circles and orange branches. (C) The haplotype network of the S genes identified in the city of Sanya. The network is built by PopART using the median-joining method. The size of the circles represents the number of corresponding haplotypes. The color indicates the corresponding sampling date.
Mutational profile
In total, the genomes from China (in the city of Sanya) had marked hypermutation, including 70 to 90 mutations (containing deletions) with a median of 80 (IQR 80–81), showing a significant difference (p < 0.05) with non-China-sourced genomes (global; Figure 4A). During the outbreak, 25 core mutations (frequency > 98%) were identified in ORF1ab (Supplementary Table S1), which also included three unique mutations (Y1064N, S2844G, and R3574K) that were exclusive or non-core to global BA.5.1.3 genomes (Table 1; Figure 4B).
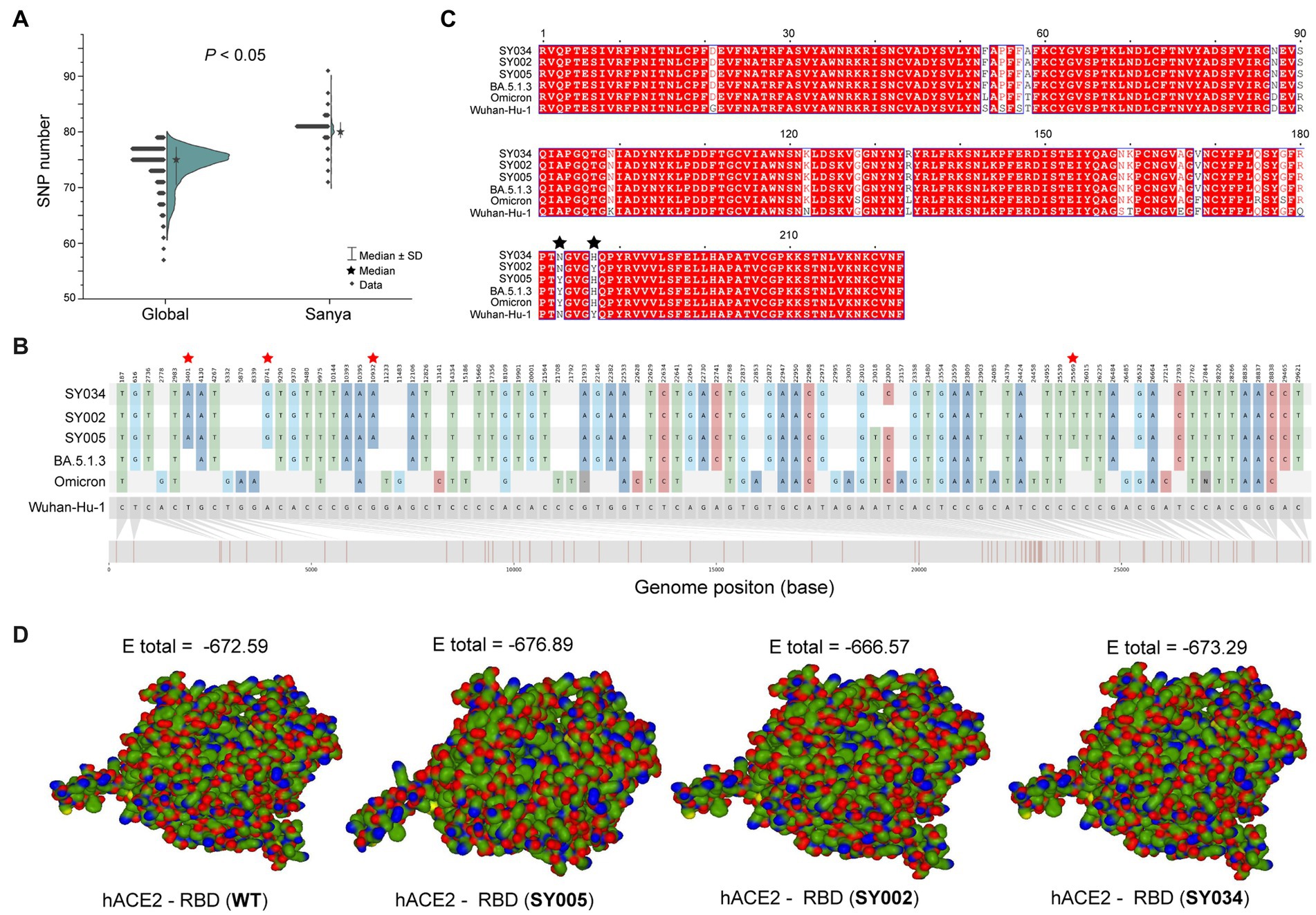
Figure 4. The mutation analyses of whole genomes identified in this study. (A) The comparison of SNP numbers of BA.5.1.3 between China (city of Sanya) and non-China (global) genomes. (B) The whole genome SNP map with Wuhan-Hu-1, Omicron, BA.5.1.3, SY005, SY002, and SY034. The red stars represent the position of unique mutations. (C) The comparison based on amino acid sequences of the RDB. The black stars represent the 501 and 505 positions of S proteins. (D) The receptor-ligand docking between hACE2 and the corresponding RBD. The E total value above each structure indicates the docking scores. The Wuhan-Hu-1, Omicron, and BA.5.1.3 represent the genomes of the original SARS-CoV-2 (MN908947.3), the first Omicron variant (EPI_ISL_6640917), and the first BA.5.1.3 variant (EPI_ISL_14026136), which are available in public databases, respectively. WT here stands for Wuhan-Hu-1.
