
Surveillance and Molecular Characterization of SARS-CoV-2 Infection in Non-Human Hosts in Gujarat, India
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
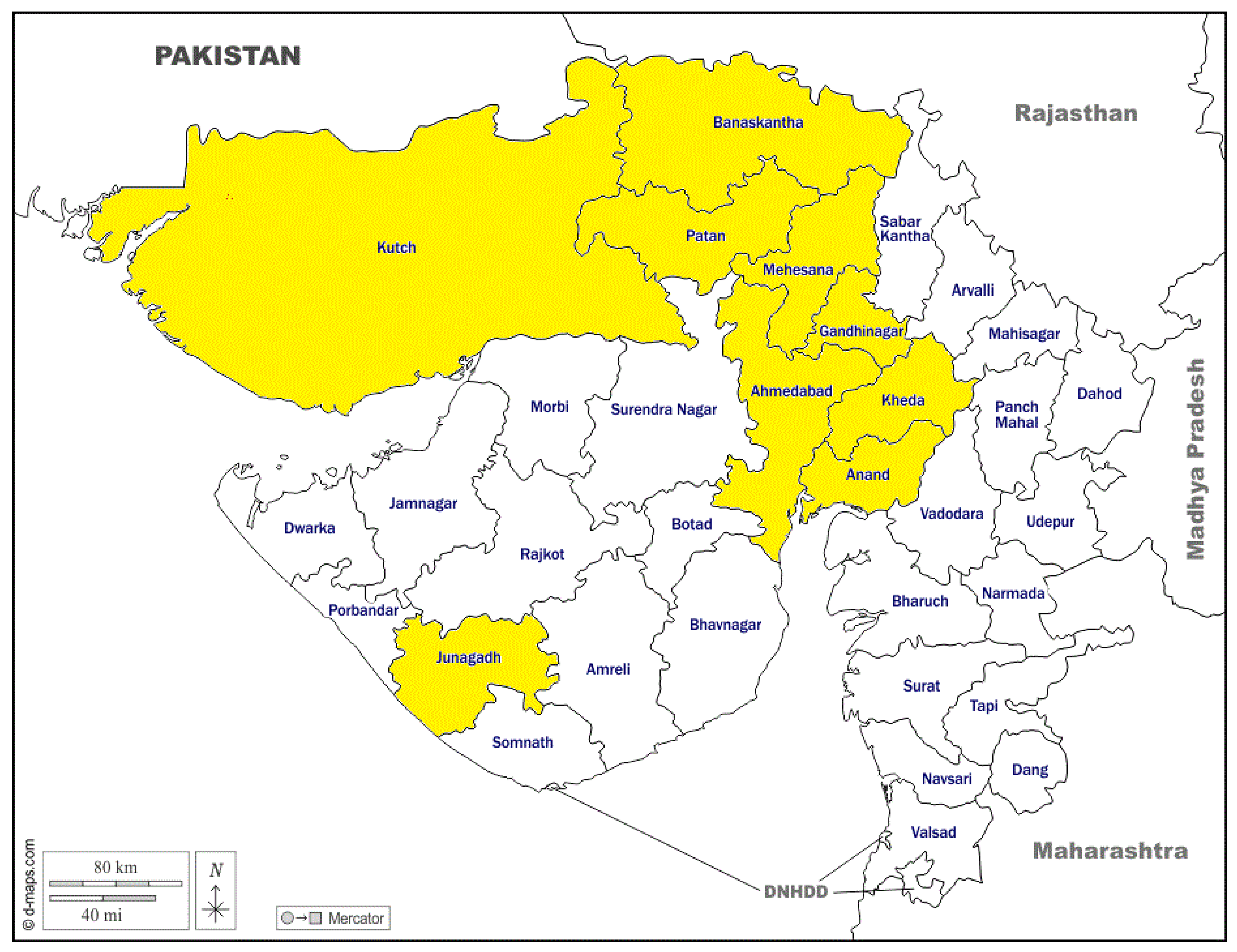
2.1. Location, Animals, and Sampling
2.2. Virus RNA Isolation and RT-qPCR
2.3. Whole-Genome Sequencing
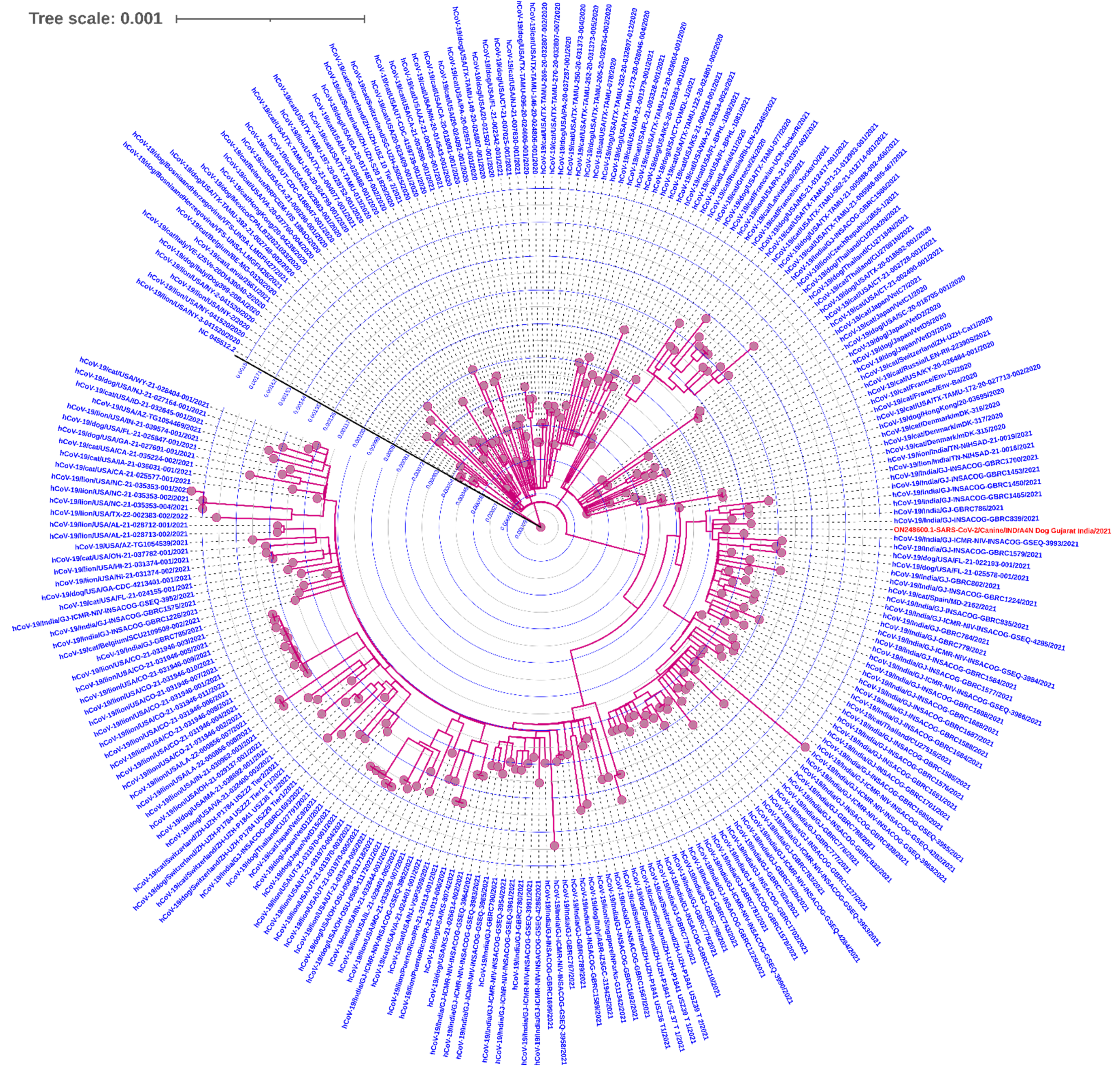
2.4. Phylogenetic Analysis
3. Results
3.1. Sampling and Prevalence Data
3.2. Analysis of RT-qPCR Results
3.3. Whole-Genome Sequencing and Variant Determination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Ethics Committee Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rodriguez-Morales, A.J.; Bonilla-Aldana, D.K.; Balbin-Ramon, G.J.; Rabaan, A.A.; Sah, R.; Paniz-Mondolfi, A.; Pagliano, P.; Esposito, S. History is repeating itself: Probable zoonotic spillover as the cause of the 2019 novel Coronavirus Epidemic. Infez. Med. 2020, 28, 3–5. [Google Scholar] [PubMed]
- Zhang, Y.Z.; Holmes, E.C. A genomic perspective on the origin and emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef]
- Lam, T.T.; Jia, N.; Zhang, Y.W.; Shum, M.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B beta coronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, M.I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Cosar, B.; Karagulleoglu, Z.Y.; Unal, S.; Ince, A.T.; Uncuoglu, D.B.; Tuncer, G.; Kilinc, B.R.; Ozkan, Y.E.; Ozkoc, H.C.; Demir, I.N.; et