Targeting Host Defense System and Rescuing Compromised Mitochondria to Increase Tolerance against Pathogens by Melatonin May Impact Outcome of Deadly Virus Infection Pertinent to COVID-19


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
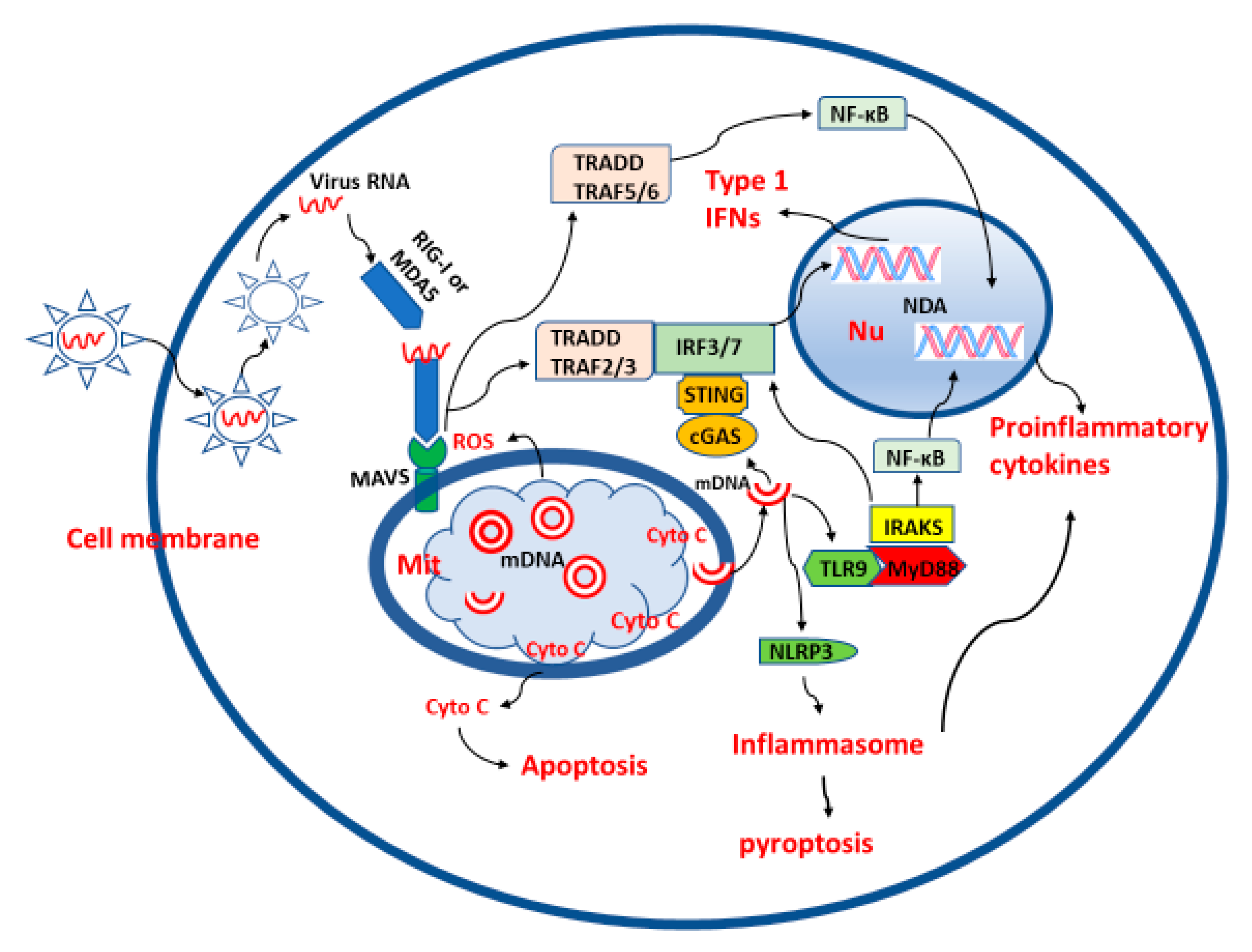
2. Roles of Mitochondria in Viral Infection
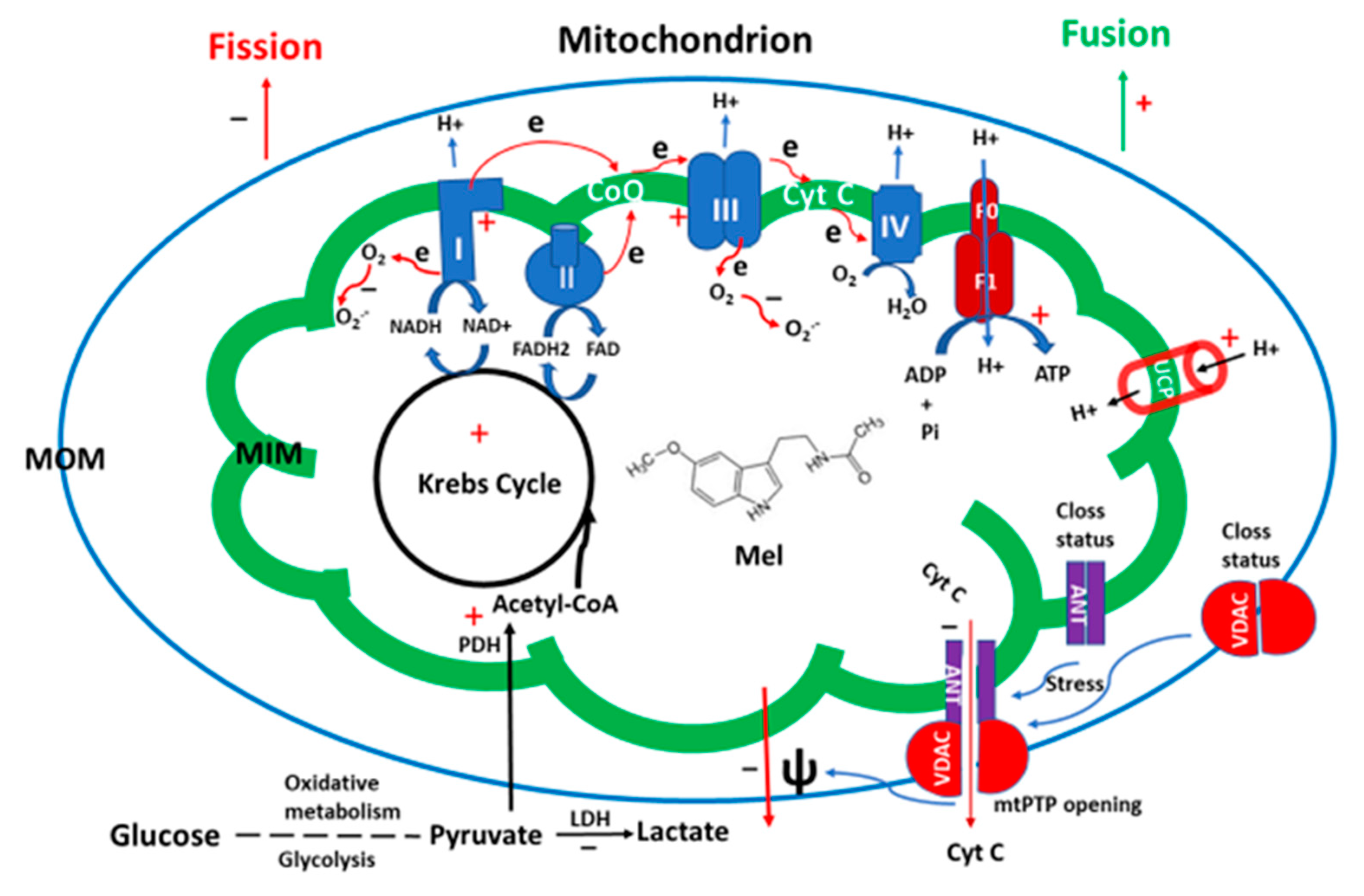
3. Mitochondria and Melatonin
4. Melatonin Synthesis: A Target of Virus Infection
5. The Outcomes of COVID-19 Are Potentially Linked to Melatonin Production
5.1. Aging
5.2. Comorbidities
5.3. Gender
5.4. Children and Pregnant Women
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tan, D.-X. Aging: An evolutionary competition between host cells and mitochondria. Med. Hypotheses 2019, 127, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.A. Vaccine-preventable diseases in pediatric patients: A review of measles, mumps, rubella, and varicella. Pediatr. Emerg. Med. Pract. 2016, 13, 1–20. [Google Scholar] [PubMed]
- Kalarikkal, S.M.; Jaishankar, G.B. Influenza Vaccine; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Kollias, A.; Kyriakoulis, K.G.; Dimakakos, E.; Poulakou, G.; Stergiou, G.S.; Syrigos, K. Thromboembolic risk and anticoagulant therapy in COVID-19 patients: Emerging evidence and call for action. Br. J. Haematol. 2020, 189. [Google Scholar] [CrossRef] [PubMed]
- Barnes, G.D.; Burnett, A.; Allen, A.; Blumenstein, M.; Clark, N.P.; Cuker, A.; Dager, W.E.; Deitelzweig, S.B.; Ellsworth, S.; Garcia, D.; et al. Thromboembolism and anticoagulant therapy during the COVID-19 pandemic: Interim clinical guidance from the anticoagulation forum. J. Thromb. Thrombolysis 2020, 50, 72–81. [Google Scholar] [CrossRef]
- Menezes-Rodrigues, F.S.; Padrão Tavares, J.G.; Pires de Oliveira, M.; Guzella de Carvalho, R.; Ruggero Errante, P.; Omar Taha, M.; José Fagundes, D.; Caricati-Neto, A. Anticoagulant and antiarrhythmic effects of heparin in the treatment of COVID-19 patients. J. Thromb. Haemost. 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117. [Google Scholar] [CrossRef]
- Klopfenstein, T.; Zayet, S.; Lohse, A.; Balblanc, J.-C.; Badie, J.; Royer, P.-Y.; Toko, L.; Mezher, C.; Kadiane-Oussou, N.J.; Bossert, M.; et al. Tocilizumab therapy reduced intensive care unit admissions and/or mortality in COVID-19 patients. Med. Mal. Infect. 2020, 50, 397–400. [Google Scholar] [CrossRef]
- Chan, K.R.; Ong, E.Z.; Mok, D.Z.L.; Ooi, E.E. Fc receptors and their influence on efficacy of therapeutic antibodies for treatment of viral diseases. Expert Rev. Anti-Infect. Ther. 2015, 13, 1351–1360. [Google Scholar] [CrossRef]
- Davidson, S. Treating influenza infection, from now and into the future. Front. Immunol. 2018, 9, 1946. [Google Scholar] [CrossRef]
- Quimque, M.T.J.; Notarte, K.I.R.; Fernandez, R.A.T.; Mendoza, M.A.O.; Liman, R.A.D.; Lim, J.A.K.; Pilapil, L.A.E.; Ong, J.K.H.; Pastrana, A.M.; Khan, A.; et al. Virtual screening-driven drug discovery of SARS-CoV2 enzyme inhibitors targeting viral attachment, replication, post-translational modification and host immunity evasion infection mechanisms. J. Biomol. Struct. Dyn. 2020, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dehelean, C.A.; Lazureanu, V.; Coricovac, D.; Mioc, M.; Oancea, R.; Marcovici, I.; Pinzaru, I.; Soica, C.; Tsatsakis, A.M.; Cretu, O. SARS-CoV-2: Repurposed drugs and novel therapeutic approaches—Insights into chemical structure—Biological activity and toxicological screening. J. Clin. Med. 2020, 9, 2084. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Estcourt, L.; Grant-Casey, J.; Dzik, S. International survey of trials of convalescent plasma to treat COVID-19 infection. Transfus. Med. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Harvala, H.; Mehew, J.; Robb, M.L.; Ijaz, S.; Dicks, S.; Patel, M.; Watkins, N.; Simmonds, P.; Brooks, T.; Johnson, R.; et al. Convalescent plasma treatment for SARS-CoV-2 infection: Analysis of the first 436 donors in England, 22 April to 12 May 2020. Euro Surveill. 2020, 25. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Kennedy, B.K. The World Goes Bats: Living longer and tolerating viruses. Cell Metab. 2020, 32, 31–43. [Google Scholar] [CrossRef]
- Pavlovich, S.S.; Lovett, S.P.; Koroleva, G.; Guito, J.C.; Arnold, C.E.; Nagle, E.R.; Kulcsar, K.; Lee, A.; Thibaud-Nissen, F.; Hume, A.J.; et al. The Egyptian rousette genome reveals unexpected features of bat antiviral immunity. Cell 2018, 173, 1098–1110.e18. [Google Scholar] [CrossRef] [Green Version]
- Shneider, A.; Kudriavtsev, A.; Vakhrusheva, A. Can melatonin reduce the severity of COVID-19 pandemic? Int. Rev. Immunol. 2020, 39, 153–162. [Google Scholar] [CrossRef]
- Tan, D.-X.; Hardeland, R.; Manchester, L.C.; Paredes, S.D.; Korkmaz, A.; Sainz, R.M.; Mayo, J.C.; Fuentes-Broto, L.; Reiter, R.J. The changing biological roles of melatonin during evolution: From an antioxidant to signals of darkness, sexual selection and fitness. Biol. Rev. Camb. Philos. Soc. 2010, 85, 607–623. [Google Scholar] [CrossRef]
- Kleszczyński, K.; Bilska, B.; Stegemann, A.; Flis, D.J.; Ziolkowski, W.; Pyza, E.; Luger, T.A.; Reiter, R.J.; Böhm, M.; Slominski, A.T. Melatonin and its metabolites ameliorate UVR-induced mitochondrial oxidative stress in human MNT-1 melanoma cells. Int. J. Mol. Sci. 2018, 19, 3786. [Google Scholar] [CrossRef] [Green Version]
- Kleszczyński, K.; Zwicker, S.; Tukaj, S.; Kasperkiewicz, M.; Zillikens, D.; Wolf, R.; Fischer, T.W. Melatonin compensates silencing of heat shock protein 70 and suppresses ultraviolet radiation-induced inflammation in human skin ex vivo and cultured keratinocytes. J. Pineal Res. 2015, 58, 117–126. [Google Scholar] [CrossRef]
- Janjetovic, Z.; Jarrett, S.G.; Lee, E.F.; Duprey, C.; Reiter, R.J.; Slominski, A.T. Melatonin and its metabolites protect human melanocytes against UVB-induced damage: Involvement of NRF2-mediated pathways. Sci. Rep. 2017, 7, 1274. [Google Scholar] [CrossRef] [Green Version]
- Shukla, M.; Sotthibundhu, A.; Govitrapong, P. Role of melatonin in regulating neurogenesis: Implications for the neurodegenerative pathology and analogous therapeutics for Alzheimer’s disease. Melatonin Res. 2020, 3, 216–242. [Google Scholar] [CrossRef]
- Mota, A.D.L.; Jardim-Perassi, B.V.; De Castro, T.B.; Colombo, J.; Sonehara, N.M.; Nishiyama, V.K.G.; Pierri, V.A.G.; de Zuccari, D.A.P. Melatonin modifies tumor hypoxia and metabolism by inhibiting HIF-1α and energy metabolic pathway in the in vitro and in vivo models of breast cancer. Melatonin Res. 2019, 2, 83–98. [Google Scholar] [CrossRef]