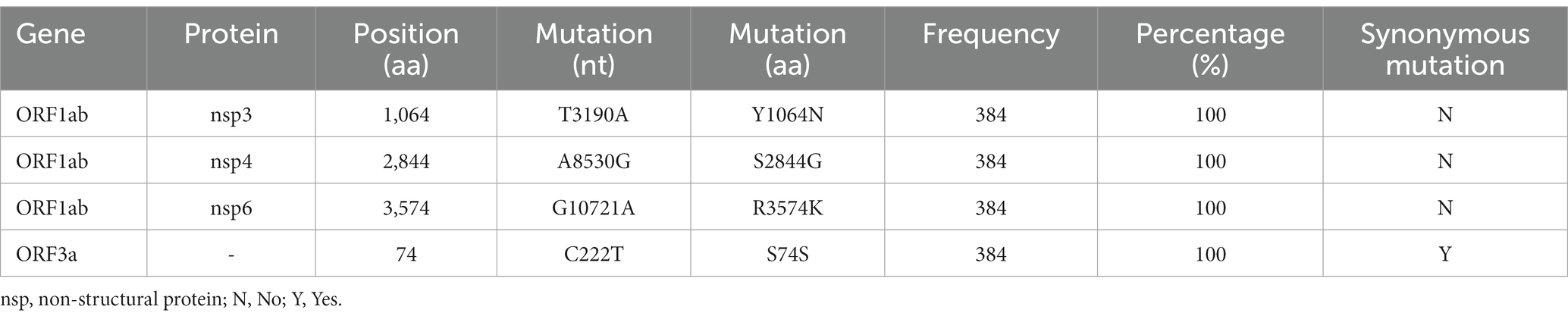
Table 1. Unique mutations identified in BA.5.1.3 genomes from China.
Obviously, the most mutations were located in the S gene, including 35 core mutations (frequency > 99% except for N501Y and Y505H) with one synonymous mutation (D1146D/C3438T). Half (17) of these core mutations of dominating genomes (H1) from the city of Sanya were located in the RBD domain (Figure 4C). The mutation profile of the S gene shared the core mutations of global Omicron BA.5.1.3 genomes (Table 2). In addition, all Sanya-sourced and globally sourced BA.5.1.3 genomes (>99.5%) shared the V289I mutation that was absent in the core mutations of any Omicron sub-lineages, while BA.5.1.3 did not contain the G142D mutation that shared by all other Omicron variants (BA.1, BA.2, BA.4, BA.5, BA.2.12.1, BQ.1, and XBB; Hodcroft, 2021). Interestingly, two revertant mutations with a relatively low frequency were found in position 501 (13.8%, 53/384) and position 505 (10.1%, 39/384), both belonging to the RBD domain (Table 2; Figure 4C).