al. SARS-CoV-2 mutations and their viral variants. Cytokine Growth Factor Rev. 2022, 63, 10–22. [Google Scholar] [CrossRef]
- Andrews, M.A.; Areekal, B.; Rajesh, K.R.; Krishnan, J.; Suryakala, R.; Krishnan, B.; Muraly, C.P.; Santhosh, P.V. First confirmed case of COVID-19 infection in India: A case report. Indian J. Med. Res. 2020, 151, 490–492. [Google Scholar] [CrossRef]
- Sharun, K.; Saied, A.A.; Tiwari, R.; Dhama, K. SARS-CoV-2 infection in domestic and feral cats: Current evidence and implications. Vet. Q. 2020, 41, 228–231. [Google Scholar] [CrossRef]
- Klaus, J.; Zini, E.; Hartmann, K.; Egberink, H.; Kipar, A.; Bergmann, M.; Palizzotto, C.; Zhao, S.; Rossi, F.; Franco, V.; et al. SARS-CoV-2 infection in dogs and cats from southern Germany and northern Italy during the first wave of the COVID-19 pandemic. Viruses 2021, 13, 1453. [Google Scholar] [CrossRef]
- Jairak, W.; Charoenkul, K.; Chamsai, E.; Udom, K.; Chaiyawong, S.; Bunpapong, N.; Boonyapisitsopa, S.; Tantilertcharoen, R.; Techakriengkrai, N.; Surachetpong, S.; et al. First cases of SARS-CoV-2 infection in dogs and cats in Thailand. Transbound. Emerg. Dis. 2022, 69, e979–e991. [Google Scholar] [CrossRef]
