
SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
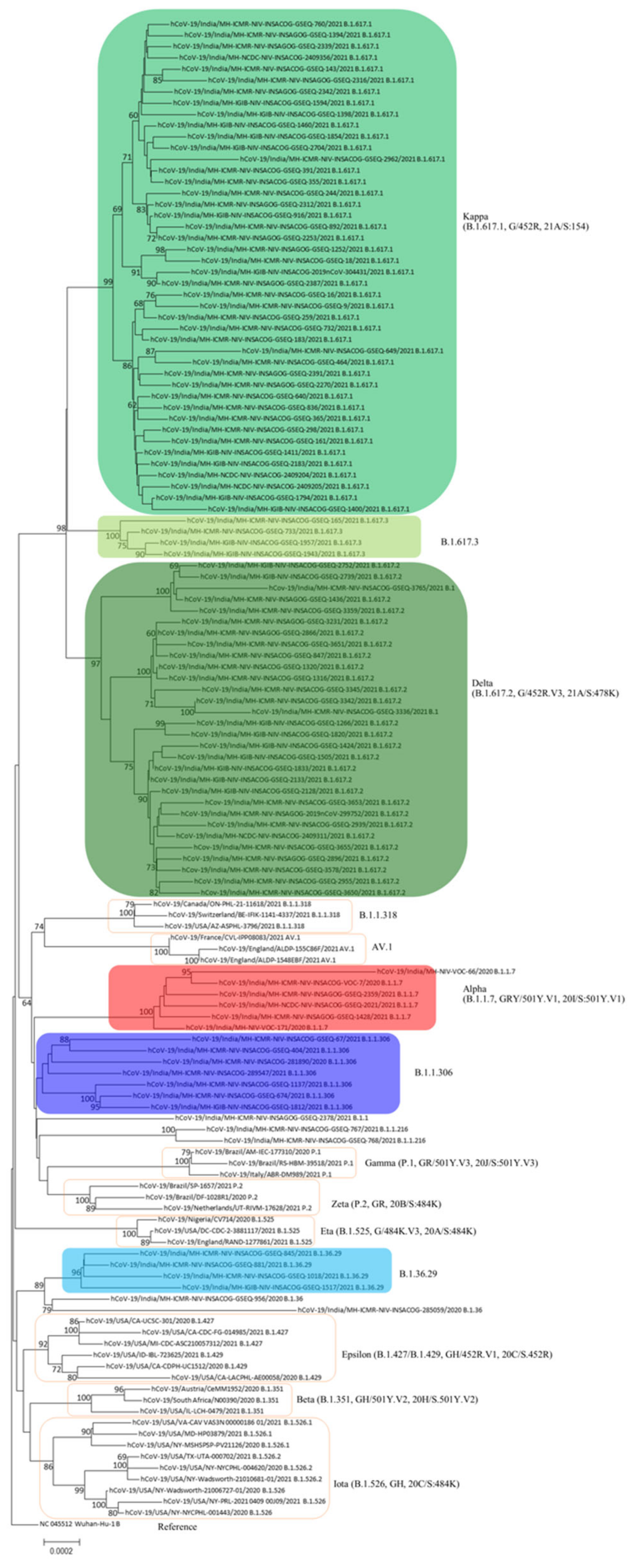
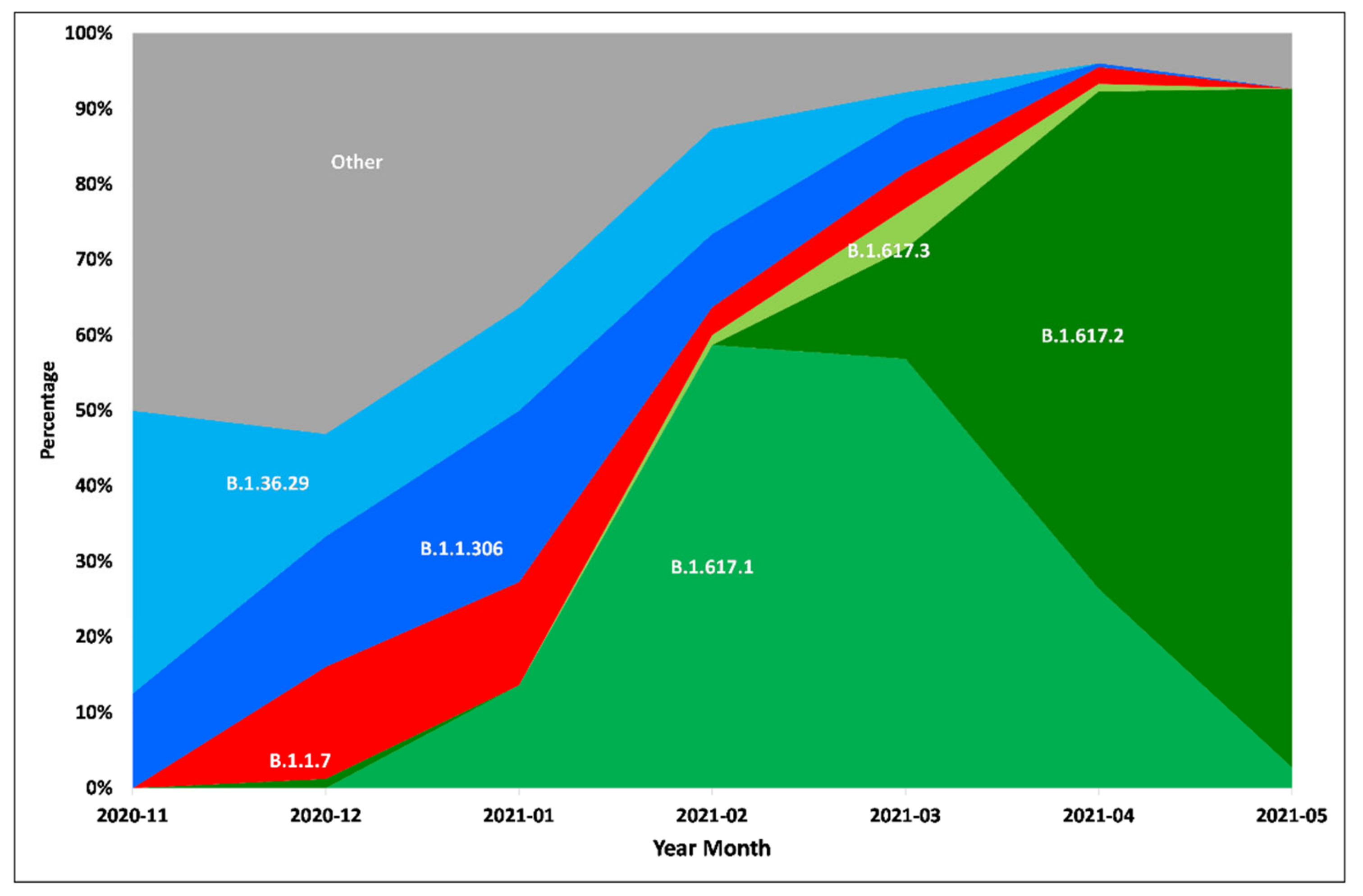
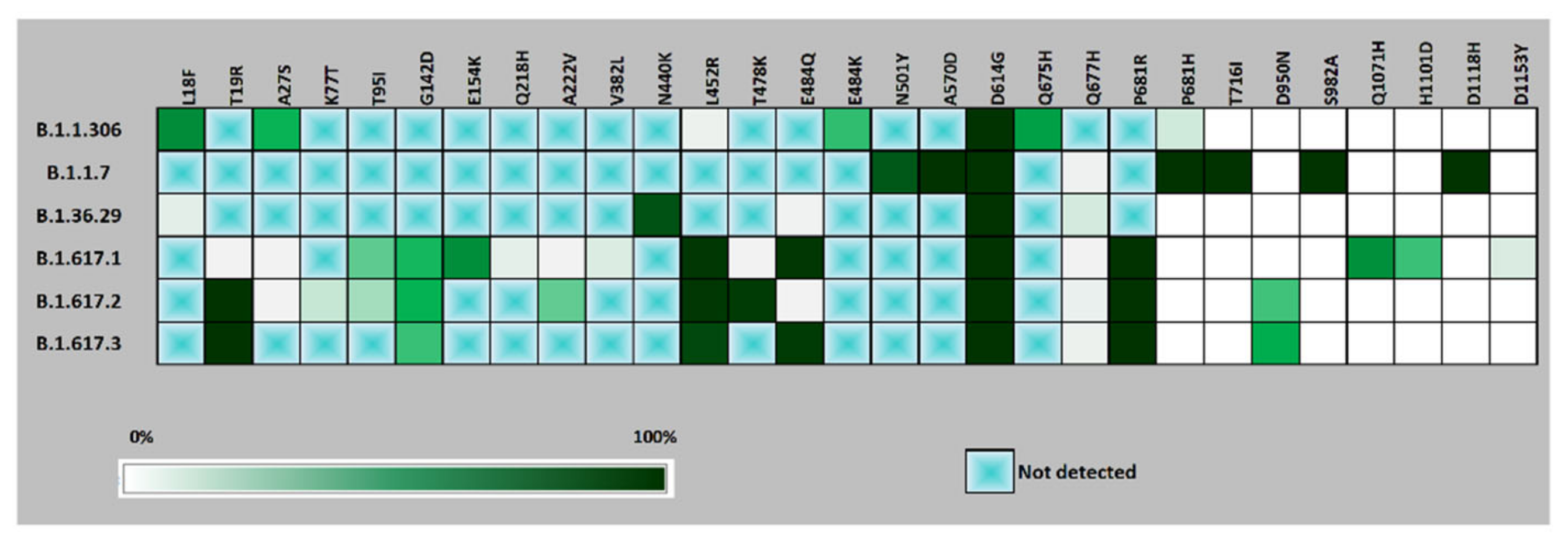
3. Results
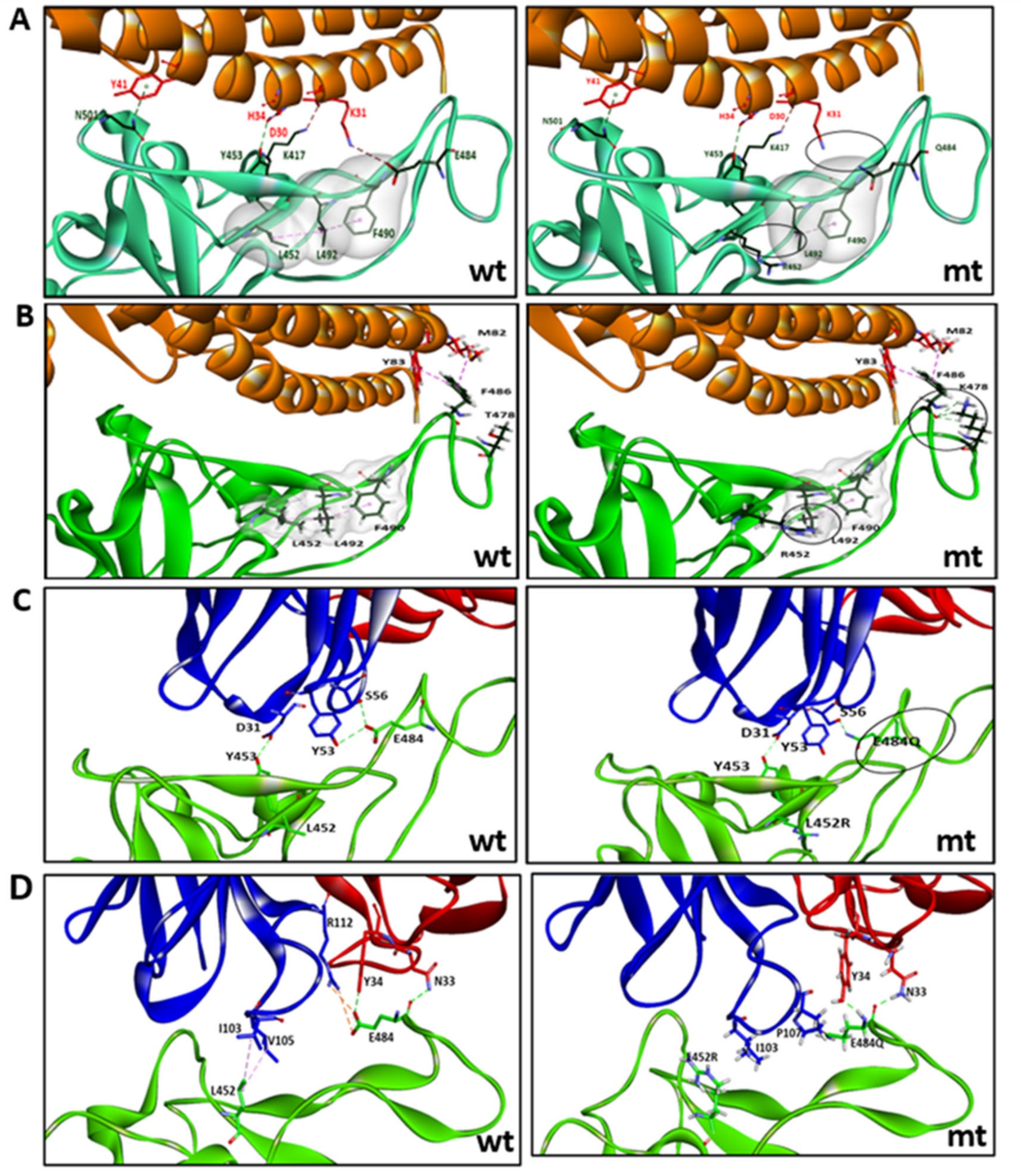
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 12 June 2021).
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Eurosurveillance 2021, 26, 2002106. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Potdar, V.; Vipat, V.; Ramdasi, A.; Jadhav, S.; Pawar-Patil, J.; Walimbe, A.; Patil, S.S.; Choudhury, M.L.; Shastri, J.; Agrawal, S.; et al. Phylogenetic classification of the whole-genome sequences of SARS-CoV-2 from India & evolutionary trends. Indian J. Med. Res. 2021, 153, 166–174. [Google Scholar]