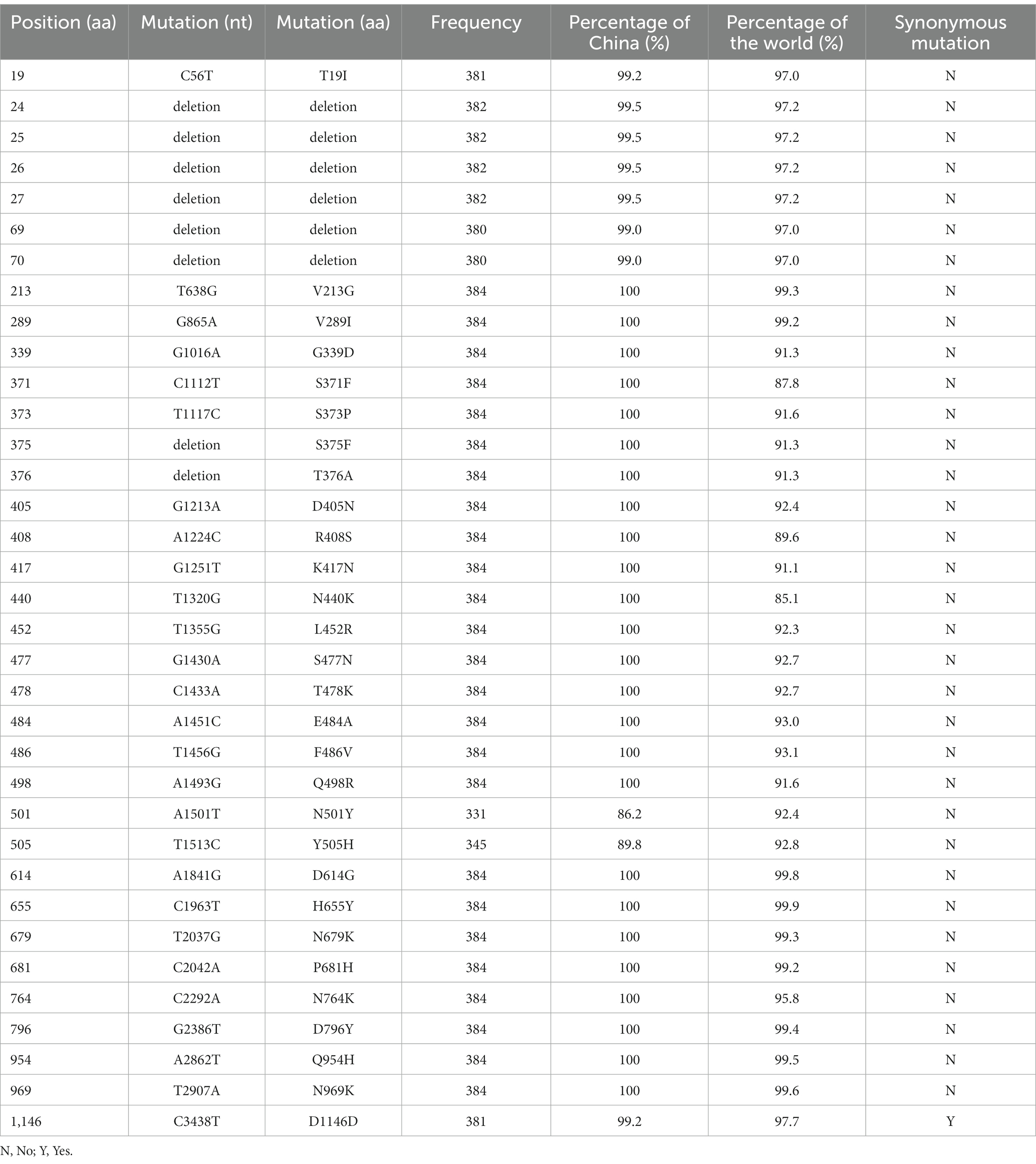
Table 2. Mutations found in S protein of BA.5.1.3 genomes from China.
We detected three (two synonymous and one non-synonymous) core (100%) mutations in ORF3a (T64T, S74S, and T223I), including one novel synonymous mutation (S74S/C222T; Table 1; Figure 4B). Additionally, 13 mutations, namely one non-synonymous mutation (T9I) in E, two non-synonymous mutations (D3N and Q19E) in M, one synonymous mutation (C15C/T45C) in ORF7a, one synonymous mutation (L18L) in ORF7b, seven non-synonymous mutations (P13L, ERS31-33del, RG203KR, and S413R) in N, and one non-synonymous mutation (L37F) in ORF10, were identified as core mutations. Meanwhile, four core mutations were identified in the non-coding regions, including C241T (5′UTR), C27889T (between ORF7b and ORF8), A28271T (between ORF8 and N), and 29734_29759del (3′UTR).
Receptor-ligand docking
The binding affinity between SARS-CoV-2 RBD and hACE2 accounted for variant infectivity and differed between variants (Kumar et al., 2022). Based on the haplotype analysis, the genome sequences of SY005, SY002, and SY034 were designated as representatives of H1, H2, and H3, respectively. We compared the docking energies of the RBD of H1 (SY005, EPI_ISL_15940718), H2 (SY002, EPI_ISL_15940715), and H3 (SY034, EPI_ISL_15940747) binding to hACE2 (Figure 4D), respectively. Results revealed that the docking energy of Wuhan-Hu-1 with hACE2 was −672.59, which was lower than that of the dominant strain SY005 (−676.89), indicating the higher infectivity and transmissibility of the virus in Sanya. The revertant mutation in position 501 (SY034) had a slightly lower docking energy (−673.29) compared with SY005, signifying that N501Y in the RBD promoted viral binding (Figure 4D). The two mutations in positions 501 and 505 reverted simultaneously (SY002) and showed more lower docking energy compared with SY005 and SY034, indicating that both N501Y and Y505H in the RBD might promote viral infectivity (Figure 4D).