- Soares, F.T.; Guimarães, H.O.; Da Silveira, P.M.S.; De Sá, A.M.A.; de Sampaio, L.F.S. Melatonin supplementation protects against the benzo(e)pyrene cytotoxicity and optic cup formation disruption in chicken embryos. Melatonin Res. 2020, 3, 210–215. [Google Scholar] [CrossRef]
- Tan, D.X.; Hardeland, R. Potential utility of melatonin in deadly infectious diseases related to the overreaction of innate immune response and destructive inflammation: Focus on COVID-19. Melatonin Res. 2020, 3, 120–143. [Google Scholar] [CrossRef]
- Pal, P.K.; Chattopadhyay, A.; Bandyopadhyay, D. Melatonin as a potential therapeutic molecule against COVID-19 associated gastrointestinal complications: An unrevealed link. Melatonin Res. 2020, 3, 417–435. [Google Scholar] [CrossRef]
- Loh, D. The potential of melatonin in the prevention and attenuation of oxidative hemolysis and myocardial injury from cd147 SARS-CoV-2 spike protein receptor binding. Melatonin Res. 2020, 3, 380–416. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Liu, C.; Manucha, W.; Abreu-Gonzalez, P.; Dominguez-Rodriguez, A. Plasticity of glucose metabolism in activated immune cells: Advantages for melatonin inhibition of COVID-19 disease. Melatonin Res. 2020, 3, 362–379. [Google Scholar] [CrossRef]
- Boga, J.A.; Coto-Montes, A. ER stress and autophagy induced by SARS-CoV-2: The targets for melatonin treatment. Melatonin Res. 2020, 3, 346–361. [Google Scholar] [CrossRef]
- Anderson, G.; Reiter, R.J. COVID-19 pathophysiology: Interactions of gut microbiome, melatonin, vitamin D, stress, kynurenine and the α-7-nicotinic receptor: Treatment implications. Melatonin Res. 2020, 3, 322–345. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, X.; Ni, L.; Di, X.; Ma, B.; Niu, S.; Liu, C.; Reiter, R.J. COVID-19: Melatonin as a potential adjuvant treatment. Life Sci. 2020, 250, 117583. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Reiter, R.J. Melatonin: Roles in influenza, Covid-19, and other viral infections. Rev. Med. Virol. 2020, 30, e2109. [Google Scholar] [CrossRef] [PubMed]
- Feitosa, E.L.; Júnior, F.T.D.S.; Neto, J.A.D.O.N.; Matos, L.F.; Moura, M.H.D.S.; Rosales, T.O.; De Freitas, G.B.L. COVID-19: Rational discovery of the therapeutic potential of melatonin as a SARS-CoV-2 main protease inhibitor. Int. J. Med. Sci. 2020, 17, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Czinn, S.J.; Reiter, R.J.; Blanchard, T.G. Crosstalk between endoplasmic reticulum stress and anti-viral activities: A novel therapeutic target for COVID-19. Life Sci. 2020, 255, 117842. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Martín Giménez, V.M.; Inserra, F.; Tajer, C.D.; Mariani, J.; Ferder, L.; Reiter, R.J.; Manucha, W. Lungs as target of COVID-19 infection: Protective common molecular mechanisms of vitamin D and melatonin as a new potential synergistic treatment. Life Sci. 2020, 254, 117808. [Google Scholar] [CrossRef]
- Kow, C.S.; Hasan, S.S. Could melatonin be used in COVID-19 patients with laryngopharyngeal reflux disease? J. Med. Virol. 2020, jmv.26150. [Google Scholar] [CrossRef]
- Reiter, R.J.; Abreu-Gonzalez, P.; Marik, P.E.; Dominguez-Rodriguez, A. Therapeutic algorithm for use of melatonin in patients with COVID-19. Front. Med. 2020, 7, 226. [Google Scholar] [CrossRef]
- Cheng, F.; Rao, S.; Mehra, R. COVID-19 treatment: Combining anti-inflammatory and antiviral therapeutics using a network-based approach. Cleve. Clin. J. Med. 2020. [Google Scholar] [CrossRef]
- Al-Zaqri, N.; Pooventhiran, T.; Alsalme, A.; Warad, I.; John, A.M.; Thomas, R. Structural and physico-chemical evaluation of melatonin and its solution-state excited properties, with emphasis on its binding with novel coronavirus proteins. J. Mol. Liq. 2020, 15. [Google Scholar] [CrossRef]
- Hazra, S.; Chaudhuri, A.G.; Tiwary, B.K.; Chakrabarti, N. Matrix metallopeptidase 9 as a host protein target of chloroquine and melatonin for immunoregulation in COVID-19: A network-based meta-analysis. Life Sci. 2020, 257, 118096. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P. High doses of melatonin as a potential therapeutic tool for the neurologic sequels of covid-19 infection. Melatonin Res. 2020, 3, 311–317. [Google Scholar] [CrossRef]
- Tan, D.-X.; Hardeland, R. Estimated doses of melatonin for treating deadly virus infections: Focus on COVID-19. Melatonin Res. 2020, 3, 276–296. [Google Scholar] [CrossRef]
- Hardeland, R.; Tan, D.-X. Protection by melatonin in respiratory diseases: Valuable information for the treatment of COVID-19. Melatonin Res. 2020, 3, 264–275. [Google Scholar] [CrossRef]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; Marik, P.E.; Reiter, R.J. Melatonin, cardiovascular disease and COVID-19: A potential therapeutic strategy? Melatonin Res. 2020, 3, 318–321. [Google Scholar] [CrossRef]
- Kleszczyński, K.; Slominski, A.T.; Steinbrink, K.; Reiter, R.J. Clinical Trials for Use of Melatonin to Fight against COVID-19 Are Urgently Needed. Nutrients 2020, 12, 2561. [Google Scholar] [CrossRef]
- Reiter, R.J.; Ma, Q.; Sharma, R. Treatment of ebola and other infectious diseases: Melatonin “goes viral”. Melatonin Res. 2020, 3, 43–57. [Google Scholar] [CrossRef]
- Tan, D.-X.; Korkmaz, A.; Reiter, R.J.; Manchester, L.C. Ebola virus disease: Potential use of melatonin as a treatment. J. Pineal Res. 2014, 57, 381–384. [Google Scholar] [CrossRef]
- Anderson, G.; Maes, M.; Markus, R.P.; Rodriguez, M. Ebola virus: Melatonin as a readily available treatment option. J. Med. Virol. 2015, 87, 537–543. [Google Scholar] [CrossRef]
- Junaid, A.; Tang, H.; van Reeuwijk, A.; Abouleila, Y.; Wuelfroth, P.; van Duinen, V.; Stam, W.; van Zonneveld, A.J.; Hankemeier, T.; Mashaghi, A. Ebola hemorrhagic shock syndrome-on-a-chip. iScience 2020, 23, 100765. [Google Scholar] [CrossRef] [Green Version]
- Castillo, R.R.; Quizon, G.R.A.; Juco, M.J.M.; Roman, A.D.E.; De Leon, D.G.; Punzalan, F.E.R.; Guingon, R.B.L.; Morales, D.D.; Tan, D.-X.; Reiter, R.J. Melatonin as adjuvant treatment for coronavirus disease 2019 pneumonia patients requiring hospitalization (MAC-19 PRO): A case series. Melatonin Res. 2020, 3, 297–310. [Google Scholar] [CrossRef]
- Smit, A.J.; Fitchett, J.M.; Engelbrecht, F.A.; Scholes, R.J.; Dzhivhuho, G.; Sweijd, N.A. Winter is coming: A southern hemisphere perspective of the environmental drivers of SARS-CoV-2 and the potential seasonality of COVID-19. Int. J. Environ. Res. Public Health 2020, 17, 5634. [Google Scholar] [CrossRef] [PubMed]
- Capone, A. Simultaneous circulation of COVID-19 and flu in Italy: Potential combined effects on the risk of death? Int. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Weishaupt, J.H.; Bartels, C.; Pölking, E.; Dietrich, J.; Rohde, G.; Poeggeler, B.; Mertens, N.; Sperling, S.; Bohn, M.; Hüther, G.; et al. Reduced oxidative damage in ALS by high-dose enteral melatonin treatment. J. Pineal Res. 2006, 41, 313–323. [Google Scholar] [CrossRef]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 255–274. [Google Scholar] [CrossRef]