- Shepard, S.S.; Meno, S.; Bahl, J.; Wilson, M.M.; Barnes, J.; Neuhaus, E. Viral deep sequencing needs an adaptive approach: IRMA, the iterative refinement meta-assembler. BMC Genom. 2016, 17, 708. [Google Scholar]
- Yadav, P.D.; Potdar, V.A.; Choudhary, M.L.; Nyayanit, D.A.; Agrawal, M.; Jadhav, S.M.; Majumdar, T.D.; Shete-Aich, A.; Basu, A.; Abraham, P.; et al. Full-genome sequences of the first two SARS-CoV-2 viruses from India. Indian J. Med. Res. 2020, 151, 200–209. [Google Scholar]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–9272. [Google Scholar] [CrossRef] [Green Version]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, A.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. Am. Assoc. Adv. Sci. 2020, 369, 1010–1014. [Google Scholar]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe. 2021, 29, 463–476.e6. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef]
- Yadav, P.D.; Nyayanit, D.A.; Majumdar, T.; Patil, S.; Kaur, H.; Gupta, N.; Shete, A.M.; Pandit, P.; Kumar, A.; Aggarwal, N.; et al. An Epidemiological Analysis of SARS-CoV-2 Genomic Sequencesfrom Different Regions of India. Viruses 2021, 13, 925. [Google Scholar] [CrossRef]
- Sarkar, R.; Mitra, S.; Chandra, P.; Saha, P.; Banerjee, A.; Dutta, S.; Chawla-Sarkar, M. Comprehensive analysis of genomic diversity of SARS-CoV-2 in different geographic regions of India: An endeavour to classify Indian SARS-CoV-2 strains on the basis of co-existing mutations. Arch. Virol. 2021, 166, 801–812. [Google Scholar] [CrossRef]
- Zeng, H.-L.; Dichio, V.; Horta, E.R.; Thorell, K.; Aurell, E. Global analysis of more than 50,000 SARS-CoV-2 genomes reveals epistasis between eight viral genes. Proc. Natl. Acad. Sci. USA 2020, 117, 31519–31526. [Google Scholar] [CrossRef] [PubMed]
- England, P.H. SARS-CoV-2 Variants of Concern and Variants under Investigation in England Technical Briefing 10. 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/984274/Variants_of_Concern_VOC_Technical_Briefing_10_England.pdf (accessed on 14 June 2021).