Molecular clock analysis
To investigate the phylogenetic history of BA.5.1.3 genomes from Sanya, the representatives of Omicron genomes were used as background to conduct a molecular clock analysis. The temporal signal test showed that the sampling dates were correlated with the root-to-tip distance, implying a significant (R2 = 1) temporal signature (Figure 5). The Bayesian analysis estimated the substitutions of Omicron BA.5.1.3 genomes at 8.896 × 10−4 nucleotide changes per site per year, which was a reliable rate compared to other lineages (Tegally et al., 2021). The time-calibrated phylogenetic tree tracked that the most recent common ancestor (tMRCA) of all BA.5.1.3 genomes from Sanya emerged in early July 2022 with 95% highest posterior density (HPD) ranging from the middle of May to the middle of September 2022 (Figure 5).
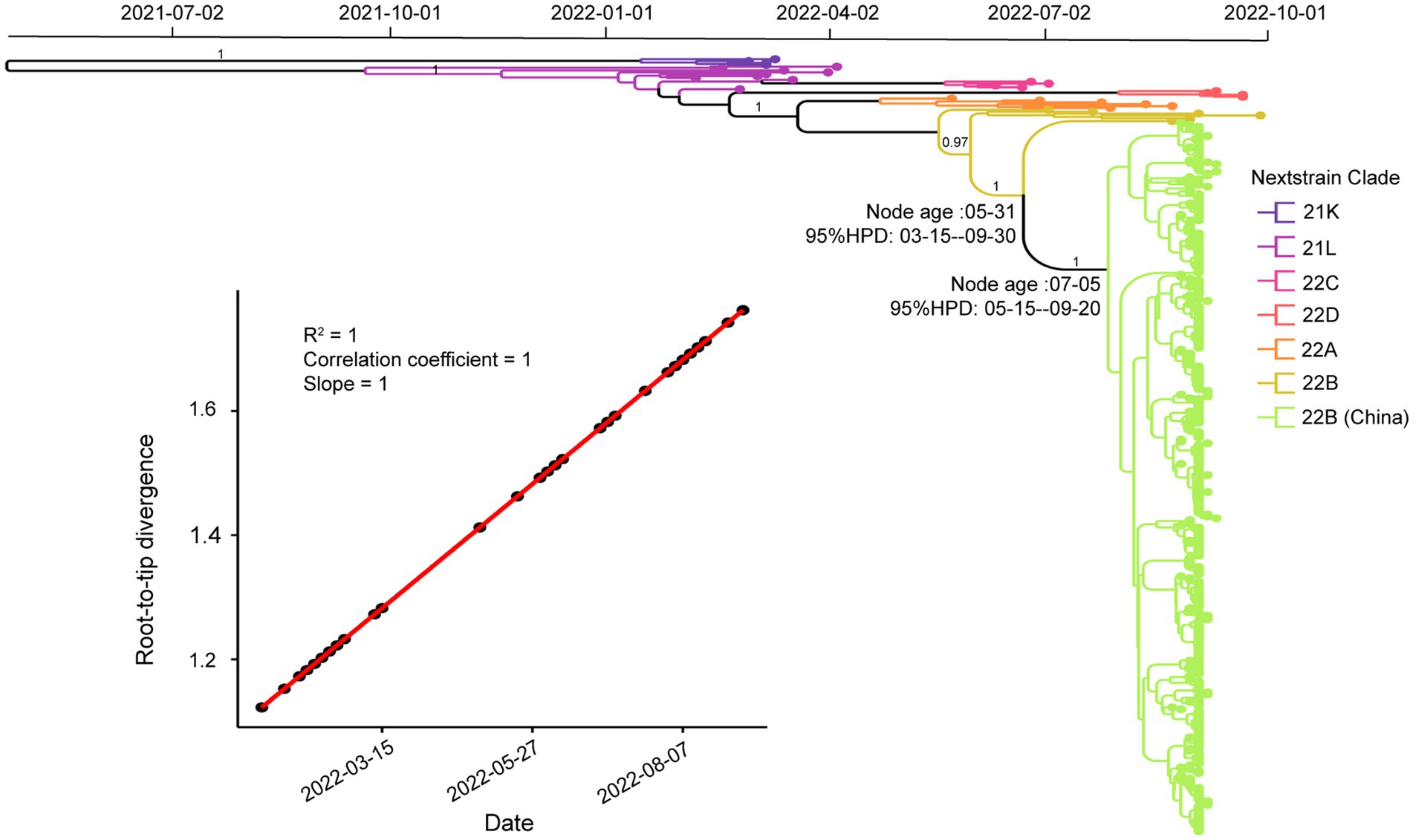
Figure 5. Temporal analysis of Omicron BA.5.1.3 genomes from China. The branches are labeled with different colors according to Nextstrain’s clades. The posterior probabilities are indicated on corresponding branches. The tMRCA of genomes from Sanya is labeled on their corresponding nodes with 95% highest probability density (HPD) values. The molecular clock signal plot revealed the correlation between sampling dates and the root-to-tip distance. The tMRCA was based on collection dates.