- Youle, R.J. Mitochondria-Striking a balance between host and endosymbiont. Science 2019, 365. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell signalling. J. Cell Sci. 2012, 125, 807–815. [Google Scholar] [CrossRef] [Green Version]
- Hill, S.; Van Remmen, H. Mitochondrial stress signaling in longevity: A new role for mitochondrial function in aging. Redox Biol. 2014, 2, 936–944. [Google Scholar] [CrossRef] [Green Version]
- Suomalainen, A.; Battersby, B.J. Mitochondrial diseases: The contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol. 2018, 19. [Google Scholar] [CrossRef]
- Molnar, M.J.; Kovacs, G.G. Mitochondrial diseases. Handb. Clin. Neurol. 2017, 145, 147–155. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawling, D.C.; Fitzgerald, M.E.; Pyle, A.M. Establishing the role of ATP for the function of the RIG-I innate immune sensor. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nuevo, A.; Zorzano, A. The sensing of mitochondrial DAMPs by non-immune cells. Cell Stress 2019, 3, 195–207. [Google Scholar] [CrossRef] [PubMed]
- de Souza Breda, C.N.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef]
- Bruns, A.M.; Horvath, C.M. LGP2 synergy with MDA5 in RLR-mediated RNA recognition and antiviral signaling. Cytokine 2015, 74, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.; Zhang, S.; Li, Y.; Wang, Y.; Peppelenbosch, M.P.; Pan, Q. Mitochondria in the biology, pathogenesis, and treatment of hepatitis virus infections. Rev. Med. Virol. 2019, 29. [Google Scholar] [CrossRef]
- Banoth, B.; Cassel, S.L. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4, 95. [Google Scholar] [CrossRef] [Green Version]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Briard, B.; Place, D.E.; Kanneganti, T.-D. DNA sensing in the innate immune response. Physiology (Bethesda) 2020, 35, 112–124. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Highton, J.; Hessian, P.A. Cell-specific expression of TLR9 isoforms in inflammation. J. Autoimmun. 2011, 36, 76–86. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Yoshizumi, T.; Imamura, H.; Taku, T.; Kuroki, T.; Kawaguchi, A.; Ishikawa, K.; Nakada, K.; Koshiba, T. RLR-mediated antiviral innate immunity requires oxidative phosphorylation activity. Sci. Rep. 2017, 7, 5379. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Lee, K.J.; Zheng, Y.; Yamaga, A.K.; Lai, M.M.C.; Ou, J.-H. Reactive oxygen species suppress hepatitis C virus RNA replication in human hepatoma cells. Hepatology 2004, 39, 81–89. [Google Scholar] [CrossRef]
- Delgado-Roche, L.; Mesta, F. Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020, 51, 384–387. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Wang, W.-H.; Che, L.; Lan, Y.; Zhang, L.-Y.; Zhan, D.-L.; Huang, Z.-Y.; Lin, Z.-N.; Lin, Y.-C. BNIP3L-dependent mitophagy promotes HBx-induced cancer stemness of hepatocellular carcinoma cells via glycolysis metabolism reprogramming. Cancers 2020, 12, 655. [Google Scholar] [CrossRef] [Green Version]
- Naifeh, J.; Jiang, J.; Varacallo, M. Biochemistry, Aerobic Glycolysis; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Polcicova, K.; Badurova, L.; Tomaskova, J. Metabolic reprogramming as a feast for virus replication. Acta Virol. 2020, 64, 201–215. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Olive, L.; Marx, W.; O’Neil, A.; Athan, E.; Carvalho, A.; Maes, M.; Walder, K.; et al. The pathophysiology of SARS-CoV-2: A suggested model and therapeutic approach. Life Sci. 2020, 118166. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Poeggeler, B.; Alvares, F.L.; Ogden, G.B.; Reiter, R.J. Melatonin immunoreactivity in the photosynthetic prokaryote Rhodospirillum rubrum: Implications for an ancient antioxidant system. Cell. Mol. Biol. Res. 1995, 41, 391–395. [Google Scholar] [PubMed]
- Tilden, A.R.; Becker, M.A.; Amma, L.L.; Arciniega, J.; McGaw, A.K. Melatonin production in an aerobic photosynthetic bacterium: An evolutionarily early association with darkness. J. Pineal Res. 1997, 22, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.-X.; Manchester, L.C.; Liu, X.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Reiter, R.J. Mitochondria and chloroplasts as the original sites of melatonin synthesis: A hypothesis related to melatonin’s primary function and evolution in eukaryotes. J. Pineal Res. 2013, 54, 127–138. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, J.; Zhang, Z.; Yang, M.; Li, Y.; Tian, X.; Ma, T.; Tao, J.; Zhu, K.; Song, Y.; et al. Mitochondria synthesize melatonin to ameliorate its function and improve mice oocyte’s quality under in vitro conditions. Int. J. Mol. Sci. 2016, 17, 939. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Feng, C.; Zheng, X.; Guo, Y.; Zhou, F.; Shan, D.; Liu, X.; Kong, J. Plant mitochondria synthesize melatonin and enhance the tolerance of plants to drought stress. J. Pineal Res. 2017, 63, e12429. [Google Scholar] [CrossRef]
- Suofu, Y.; Li, W.; Jean-Alphonse, F.G.; Jia, J.; Khattar, N.K.; Li, J.; Baranov, S.V.; Leronni, D.; Mihalik, A.C.; He, Y.; et al. Dual role of mitochondria in producing melatonin and driving GPCR signaling to block cytochrome c release. Proc. Natl. Acad. Sci. USA 2017, 114, E7997–E8006. [Google Scholar] [CrossRef] [Green Version]
- Quintela, T.; Gonçalves, I.; Silva, M.; Duarte, A.C.; Guedes, P.; Andrade, K.; Freitas, F.; Talhada, D.; Albuquerque, T.; Tavares, S.; et al. Choroid plexus is an additional source of melatonin in the brain. J. Pineal Res. 2018, e12528. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef]
- Agrimi, G.; Di Noia, M.A.; Marobbio, C.M.T.; Fiermonte, G.; Lasorsa, F.M.; Palmieri, F. Identification of the human mitochondrial S-adenosylmethionine transporter: Bacterial expression, reconstitution, functional characterization and tissue distribution. Biochem. J. 2004, 379, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Macías, M.; Escames, G.; Reiter, R.J.; Agapito, M.T.; Ortiz, G.G.; Acuña-Castroviejo, D. Melatonin-induced increased activity of the respiratory chain complexes I and IV can prevent mitochondrial damage induced by ruthenium red in vivo. J. Pineal Res. 2000, 28, 242–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria-targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Chattopadhyay, A.; Pal, P.K.; Bandyopadhyay, D. Melatonin is a potential therapeutic molecule for oxidative stress induced red blood cell (RBC) injury: A review. Melatonin Res. 2020, 3, 1–31. [Google Scholar] [CrossRef]