- Tada, T.; Zhou, H.; Dcosta, B.M.; Samanovic, M.I.; Mulligan, M.J.; Landau, N.R. The Spike Proteins of SARS-CoV-2 B.1.617 and B.1.618 Variants Identified in India Provide Partial Resistance to Vaccine-elicited and Therapeutic Monoclonal Antibodies. bioRxiv 2021. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by variant B.1.427/B.1.429. bioRxiv 2021. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Ikeda, T.; Saito, A.; Tan, T.S.; Ngare, I.; Nasser, H.; Kimura, I.; Uriu, K.; et al. An emerging SARS-CoV-2 mutant evading cellular immunity and increasing viral infectivity. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.; Patil, D.Y.; Nyayanit, D.A.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. Neutralization of variant under investigation B.1.617 with sera of BBV152 vaccinees. Clin. Infect. Dis. 2021, ciab411. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Deshpande, G.; Nyayanit, D.A.; Patil, D.Y.; Gupta, N.; Sahay, R.R.; Shete, A.M.; Kumar, S.; et al. Neutralization potential of Covishield vaccinated individuals sera against B.1.617.1. Clin. Infect. Dis. 2021, ciab483. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sapkal, G.N.; Ella, R.; Sahay, R.R.; Nyayanit, D.A.; Patil, D.Y.; Deshpande, G.; Shete, A.M.; Gupta, N.; Mohan, V.K.; et al. Neutralization against B.1.351 and B.1.617.2 with sera of COVID-19 recovered cases and vaccinees of BBV152. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, S.S.; Thelwall, S. Effectiveness of COVID-19 vaccines against the B.1.617.2 variant. medRxiv 2021. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gao, K.; Wang, R.; Wei, G.-W. Revealing the threat of emerging SARS-CoV-2 mutations to antibody therapies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Errico, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe. 2021, 29, 477–488.e4. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and neutralization of a spike L452R SARS-CoV-2 variant. Cell 2021, 184, 3426–3437.e8. [Google Scholar] [CrossRef] [PubMed]
- Tchesnokova, V.; Kulakesara, H.; Larson, L.; Bowers, V.; Rechkina, E.; Kisiela, D.; Sledneva, Y.; Choudhury, D.; Maslova, I.; Deng, K.; et al. Acquisition of the L452R mutation in the ACE2-binding interface of Spike protein triggers recent massive expansion of SARS-CoV-2 variants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Krüger, N.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Sidarovich, A.; Moldenhauer, A.-S.; Winkler, M.S.; Schulz, S.; et al. SARS-CoV-2 variant B.1.617 is resistant to Bamlanivimab and evades antibodies induced by infection and vaccination. bioRxiv 2021. [Google Scholar] [CrossRef]
- Di Giacomo, S.; Mercatelli, D.; Rakhimov, A.; Giorgi, F.M. Preliminary report on severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) Spike mutation T478K. J. Med. Virol. 2021. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. https://doi.org/10.3390/microorganisms9071542
Cherian S, Potdar V, Jadhav S, Yadav P, Gupta N, Das M, Rakshit P, Singh S, Abraham P, Panda S, et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms. 2021; 9(7):1542. https://doi.org/10.3390/microorganisms9071542
Chicago/Turabian StyleCherian, Sarah, Varsha Potdar, Santosh Jadhav, Pragya Yadav, Nivedita Gupta, Mousumi Das, Partha Rakshit, Sujeet Singh, Priya Abraham, Samiran Panda, and et al. 2021. "SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India" Microorganisms 9, no. 7: 1542. https://doi.org/10.3390/microorganisms9071542