Spatial dynamics
To illustrate the population genetics, the Bayesian skyline analysis was conducted based on global BA.5.1.3 genomes, including those from SARS-CoV-2 Omicron BA.5.1.3 outbreak in Sanya. The result showed that the population size of Omicron BA.5.1.3 grew steadily since its emergence in February 2022 and reached the relatively peak in mid-July 2022 (Supplementary Figure S2). The population size reached the summit at the end of August 2022 and decreased starting from that time (Supplementary Figure S2). To infer their spatial spread, the Omicron BA.5.1.3 genomes were first split into different groups based on their corresponding countries and territories. We performed Bayesian phylogeographic analysis according to the BSSVS method, detecting the presence of 18 migration links in the transmission processes of BA.5.1.3 worldwide, with posterior probability (PP) values of 1 (Supplementary Figure S3). Obviously, certain European and European-adjacent countries, especially Germany and United Kingdom, may have played a pivotal role in BA.5.1.3 transmission (Supplementary Figure S3; Supplementary Table S2).
Discussion
The failure to control the transmission of SARS-CoV-2 in many countries and territories around the world has led to the development of hypotheses suggesting that substantial evolutionary mutations accounting for the emergence of virus variants, particularly the Omicron (Fontanet et al., 2021; Mascola et al., 2021; Tegally et al., 2021). In this study, we investigated the China’s first SARS-CoV-2 Omicron BA.5.1.3 outbreak, which has caused more than 8,500 confirmed cases and led to the lockdown of the tropical island known as Hainan Province.
The number of confirmed cases rose quickly in the initial stages of this outbreak. More than half these cases are female (58%), differing from those of the WT (<50%), Alpha (39%), Beta (27%), and Delta (49%) variants (Huang et al., 2020; Ong et al., 2022). In this cohort, we also unexpectedly observed three infants (≤1 years old) and 11 persons of advanced age (≥70 years old), suggesting that people of all ages may be susceptible to Omicron BA.5.1.3. The viral loads in the upper respiratory tract caused by BA.5.1.3 among different age and sex groups show no obvious difference. Surprisingly, the BA.5.1.3 in this study seems to result in higher overall viral load (with a median Ct value of 25.2 for N gene from here) compared with BA.1 (median = 26.29) and BA.2 (median = 25.48) in symptomatic respondents (Whitaker et al., 2022). Meanwhile, the overall median Ct value of 26.4 from large-community surveillance study (Walker et al., 2021) indicated potentially faster transmission and enhanced viral fitness of BA.5.1.3.
The highly mutated SRAS-CoV-2 Omicron BA.5.1.3 have accumulated 35 core mutations in the S protein, higher than that found in the S proteins of Alpha (10), Beta (11), Delta (12), Gamma (9), and Omicron BA.2 (31), though lower than that found in the S protein of Omicron BA.1 (36; Elbe and Buckland-Merrett, 2017). Furthermore, this finding identically reflected the mutational pattern of BA.5.1.3 as it circulated around the world. Half of these mutations are located in the RBD, resulting in higher binding affinity and selective advantage. The receptor-ligand docking estimation confirmed that N501Y and Y505H mutations of the S protein in SARS-CoV-2 enhance its binding affinity to hACE2, suggesting a higher rate of potential immune escape (Tian et al., 2021; Kumar et al., 2022). The S:V289I mutation is detected in all genomes from Sanya and > 99.5% BA.5.1.3 genomes from the GISAID database. Globally, genomes containing the S:V289I mutation accounted for 0.36% of all SARS-CoV-2 genomes, primarily dominated and prevalent in AY.47 (73.55%) and BA.5.1.3 (20.73%; Elbe and Buckland-Merrett, 2017; Chen et al., 2021). The S:G142D mutation is in about half (48.96%) of all SARS-CoV-2 genomes, mainly in BA.2 (18.00%), AY.4 (9.35%), BA.2.12.1 (4.08%), BA.5.2.1 (3.76%), BA.5.2 (3.37%), AY.103 (3.11%), BA.2.9 (3.11%), and BA.5.1 (3.05%; 2 October 2022; Chen et al., 2021). Thus, the absence of S:G142D mutation and presence of S:V289I mutation could be potential markers of BA.5.1.3 sub-lineage when compared with other sub-lineages (Chen et al., 2021; Hodcroft, 2021).