- Pal, P.K.; Bhattacharjee, B.; Chattopadhyay, A.; Bandyopadhyay, D. Pleiotropic roles of melatonin against oxidative stress mediated tissue injury in the gastrointestinal tract: An overview. Melatonin Res. 2019, 2, 158–184. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Fuentes-Broto, L.; Paredes, S.D.; Reiter, R.J. Significance and application of melatonin in the regulation of brown adipose tissue metabolism: Relation to human obesity. Obes. Rev. 2011, 12, 167–188. [Google Scholar] [CrossRef]
- Fernández Vázquez, G.; Reiter, R.J.; Agil, A. Melatonin increases brown adipose tissue mass and function in Zücker diabetic fatty rats: Implications for obesity control. J. Pineal Res. 2018, 64, e12472. [Google Scholar] [CrossRef]
- Aslan, G.; Gül, H.F.; Tektemur, A.; Sahna, E. Ischemic postconditioning reduced myocardial ischemia-reperfusion injury: The roles of melatonin and uncoupling protein 3. Anatol. J. Cardiol. 2020, 23, 19–27. [Google Scholar] [CrossRef]
- Hardeland, R. Neuroprotection by radical avoidance: Search for suitable agents. Molecules 2009, 14, 5054–5102. [Google Scholar] [CrossRef]
- Berger, H.R.; Nyman, A.K.G.; Morken, T.S.; Widerøe, M. Transient effect of melatonin treatment after neonatal hypoxic-ischemic brain injury in rats. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Erkanli, K.; Kayalar, N.; Erkanli, G.; Ercan, F.; Sener, G.; Kirali, K. Melatonin protects against ischemia/reperfusion injury in skeletal muscle. J. Pineal Res. 2005, 39, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.A.; Gong, L.R.; Yu, J.B.; Kan, Y.X. The role of melatonin in electroacupuncture alleviating lung injury induced by limb ischemia-reperfusion in rabbits. Med. Sci. Monit. 2020, 26. [Google Scholar] [CrossRef]
- Liu, L.; Chen, H.; Jin, J.; Tang, Z.; Yin, P.; Zhong, D.; Li, G. Melatonin ameliorates cerebral ischemia/reperfusion injury through SIRT3 activation. Life Sci. 2019, 239. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhang, L.Y.; Chen, Y.; Bai, Y.P.; Jia, J.; Feng, J.G.; Liu, K.X.; Zhou, J. Melatonin alleviates intestinal injury, neuroinflammation and cognitive dysfunction caused by intestinal ischemia/reperfusion. Int. Immunopharmacol. 2020, 85. [Google Scholar] [CrossRef]
- Qi, X.; Wang, J. Melatonin improves mitochondrial biogenesis through the AMPK/PGC1α pathway to attenuate ischemia/reperfusion-induced myocardial damage. Aging 2020, 12, 7299–7312. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Macías, M.; León, J.; Escames, G.; Khaldy, H.; Acuña-Castroviejo, D. Melatonin increases the activity of the oxidative phosphorylation enzymes and the production of ATP in rat brain and liver mitochondria. Int. J. Biochem. Cell Biol. 2002, 34, 348–357. [Google Scholar] [CrossRef]
- López, A.; Ortiz, F.; Doerrier, C.; Venegas, C.; Fernández-Ortiz, M.; Aranda, P.; Díaz-Casado, M.E.; Fernández-Gil, B.; Barriocanal-Casado, E.; Escames, G.; et al. Mitochondrial impairment and melatonin protection in parkinsonian mice do not depend of inducible or neuronal nitric oxide synthases. PLoS ONE 2017, 12, e0183090. [Google Scholar] [CrossRef]
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Investig. 2020, 130, 3124–3136. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zhao, C.; Xiang, H.; Zhao, X.; Zhong, R. Melatonin inhibits formation of mitochondrial permeability transition pores and improves oxidative phosphorylation of frozen-thawed ram sperm. Front. Endocrinol. 2019, 10, 896. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Tao, J.; Chai, M.; Wu, H.; Wang, J.; Li, G.; He, C.; Xie, L.; Ji, P.; Dai, Y.; et al. Melatonin improves the quality of inferior bovine oocytes and promoted their subsequent IVF embryo development: Mechanisms and results. Molecules 2017, 22, 2059. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.-X.; Reiter, R.J. Mitochondria: The birth place, battle ground and the site of melatonin metabolism in cells. Melatonin Res. 2019, 2, 44–66. [Google Scholar] [CrossRef]
- Liu, R.Y.; Zhou, J.N.; van Heerikhuize, J.; Hofman, M.A.; Swaab, D.F. Decreased melatonin levels in postmortem cerebrospinal fluid in relation to aging, Alzheimer’s disease, and apolipoprotein E-epsilon4/4 genotype. J. Clin. Endocrinol. Metab. 1999, 84, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Barceló, E.J.; Cos, S.; Fernández, R.; Mediavilla, M.D. Melatonin and mammary cancer: A short review. Endocr. Relat. Cancer 2003, 10. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Chen, T.; Li, J.; Xiao, J.; Wang, S.; Guo, X.; Bu, P. Correlations of serum cyclophilin a and melatonin concentrations with hypertension-induced left ventricular hypertrophy. Arch. Med. Res. 2017, 48. [Google Scholar] [CrossRef] [PubMed]
- Tamtaji, O.R.; Reiter, R.J.; Alipoor, R.; Dadgostar, E.; Kouchaki, E.; Asemi, Z. Melatonin and Parkinson disease: Current status and future perspectives for molecular mechanisms. Cell. Mol. Neurobiol. 2020, 40. [Google Scholar] [CrossRef]
- Cardinali, D.P. Melatonin: Clinical perspectives in neurodegeneration. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef]
- Hardeland, R.; Cardinali, D.P.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin and brain inflammaging. Prog. Neurobiol. 2015, 127–128. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Barcelo, E.J.; Rueda, N.; Mediavilla, M.D.; Martinez-Cue, C.; Reiter, R.J. Clinical uses of melatonin in neurological diseases and mental and behavioural disorders. Curr. Med. Chem. 2017, 24. [Google Scholar] [CrossRef]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Cho, H.M.; Sun, W. The coordinated regulation of mitochondrial structure and function by Drp1 for mitochondrial quality surveillance. BMB Rep. 2019, 52. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Lendahl, U.; Nistér, M.; Zhao, J. Regulation of mammalian mitochondrial dynamics: Opportunities and challenges. Front. Endocrinol. 2020, 11, 374. [Google Scholar] [CrossRef] [PubMed]
- Kalia, R.; Wang, R.Y.-R.; Yusuf, A.; Thomas, P.V.; Agard, D.A.; Shaw, J.M.; Frost, A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Fayaz, S.M.; Raj, Y.V.; Krishnamurthy, R.G. CypD: The Key to the death door. CNS Neurol. Disord. Drug Targets 2015, 14, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Stevens, M.V.; Kohr, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011, 286, 40184–40192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Zhang, Y.; Hu, S.; Shi, C.; Zhu, P.; Ma, Q.; Jin, Q.; Cao, F.; Tian, F.; Chen, Y. Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, D.; Zhu, P.; Ma, Q.; Toan, S.; Wang, J.; Hu, S.; Chen, Y.; Zhang, Y. Inhibitory effect of melatonin on necroptosis via repressing the Ripk3-PGAM5-CypD-mPTP pathway attenuates cardiac microvascular ischemia-reperfusion injury. J. Pineal Res. 2018, 65. [Google Scholar] [CrossRef]
- Asghari, M.H.; Abdollahi, M.; de Oliveira, M.R.; Nabavi, S.M. A review of the protective role of melatonin during phosphine-induced cardiotoxicity: Focus on mitochondrial dysfunction, oxidative stress and apoptosis. J. Pharm. Pharmacol. 2017, 69, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Jou, M.-J.; Peng, T.-I.; Yu, P.-Z.; Jou, S.-B.; Reiter, R.J.; Chen, J.-Y.; Wu, H.-Y.; Chen, C.-C.; Hsu, L.-F. Melatonin protects against common deletion of mitochondrial DNA-augmented mitochondrial oxidative stress and apoptosis. J. Pineal Res. 2007, 43, 389–403. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Y.; Shi, H.; Wang, J.; Zheng, Z.; Chen, J.; Chen, X.; Zhang, Z.; Xu, D.; Wang, X.; et al. Melatonin ameliorates intervertebral disc degeneration via the potential mechanisms of mitophagy induction and apoptosis inhibition. J. Cell. Mol. Med. 2019, 23, 2136–2148. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.M.; Kim, H.J.; Lee, J.H.; Lee, S.H. Melatonin enhances mitophagy by upregulating expression of heat shock 70 kDa protein 1L in human mesenchymal stem cells under oxidative stress. Int. J. Mol. Sci. 2019, 20, 4545. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.R.; Zhou, Y.J.; Yang, J.Q.; Liu, F.; Wu, X.P.; Sha, Y. Melatonin attenuates calcium deposition from vascular smooth muscle cells by activating mitochondrial fusion and mitophagy via an AMPK/OPA1 signaling pathway. Oxid. Med. Cell. Longev. 2020, 2020, 5298483. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Fernández-Palanca, P.; San-Miguel, B.; Álvarez, M.; González-Gallego, J.; Tuñón, M.J. Melatonin modulates mitophagy, innate immunity and circadian clocks in a model of viral-induced fulminant hepatic failure. J. Cell. Mol. Med. 2020, 24. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Qiu, X.; Wang, Y.; Liu, J.; Li, Q.; Jiang, H.; Li, S.; Song, C. Long-term oral melatonin alleviates memory deficits, reduces amyloid-β deposition associated with downregulation of BACE1 and mitophagy in APP/PS1 transgenic mice. Neurosci. Lett. 2020, 735, 135192. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yang, Y.; Gao, Y.; Wang, Z.; Ma, J. Melatonin attenuates anoxia/reoxygenation injury by inhibiting excessive mitophagy through the MT2/SIRT3/FoxO3a signaling pathway in h9c2 cells. Drug Des. Devel. Ther. 2020, 14, 2047–2060. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M. Neuronal mitophagy in neurodegenerative diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178. [Google Scholar] [CrossRef]
- Hardeland, R. Recent findings in melatonin research and their relevance to the CNS. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18. [Google Scholar] [CrossRef]
- Hardeland, R.; Balzer, I.; Poeggeler, B.; Fuhrberg, B.; Una, H.; Behrmann, G.; Wolf, R.; Meyer, T.J.; Reiter, R.J. On the primary functions of melatonin in evolution: Mediation of photoperiodic signals in a unicell, photooxidation, and scavenging of free radicals. J. Pineal Res. 1995, 18, 104–111. [Google Scholar] [CrossRef]
- Fuhrberg, B.; Hardeland, R.; Poeggeler, B.; Behrmann, C. Dramatic rises of melatonin and 5-methoxytryptamine in gonyaulax exposed to decreased temperature. Biol. Rhythm Res. 1997, 28, 144–150. [Google Scholar] [CrossRef]
- Liu, T.; Zhao, F.; Liu, Z.; Zuo, Y.; Hou, J.; Wang, Y. Identification of melatonin in Trichoderma spp. and detection of melatonin content under controlled-stress growth conditions from T. asperellum. J. Basic Microbiol. 2016, 56, 838–843. [Google Scholar] [CrossRef]
- Tan, D.-X.; Manchester, L.C.; Terron, M.P.; Flores, L.J.; Reiter, R.J. One molecule, many derivatives: A never-ending interaction of melatonin with reactive oxygen and nitrogen species? J. Pineal Res. 2007, 42, 28–42. [Google Scholar] [CrossRef]
- Shi, H.; Chen, Y.; Tan, D.-X.; Reiter, R.J.; Chan, Z.; He, C. Melatonin induces nitric oxide and the potential mechanisms relate to innate immunity against bacterial pathogen infection in Arabidopsis. J. Pineal Res. 2015, 59, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Back, K. Melatonin is required for H2O2-And NO-mediated defense signaling through MAPKKK3 and OXI1 in Arabidopsis thaliana. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef] [PubMed]
- Valero, N.; Mosquera, J.; Alcocer, S.; Bonilla, E.; Salazar, J.; Álvarez-Mon, M. Melatonin, minocycline and ascorbic acid reduce oxidative stress and viral titers and increase survival rate in experimental Venezuelan equine encephalitis. Brain Res. 2015, 1622, 368–376. [Google Scholar] [CrossRef]
- Nehela, Y.; Killiny, N. Infection with phytopathogenic bacterium inhibits melatonin biosynthesis, decreases longevity of its vector, and suppresses the free radical-defense. J. Pineal Res. 2018, 65, e12511. [Google Scholar] [CrossRef] [PubMed]
- Mehraj, V.; Routy, J.-P. Tryptophan catabolism in chronic viral infections: Handling uninvited guests. Int. J. Tryptophan Res. 2015, 8, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Hardeland, R. Melatonin, hormone of darkness and more: Occurrence, control mechanisms, actions and bioactive metabolites. Cell. Mol. Life Sci. 2008, 65. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, M.; Higuchi, K.; Aouini, A.; Ezura, H. Lowering intercellular melatonin levels by transgenic analysis of indoleamine 2,3-dioxygenase from rice in tomato plants. J. Pineal Res. 2010, 49, 239–247. [Google Scholar] [CrossRef]
- Back, K. Melatonin metabolism, signaling and possible roles in plants. Plant J. 2020. [Google Scholar] [CrossRef]
- Anand, S.K.; Singh, J.; Gaba, A.; Tikoo, S.K. Effect of bovine adenovirus 3 on mitochondria. Vet. Res. 2014, 45, 45. [Google Scholar] [CrossRef] [Green Version]
- Zan, J.; Liu, J.; Zhou, J.-W.; Wang, H.-L.; Mo, K.-K.; Yan, Y.; Xu, Y.-B.; Liao, M.; Su, S.; Hu, R.-L.; et al. Rabies virus matrix protein induces apoptosis by targeting mitochondria. Exp. Cell Res. 2016, 347, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Jassey, A.; Liu, C.-H.; Changou, C.A.; Richardson, C.D.; Hsu, H.-Y.; Lin, L.-T. Hepatitis C virus non-structural protein 5A (NS5A) disrupts mitochondrial dynamics and induces mitophagy. Cells 2019, 8, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, P.; Cheng, Y.; Song, S.; Qiu, J.; Yi, L.; Cao, Z.; Li, J.; Cheng, S.; Wang, J. Viral nonstructural protein 1 induces mitochondrion-mediated apoptosis in mink enteritis virus infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.E.; Fazal, F.M.; Parker, K.R.; Zou, J.; Chang, H.Y. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst. 2020, 11, 102–108.e3. [Google Scholar] [CrossRef]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Cell Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef]