Three (Y1064N, S2844G, and R3574K) unique core (100%) mutations in ORF1ab are identified in BA.5.1.3 genomes from this study. Before 1 August 2022, there are relatively few genomes (94 belonging to 30 sub-lineages) harboring the ORF1a:Y1064N mutation, with only 5 genomes in BA.5.1.3, and 2,754 genomes (belonging to 46 sub-lineages) possessing the ORF1a:R3574K mutation with only 670 genomes in BA.5.1.3 from public database (Chen et al., 2021). However, the ORF1a:S2844G has not so far been detected in BA.5.13 genomes. Given that other core mutations are identical to publicly available BA.5.1.3 genomes, these unique core mutations of genomes from Sanya indicate that the Omicron BA.5.1.3 outbreak shares the same transmission chain. In addition, its three unique core mutations are within non-structural protein (nsp) 3, nsp4, and nsp6, respectively, which play different but equally essential roles in the viral life cycle (Wu et al., 2020; Armstrong et al., 2021; Wang and Cheng, 2022). As the largest non-structural protein, nsp3 plays a pivotal role in SARS-CoV-2 replication and transcription complexes assembly (Armstrong et al., 2021). When nsp4 binds with nsp3, this can cause membrane rearrangement and generate double-membrane vesicles with nsp6 protein as the connectors (Sakai et al., 2017; Ricciardi et al., 2022). Thus, the three unique core mutations may enhance the interaction of these non-structural proteins, resulting in more efficient viral replication.
The time-resolved maximum clade credibility (MCC) analysis estimated that the emergence of tMRCA was 1 month before the Hainan Province outbreak. Due to the earlier use of regular and large-scale nucleic acid amplification tests earlier in China, it seems impossible that the virus could circulate in Sanya City for so long without being detected, suggesting that this Omicron BA.5.1.3 strain might have evolved and adapted for some time before spreading to China. However, the detailed transmission routine of the Omicron BA.5.1.3 to China is still unclear.
This study also has some limitations. First, the clinical symptoms were unavailable to investigate the symptomatic differences from other lineages. Second, the nasopharyngeal swab specimens were only collected once, thus limiting us from monitoring long-term viral load changes. In addition, we were unable to sequence the whole genomes from all positive specimens, indicating of a potential underestimation of population size and mutations.
Conclusion
Our study tracks the BA.5.1.3 outbreak in the city of Sanya in Hainan Province in China, delineates its mutational profile (including unique mutation), and estimates the timing of its emergence. Our results highlight the importance of genomic surveillance and of carrying out collaboration and sharing of genomic data worldwide for an effective response to the COVID-19 pandemic.
Data availability statement
The datasets presented in this study have been deposited in the GISAID database (https://www.gisaid.org/) under accession numbers EPI_ISL_15940714 to EPI_ISL_15941103, and EPI_ISL_15942295.
Author contributions
WZ and CW conceived and designed this study. XW, XZ, YL, and LH collected samples and conducted the experiments. WZ analyzed the data and prepared all the figures. WZ and XW wrote the first manuscript. WZ, CW, and JY revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by Golden Seed Program of Beijing Chaoyang Hospital (CYJZ202220) and also supported by the innovation platform for Jianguo Xu Academicians of Hainan Province.
Acknowledgments
We would like to thank the authors from the originating laboratories that generated SARS-CoV-2 sequences used as reference dataset in this study, as well as the GISAID database (https://www.gisaid.org/) for providing their open-sharing platform.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1183633/full#supplementary-material
Footnotes
2. ^https://github.com/s-andrews/FastQC
3. ^https://github.com/cov-lineages/pangoLEARN
References
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc
Araf, Y., Akter, F., Tang, Y. D., Fatemi, R., Parvez, M. S. A., Zheng, C., et al. (2022). Omicron variant of SARS-CoV-2: genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 94, 1825–1832. doi: 10.1002/jmv.27588
Armstrong, L. A., Lange, S. M., Dee Cesare, V., Matthews, S. P., Nirujogi, R. S., Cole, I., et al. (2021). Biochemical characterization of protease activity of Nsp3 from SARS-CoV-2 and its inhibition by nanobodies. PLoS One 16:e0253364. doi: 10.1371/journal.pone.0253364
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Berkhout, B., and Herrera-Carrillo, E. (2022). SARS-CoV-2 evolution: on the sudden appearance of the omicron variant. J. Virol. 96:e0009022. doi: 10.1128/jvi.00090-22
Bielejec, F., Baele, G., Vrancken, B., Suchard, M. A., Rambaut, A., and Lemey, P. (2016). SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 33, 2167–2169. doi: 10.1093/molbev/msw082
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen, C., Nadeau, S., Yared, M., Voinov, P., Xie, N., Roemer, C., et al. (2021). CoV-Spectrum: analysis of globally shared SARS-CoV-2 data to identify and characterize new variants. Bioinformatics 38, 1735–1737. doi: 10.1093/bioinformatics/btab856
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92. doi: 10.4161/fly.19695
Elbe, S., and Buckland-Merrett, G. (2017). Data, disease and diplomacy: GISAID's innovative contribution to global health. Glob Chall 1, 33–46. doi: 10.1002/gch2.1018
Farkas, C., Mella, A., Turgeon, M., and Haigh, J. J. (2021). A novel SARS-CoV-2 viral sequence Bioinformatic pipeline has found genetic evidence that the viral 3' untranslated region (UTR) is evolving and generating increased viral diversity. Front. Microbiol. 12:665041. doi: 10.3389/fmicb.2021.665041
Fontanet, A., Autran, B., Lina, B., Kieny, M. P., Karim, S. S. A., and Sridhar, D. (2021). SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 397, 952–954. doi: 10.1016/s0140-6736(21)00370-6
Hadfield, J., Megill, C., Bell, S. M., Huddleston, J., Potter, B., Callender, C., et al. (2018). Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123. doi: 10.1093/bioinformatics/bty407
He, X., Hong, W., Pan, X., Lu, G., and Wei, X. (2021). SARS-CoV-2 omicron variant: characteristics and prevention. MedComm 2, 838–845. doi: 10.1002/mco2.110
Hodcroft, E. (2021). CoVariants: SARS-CoV-2 mutations and variants of interest. Available at: https://covariants.org/.