- Boodhoo, N.; Kamble, N.; Sharif, S.; Behboudi, S. Glutaminolysis and glycolysis are essential for optimal replication of marek’s disease virus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Clippinger, A.J.; Alwine, J.C. Viral effects on metabolism: Changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 2011, 19, 360–367. [Google Scholar] [CrossRef] [Green Version]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef]
- Rocha, C.S.; Martins, A.D.; Rato, L.; Silva, B.M.; Oliveira, P.F.; Alves, M.G. Melatonin alters the glycolytic profile of Sertoli cells: Implications for male fertility. Mol. Hum. Reprod. 2014, 20, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Zhou, C.; Lu, Y.; Mao, L.; Xi, Y.; Mei, X.; Wang, X.; Zhang, L.; Yu, Z.; Zhou, Z. Melatonin antagonizes nickel-induced aerobic glycolysis by blocking ROS-mediated HIF-1α/miR210/ISCU axis activation. Oxid. Med. Cell. Longev. 2020, 2020, 5406284. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Dominquez-Rodriguez, A.; Marik, P.E.; Abreu-Gonzalez, P. Melatonin inhibits COVID-19-induced cytokine storm by reversing aerobic glycolysis in immune cells: A mechanistic analysis. Med. Drug Discov. 2020, 6, 100044. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Sharma, R.; Ma, Q.M. Switching diseased cells from cytosolic aerobic glycolysis to mitochondrial oxidative phosphorylation: A metabolic rhythm regulated by melatonin? J. Pineal Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Ramos, E.; López-Muñoz, F.; Gil-Martín, E.; Escames, G.; Reiter, R.J. Coronavirus Disease 2019 (COVID-19) and Its Neuroinvasive Capacity: Is It Time for Melatonin? Cell. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi-Motamayel, F.; Vaziri-Amjad, S.; Goodarzi, M.T.; Samie, L.; Poorolajal, J. Evaluation of salivary melatonin levels in HIV-positive patients: A historical cohort study. Rev. Recent Clin. Trials 2017, 12, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, G.; Nigro, L.; Palermo, F.; Leto, D.; Pomerantz, R.J.; Cacopardo, B. Reduction of serum melatonin levels in HIV-1-infected individuals’ parallel disease progression: Correlation with serum interleukin-12 levels. Infection 2003, 31, 379–382. [Google Scholar] [CrossRef]
- Cortis, D. On determining the age distribution of COVID-19 pandemic. Front. Public Heal. 2020, 8, 202. [Google Scholar] [CrossRef]
- Berenguer, J.; Ryan, P.; Rodríguez-Baño, J.; Jarrín, I.; Carratalà, J.; Pachón, J.; Yllescas, M.; Arribas, J.R. COVID-19@Spain study group characteristics and predictors of death among 4,035 consecutively hospitalized patients with COVID-19 in Spain. Clin. Microbiol. Infect. 2020. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and the theories of aging: A critical appraisal of melatonin’s role in antiaging mechanisms. J. Pineal Res. 2013, 55, 325–356. [Google Scholar] [CrossRef]
- Strindhall, J.; Nilsson, B.O.; Löfgren, S.; Ernerudh, J.; Pawelec, G.; Johansson, B.; Wikby, A. No immune risk profile among individuals who reach 100 years of age: Findings from the Swedish NONA immune longitudinal study. Exp. Gerontol. 2007, 42. [Google Scholar] [CrossRef] [Green Version]
- Salvioli, S.; Capri, M.; Valensin, S.; Tieri, P.; Monti, D.; Ottaviani, E.; Franceschi, C. Inflamm-aging, cytokines and aging: State of the art, new hypotheses on the role of mitochondria and new perspectives from systems biology. Curr. Pharm. Des. 2006, 12. [Google Scholar] [CrossRef] [PubMed]
- Boren, E.; Gershwin, M.E. Inflamm-aging: Autoimmunity, and the immune-risk phenotype. Autoimmun. Rev. 2004, 3. [Google Scholar] [CrossRef] [PubMed]
- Cevenini, E.; Monti, D.; Franceschi, C. Inflamm-ageing. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Huether, G.; Poeggeler, B.; Reimer, A.; George, A. Effect of tryptophan administration on circulating melatonin levels in chicks and rats: Evidence for stimulation of melatonin synthesis and release in the gastrointestinal tract. Life Sci. 1992, 51, 945–953. [Google Scholar] [CrossRef]
- Tan, D.; Xu, B.; Zhou, X.; Reiter, R. Pineal calcification, melatonin production, aging, associated health consequences and rejuvenation of the pineal gland. Molecules 2018, 23, 301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardeland, R.; Pandi-Perumal, S.R. Melatonin, a potent agent in antioxidative defense: Actions as a natural food constituent, gastrointestinal factor, drug and prodrug. Nutr. Metab. 2005, 2, 22. [Google Scholar] [CrossRef] [Green Version]
- Waldhauser, F.; Ehrhart, B.; Förster, E. Clinical aspects of the melatonin action: Impact of development, aging, and puberty, involvement of melatonin in psychiatric disease and importance of neuroimmunoendocrine interactions. Experientia 1993, 49. [Google Scholar] [CrossRef]
- Waldhauser, F.; Weiszenbacher, G.; Tatzer, E.; Gisinger, B.; Waldhauser, M.; Schemper, M.; Frisch, H. Alterations in nocturnal serum melatonin levels in humans with growth and aging. J. Clin. Endocrinol. Metab. 1988, 66, 648–652. [Google Scholar] [CrossRef]
- Hardeland, R.; Cardinali, D.P.; Srinivasan, V.; Spence, D.W.; Brown, G.M.; Pandi-Perumal, S.R. Melatonin--a pleiotropic, orchestrating regulator molecule. Prog. Neurobiol. 2011, 93, 350–384. [Google Scholar] [CrossRef] [Green Version]
- Caci, G.; Albini, A.; Malerba, M.; Noonan, D.M.; Pochetti, P.; Polosa, R. COVID-19 and obesity: Dangerous liaisons. J. Clin. Med. 2020, 9, 2511. [Google Scholar] [CrossRef] [PubMed]
- Mai, F.; Del Pinto, R.; Ferri, C. COVID-19 and cardiovascular diseases. J. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, M.; Sansom, S.; Frankenberger, C.; Ward, E.; Hota, B. Clinical course and factors associated with hospitalization and critical illness among COVID-19 patients in Chicago, Illinois. Acad. Emerg. Med. 2020. [Google Scholar] [CrossRef]
- Simko, F.; Reiter, R.J. Is melatonin deficiency a unifying pathomechanism of high risk patients with COVID-19? Life Sci. 2020, 256, 117902. [Google Scholar] [CrossRef] [PubMed]
- Mesri Alamdari, N.; Mahdavi, R.; Roshanravan, N.; Lotfi Yaghin, N.; Ostadrahimi, A.R.; Faramarzi, E. A double-blind, placebo-controlled trial related to the effects of melatonin on oxidative stress and inflammatory parameters of obese women. Horm. Metab. Res. 2015, 47, 504–508. [Google Scholar] [CrossRef] [Green Version]
- Szewczyk-Golec, K.; Rajewski, P.; Gackowski, M.; Mila-Kierzenkowska, C.; Wesołowski, R.; Sutkowy, P.; Pawłowska, M.; Woźniak, A. Melatonin supplementation lowers oxidative stress and regulates adipokines in obese patients on a calorie-restricted diet. Oxid. Med. Cell. Longev. 2017, 2017, 8494107. [Google Scholar] [CrossRef]
- Malazonia, A.; Zerekidze, T.; Giorgadze, E.; Chkheidze, N.; Asatiani, K. Melatonin level variations with different behavioural risk factors in obese female patients. Open Access Maced. J. Med. Sci. 2017, 5, 613–617. [Google Scholar] [CrossRef] [Green Version]
- Ostrowska, O.; Zwirska-Korczala, Z.-K.; Buntner, B.; Pardela, P.; Drozdz, D. Association of body mass and body fat distribution with serum melatonin levels in obese women either non-operated or after jejunoileostomy. Endocr. Regul. 1996, 30, 33–40. [Google Scholar]
- Grosshans, M.; Vollmert, C.; Vollstaedt-Klein, S.; Nolte, I.; Schwarz, E.; Wagner, X.; Leweke, M.; Mutschler, J.; Kiefer, F.; Bumb, J.M. The association of pineal gland volume and body mass in obese and normal weight individuals: A pilot study. Psychiatr. Danub. 2016, 28, 220–224. [Google Scholar]
- El-Missiry, M.A.; El-Missiry, Z.M.A.; Othman, A.I. Melatonin is a potential adjuvant to improve clinical outcomes in individuals with obesity and diabetes with coexistence of Covid-19. Eur. J. Pharmacol. 2020, 882, 173329. [Google Scholar] [CrossRef]
- Dominguez-Rodriguez, A.; Abreu-Gonzalez, P.; Garcia-Gonzalez, M.J.; Samimi-Fard, S.; Reiter, R.J.; Kaski, J.C. Association of ischemia-modified albumin and melatonin in patients with ST-elevation myocardial infarction. Atherosclerosis 2008, 199, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Lorente, L.; Martín, M.M.; Abreu-González, P.; Pérez-Cejas, A.; Ramos, L.; Argueso, M.; Solé-Violán, J.; Cáceres, J.J.; Jiménez, A.; García-Marín, V. Serum melatonin levels are associated with mortality in patients with malignant middle cerebral artery infarction. J. Int. Med. Res. 2018, 46, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.E.; Stone, P.H.; Turi, Z.G.; Rutherford, J.D.; Czeisler, C.A.; Parker, C.; Poole, W.K.; Passamani, E.; Roberts, R.; Robertson, T.; et al. Circadian variation in the frequency of onset of acute myocardial infarction. N. Engl. J. Med. 1985, 313, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Willich, S.N.; Goldberg, R.J.; Maclure, M.; Perriello, L.; Muller, J.E. Increased onset of sudden cardiac death in the first three hours after awakening. Am. J. Cardiol. 1992, 70, 65–68. [Google Scholar] [CrossRef]
- Conway, E.M.; Pryzdial, E.L.G. Is the COVID-19 thrombotic catastrophe complement-connected? J. Thromb. Haemost. 2020. [Google Scholar] [CrossRef]
- Coccheri, S. COVID-19: The crucial role of blood coagulation and fibrinolysis. Intern. Emerg. Med. 2020. [Google Scholar] [CrossRef]
- Osterud, B. Tissue factor/TFPI and blood cells. Thromb. Res. 2012, 129, 274–278. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T. Inflammation and coagulation. Crit. Care Med. 2010, 38, S26–S34. [Google Scholar] [CrossRef]
- Zhou, H.; Li, D.; Zhu, P.; Hu, S.; Hu, N.; Ma, S.; Zhang, Y.; Han, T.; Ren, J.; Cao, F.; et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef]
- NaveenKumar, S.K.; Hemshekhar, M.; Kemparaju, K.; Girish, K.S. Hemin-induced platelet activation and ferroptosis is mediated through ROS-driven proteasomal activity and inflammasome activation: Protection by Melatonin. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2303–2316. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Khotina, V.; Ivanova, E.A.; Orekhov, A.N. NADPH oxidases and their role in atherosclerosis. Biomedicines 2020, 8, 206. [Google Scholar] [CrossRef]
- Iversen, P.O.; Dahm, A.; Skretting, G.; Mowinckel, M.-C.; Stranda, A.; Østerud, B.; Sandset, P.M.; Kostovski, E. Reduced peak, but no diurnal variation, in thrombin generation upon melatonin supplementation in tetraplegia. A randomised, placebo-controlled study. Thromb. Haemost. 2015, 114, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Anu, K.R.; Birangal, S.R.; Nikam, A.N.; Pandey, A.; Mutalik, S.; Joseph, A. Role of comorbidities like diabetes on severe acute respiratory syndrome coronavirus-2: A review. Life Sci. 2020, 118202. [Google Scholar] [CrossRef]
- Liu, Z.; Li, J.; Huang, J.; Guo, L.; Gao, R.; Luo, K.; Zeng, G.; Zhang, T.; Yi, M.; Huang, Y.; et al. Association Between Diabetes and COVID-19: A retrospective observational study with a large sample of 1,880 cases in Leishenshan hospital, Wuhan. Front. Endocrinol. 2020, 11, 478. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Vella, V.; Belfiore, A. COVID-19 and Diabetes: The importance of controlling RAGE. Front. Endocrinol. 2020, 11, 526. [Google Scholar] [CrossRef]
- Peschke, E.; Mühlbauer, E. New evidence for a role of melatonin in glucose regulation. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 829–841. [Google Scholar] [CrossRef]
- Doosti-Irani, A.; Ostadmohammadi, V.; Mirhosseini, N.; Mansournia, M.A.; Reiter, R.J.; Kashanian, M.; Rahimi, M.; Razavi, M.; Asemi, Z. The effects of melatonin supplementation on glycemic control: A systematic review and meta-analysis of randomized controlled trials. Horm. Metab. Res. 2018, 50, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Ostadmohammadi, V.; Soleimani, A.; Bahmani, F.; Aghadavod, E.; Ramezani, R.; Reiter, R.J.; Mansournia, M.A.; Banikazemi, Z.; Soleimani, M.; Zaroudi, M.; et al. The effects of melatonin supplementation on parameters of mental health, glycemic control, markers of cardiometabolic risk, and oxidative stress in diabetic hemodialysis patients: A randomized, double-blind, placebo-controlled trial. J. Ren. Nutr. 2020, 30, 242–250. [Google Scholar] [CrossRef]
- Kasradze, D.; Tavartkiladze, A.; Kasradze, M.; Nozadze, P. The study of melatonin protective activity on pancreatic β-cells under the condition of alloxan-induced diabetes during aging. Georgian Med. News 2010, 189, 56–63. [Google Scholar]
- Park, J.-H.; Shim, H.-M.; Na, A.-Y.; Bae, K.-C.; Bae, J.-H.; Im, S.-S.; Cho, H.-C.; Song, D.-K. Melatonin prevents pancreatic β-cell loss due to glucotoxicity: The relationship between oxidative stress and endoplasmic reticulum stress. J. Pineal Res. 2014, 56, 143–153. [Google Scholar] [CrossRef]