Huang, C., Wang, Y., Li, X., Ren, L., Zhao, J., Hu, Y., et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. doi: 10.1016/s0140-6736(20)30183-5
Jackson, C. B., Farzan, M., Chen, B., and Choe, H. (2022). Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 23, 3–20. doi: 10.1038/s41580-021-00418-x
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Korber, B., Fischer, W. M., Gnanakaran, S., Yoon, H., Theiler, J., Abfalterer, W., et al. (2020). Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cells 182, 812–827.e19. doi: 10.1016/j.cell.2020.06.043
Kumar, S., Thambiraja, T. S., Karuppanan, K., and Subramaniam, G. (2022). Omicron and Delta variant of SARS-CoV-2: a comparative computational study of spike protein. J. Med. Virol. 94, 1641–1649. doi: 10.1002/jmv.27526
Lan, J., Ge, J., Yu, J., Shan, S., Zhou, H., Fan, S., et al. (2020). Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581, 215–220. doi: 10.1038/s41586-020-2180-5
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leigh, J. W., and Bryant, D. (2015). Popart: full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210x.12410
Macindoe, G., Mavridis, L., Venkatraman, V., Devignes, M. D., and Ritchie, D. W. (2010). HexServer: an FFT-based protein docking server powered by graphics processors. Nucleic Acids Res. 38, W445–W449. doi: 10.1093/nar/gkq311
Mascola, J. R., Graham, B. S., and Fauci, A. S. (2021). SARS-CoV-2 viral variants-tackling a moving target. JAMA 325, 1261–1262. doi: 10.1001/jama.2021.2088
Maxmen, A. (2022). Are new omicron subvariants a threat? Here's how scientists are keeping watch. Nature 604, 605–606. doi: 10.1038/d41586-022-01069-4
O’Toole, Á., Scher, E., Underwood, A., Jackson, B., Hill, V., McCrone, J. T., et al. (2021). Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol 7:veab064. doi: 10.1093/ve/veab064
Ong, S. W. X., Chiew, C. J., Ang, L. W., Mak, T. M., Cui, L., Toh, M., et al. (2022). Clinical and Virological features of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern: a retrospective cohort study comparing B.1.1.7 (alpha), B.1.351 (Colonia et al.), and B.1.617.2 (Delta). Clin. Infect. Dis. 75, e1128–e1136. doi: 10.1093/cid/ciab721
Plante, J. A., Liu, Y., Liu, J., Xia, H., Johnson, B. A., Lokugamage, K. G., et al. (2021). Spike mutation D614G alters SARS-CoV-2 fitness. Nature 592, 116–121. doi: 10.1038/s41586-020-2895-3
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian Phylogenetics using tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rambaut, A., Lam, T. T., Max Carvalho, L., and Pybus, O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly path-O-gen). Virus Evol 2:vew007. doi: 10.1093/ve/vew007
Ricciardi, S., Guarino, A. M., Giaquinto, L., Polishchuk, E. V., Santoro, M., di Tullio, G., et al. (2022). The role of NSP6 in the biogenesis of the SARS-CoV-2 replication organelle. Nature 606, 761–768. doi: 10.1038/s41586-022-04835-6
Robert, X., and Gouet, P. (2014). Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324. doi: 10.1093/nar/gku316
Roychoudhury, S., Das, A., Sengupta, P., Dutta, S., Roychoudhury, S., Choudhury, A. P., et al. (2020). Viral pandemics of the last four decades: pathophysiology, health impacts and perspectives. Int. J. Environ. Res. Public Health 17:9411. doi: 10.3390/ijerph17249411
Roychoudhury, S., das, A., Sengupta, P., Dutta, S., Roychoudhury, S., Kolesarova, A., et al. (2021). Viral pandemics of twenty-first century. J. Microbiol. Biotechnol. Food Sci. 10, 711–716. doi: 10.15414/jmbfs.2021.10.4.711-716
Sakai, Y., Kawachi, K., Terada, Y., Omori, H., Matsuura, Y., and Kamitani, W. (2017). Two-amino acids change in the nsp4 of SARS coronavirus abolishes viral replication. Virology 510, 165–174. doi: 10.1016/j.virol.2017.07.019