- Lee, Y.H.; Jung, H.S.; Kwon, M.J.; Jang, J.E.; Kim, T.N.; Lee, S.H.; Kim, M.-K.; Park, J.H. Melatonin protects INS-1 pancreatic β-cells from apoptosis and senescence induced by glucotoxicity and glucolipotoxicity. Islets 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- McMullan, C.J.; Curhan, G.C.; Schernhammer, E.S.; Forman, J.P. Association of nocturnal melatonin secretion with insulin resistance in nondiabetic young women. Am. J. Epidemiol. 2013, 178, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyssenko, V.; Nagorny, C.L.; Erdos, M.R.; Wierup, N.; Jonsson, A.; Spégel, P.; Bugliani, M.; Saxena, R.; Fex, M.; Pulizzi, N.; et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat. Genet. 2009, 41. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Nagorny, C.L.F.; Singh, P.; Bennet, H.; Yu, Q.; Alenkvist, I.; Isomaa, B.; Östman, B.; Söderström, J.; Pesonen, A.K.; et al. Increased melatonin signaling is a risk factor for type 2 diabetes. Cell Metab. 2016, 23. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and the pathologies of weakened or dysregulated circadian oscillators. J. Pineal Res. 2017, 62. [Google Scholar] [CrossRef]
- Kor, Y.; Geyikli, I.; Keskin, M.; Akan, M. Preliminary study: Evaluation of melatonin secretion in children and adolescents with type 1 diabetes mellitus. Indian J. Endocrinol. Metab. 2014, 18, 565–568. [Google Scholar] [CrossRef]
- Abdolsamadi, H.; Goodarzi, M.T.; Ahmadi Motemayel, F.; Jazaeri, M.; Feradmal, J.; Zarabadi, M.; Hoseyni, M.; Torkzaban, P. Reduction of melatonin level in patients with type ii diabetes and periodontal diseases. J. Dent. Res. Dent. Clin. Dent. Prospects 2014, 8, 160–165. [Google Scholar] [CrossRef]
- Reutrakul, S.; Sumritsopak, R.; Saetung, S.; Chanprasertyothin, S.; Chailurkit, L.-O.; Anothaisintawee, T. Lower nocturnal urinary 6-sulfatoxymelatonin is associated with more severe insulin resistance in patients with prediabetes. Neurobiol. Sleep Circadian Rhythm. 2018, 4, 10–16. [Google Scholar] [CrossRef]
- McMullan, C.J.; Schernhammer, E.S.; Rimm, E.B.; Hu, F.B.; Forman, J.P. Melatonin secretion and the incidence of type 2 diabetes. JAMA 2013, 309, 1388–1396. [Google Scholar] [CrossRef]
- Qiu, P.; Zhou, Y.; Wang, F.; Wang, H.; Zhang, M.; Pan, X.; Zhao, Q.; Liu, J. Clinical characteristics, laboratory outcome characteristics, comorbidities, and complications of related COVID-19 deceased: A systematic review and meta-analysis. Aging Clin. Exp. Res. 2020. [Google Scholar] [CrossRef]
- Alkhouli, M.; Nanjundappa, A.; Annie, F.; Bates, M.C.; Bhatt, D.L. Sex differences in case fatality rate of COVID-19: Insights from a multinational registry. Mayo Clin. Proc. 2020, 95, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Ghobrial, R.M.; Kubiak, J.Z. The role of genetic sex and mitochondria in response to COVID-19 infection. Int. Arch. Allergy Immunol. 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan-Radha, V.; Aitkenhead, I.; Clancy, D.J.; Chown, S.L.; Dowling, D.K. Sex-specific effects of mitochondrial haplotype on metabolic rate in Drosophila melanogaster support predictions of the Mother’s Curse hypothesis. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190178. [Google Scholar] [CrossRef] [Green Version]
- Claypool, L.E.; Wood, R.I.; Yellon, S.M.; Foster, D.L. The ontogeny of melatonin secretion in the lamb. Endocrinology 1989, 124, 2135–2143. [Google Scholar] [CrossRef]
- Guzzaloni, G.; Grugni, G.; Tonelli, E.; Ardizzi, A.; Caló, G.; Moro, D.; Mazzilli, G.; Morabito, F. Melatonin response to TRH in prepubertal and pubertal healthy subjects. Horm. Metab. Res. 1993, 25, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rodríguez, P.; de Pablo, A.L.L.; Condezo-Hoyos, L.; Martín-Cabrejas, M.A.; Aguilera, Y.; Ruiz-Hurtado, G.; Gutierrez-Arzapalo, P.Y.; Ramiro-Cortijo, D.; Fernández-Alfonso, M.S.; González, M.D.C.; et al. Fetal undernutrition is associated with perinatal sex-dependent alterations in oxidative status. J. Nutr. Biochem. 2015, 26, 1650–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, A.; Chorath, K.; Moreira, A.; Evans, M.; Burmeister-Morton, F.; Burmeister, F.; Naqvi, R.; Petershack, M.; Moreira, A. COVID-19 in 7780 pediatric patients: A systematic review. EClinicalMedicine 2020, 24. [Google Scholar] [CrossRef]
- NA, P. Pediatric COVID-19: Systematic review of the literature. Am. J. Otolaryngol. 2020, 41. [Google Scholar] [CrossRef]
- Cheng, B.; Jiang, T.; Zhang, L.; Hu, R.; Tian, J.; Jiang, Y.; Huang, B.; Li, J.; Wei, M.; Yang, J.; et al. Clinical characteristics of pregnant women with coronavirus disease 2019 in Wuhan, China. Open Forum Infect. Dis. 2020, 7. [Google Scholar] [CrossRef]
- Wierrani, F.; Grin, W.; Hlawka, B.; Kroiss, A.; Grünberger, W. Elevated serum melatonin levels during human late pregnancy and labour. J. Obstet. Gynaecol. 1997, 17, 449–451. [Google Scholar] [CrossRef]
- Kivelä, A. Serum melatonin during human pregnancy. Acta Endocrinol. (Copenh.) 1991, 124, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Lanoix, D.; Beghdadi, H.; Lafond, J.; Vaillancourt, C. Human placental trophoblasts synthesize melatonin and express its receptors. J. Pineal Res. 2008, 45, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.J.; Lee, A.; Xia, Y.; Lin, L.H.; Black, M.; Cotzia, P.; Hochman, J.S.; Berger, J.S. Biomarkers of platelet activity and vascular health associate with thrombosis and mortality in patients with COVID-19. Circ. Res. 2020. [Google Scholar] [CrossRef]
- Sagrillo-Fagundes, L.; Soliman, A.; Vaillancourt, C. Maternal and placental melatonin: Actions and implication for successful pregnancies. Minerva Ginecol. 2014, 66, 251–266. [Google Scholar] [PubMed]
- Drozdova, A.; Okuliarova, M.; Zeman, M. The effect of different wavelengths of light during incubation on the development of rhythmic pineal melatonin biosynthesis in chick embryos. Animal 2019, 13, 1635–1640. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Tao, J.; Wu, H.; Guan, S.; Liu, L.; Zhang, L.; Deng, S.; He, C.; Ji, P.; Liu, J.; et al. Aanat knockdown and melatonin supplementation in embryo development: Involvement of mitochondrial function and DNA methylation. Antioxid. Redox Signal. 2019, 30, 2050–2065. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, D.-X.; Hardeland, R. Targeting Host Defense System and Rescuing Compromised Mitochondria to Increase Tolerance against Pathogens by Melatonin May Impact Outcome of Deadly Virus Infection Pertinent to COVID-19. Molecules 2020, 25, 4410. https://doi.org/10.3390/molecules25194410
Tan D-X, Hardeland R. Targeting Host Defense System and Rescuing Compromised Mitochondria to Increase Tolerance against Pathogens by Melatonin May Impact Outcome of Deadly Virus Infection Pertinent to COVID-19. Molecules. 2020; 25(19):4410. https://doi.org/10.3390/molecules25194410
Chicago/Turabian StyleTan, Dun-Xian, and Ruediger Hardeland. 2020. "Targeting Host Defense System and Rescuing Compromised Mitochondria to Increase Tolerance against Pathogens by Melatonin May Impact Outcome of Deadly Virus Infection Pertinent to COVID-19" Molecules 25, no. 19: 4410. https://doi.org/10.3390/molecules25194410