Saxena, S. K., Kumar, S., Ansari, S., Paweska, J. T., Maurya, V. K., Tripathi, A. K., et al. (2022). Characterization of the novel SARS-CoV-2 omicron (B.1.1.529) variant of concern and its global perspective. J. Med. Virol. 94, 1738–1744. doi: 10.1002/jmv.27524
Shrestha, L. B., Foster, C., Rawlinson, W., Tedla, N., and Bull, R. A. (2022). Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: implications for immune escape and transmission. Rev. Med. Virol. 32:e2381. doi: 10.1002/rmv.2381
Suchard, M. A., Lemey, P., Baele, G., Ayres, D. L., Drummond, A. J., and Rambaut, A. (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol 4:vey016. doi: 10.1093/ve/vey016
Tegally, H., Moir, M., Everatt, J., Giovanetti, M., Scheepers, C., Wilkinson, E., et al. (2022a). Emergence of SARS-CoV-2 omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 28, 1785–1790. doi: 10.1038/s41591-022-01911-2
Tegally, H., Moir, M., Everatt, J., Giovanetti, M., Scheepers, C., Wilkinson, E., et al. (2022b). Continued emergence and evolution of omicron in South Africa: new BA.4 and BA.5 lineages. medRxiv [Preprint].
Tegally, H., Wilkinson, E., Giovanetti, M., Iranzadeh, A., Fonseca, V., Giandhari, J., et al. (2021). Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 592, 438–443. doi: 10.1038/s41586-021-03402-9
Tian, F., Tong, B., Sun, L., Shi, S., Zheng, B., Wang, Z., et al. (2021). N501Y mutation of spike protein in SARS-CoV-2 strengthens its binding to receptor ACE2. Elife 10: e69091. doi: 10.7554/eLife.69091
Viana, R., Moyo, S., Amoako, D. G., Tegally, H., Scheepers, C., Althaus, C. L., et al. (2022). Rapid epidemic expansion of the SARS-CoV-2 omicron variant in southern Africa. Nature 603, 679–686. doi: 10.1038/s41586-022-04411-y
Walker, A. S., Pritchard, E., House, T., Robotham, J. V., Birrell, P. J., Bell, I., et al. (2021). Ct threshold values, a proxy for viral load in community SARS-CoV-2 cases, demonstrate wide variation across populations and over time. Elife 10:e64683. doi: 10.7554/eLife.64683
Wang, L., and Cheng, G. (2022). Sequence analysis of the emerging SARS-CoV-2 variant omicron in South Africa. J. Med. Virol. 94, 1728–1733. doi: 10.1002/jmv.27516
Wang, L., Zhou, H. Y., Li, J. Y., Cheng, Y. X., Zhang, S., Aliyari, S., et al. (2022). Potential intervariant and intravariant recombination of Delta and omicron variants. J. Med. Virol. 94, 4830–4838. doi: 10.1002/jmv.27939
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–w303. doi: 10.1093/nar/gky427
Whitaker, M., Elliott, J., Bodinier, B., Barclay, W., Ward, H., Cooke, G., et al. (2022). Variant-specific symptoms of COVID-19 in a study of 1,542,510 adults in England. Nat. Commun. 13:6856. doi: 10.1038/s41467-022-34244-2
WHO (2022). Tracking SARS-CoV-2 variants. Available at: https://www.who.int/activities/tracking-SARS-CoV-2-variants.
Wu, A., Peng, Y., Huang, B., Ding, X., Wang, X., Niu, P., et al. (2020). Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 27, 325–328. doi: 10.1016/j.chom.2020.02.001
Zhu, W., Yang, J., Lu, S., Huang, Y., Jin, D., Pu, J., et al. (2022). Novel pegiviruses infecting wild birds and rodents. Virol. Sin. 37, 208–214. doi: 10.1016/j.virs.2022.01.013
Keywords: SARS-CoV-2, Omicron BA.5.1.3, outbreak, mutation, Sanya
Citation: Wang X, Zhu X, Lin Y, He L, Yang J, Wang C and Zhu W (2023) Tracking the first SARS-CoV-2 Omicron BA.5.1.3 outbreak in China. Front. Microbiol. 14:1183633. doi: 10.3389/fmicb.2023.1183633
Edited by:
Wenshi Wang, Xuzhou Medical University, ChinaReviewed by:
Jianhui Wang, Yale University, United StatesPetr Sláma, Mendel University in Brno, Czechia
Copyright © 2023 Wang, Zhu, Lin, He, Yang, Wang and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chuan Wang, wangchuan@scu.edu.cn; Wentao Zhu, wentaozhu@126.com
†These authors have contributed equally to this work