- Barroso-Arévalo, S.; Rivera, B.; Domínguez, L.; Sánchez-Vizcaíno, J.M. First detection of SARS-CoV-2 B.1.1.7 variant of concern in an asymptomatic dog in Spain. Viruses 2021, 13, 1379. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Kumar, N.; Bhatia, S.; Aasdev, A.; Kanniappan, S.; Sekhar, A.; Aparna, G.; Silambarasan, R.; Sreekumar, C.; Dubey, C.K.; et al. SARS-CoV-2 delta variant among Asiatic lions, India. Emerg. Infect. Dis. 2021, 27, 2723. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS–coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matta, S.; Rajpal, S.; Chopra, K.K.; Arora, V.K. COVID-19 vaccines and new mutant strains impacting the pandemic. Indian J. Tuberc. 2021, 68, 171–173. [Google Scholar] [CrossRef]
- Joshi, M.; Kumar, M.; Srivastava, V.; Kumar, D.; Rathore, D.S.; Pandit, R.; Graham, D.W.; Joshi, C.G. Genetic sequencing detected the SARS-CoV-2 delta variant in wastewater a month prior to the first COVID-19 case in Ahmedabad (India). Environ. Pollut. 2022, 310, 119757. [Google Scholar] [CrossRef]
- Huddleston, J.; Hadfield, J.; Sibley, T.R.; Lee, J.; Fay, K.; Ilcisin, M.; Harkins, E.; Bedford, T.; Neher, R.A.; Hodcroft, E.B. Augur: A bioinformatics toolkit for phylogenetic analyses of human pathogens. J. Open Source Softw. 2021, 6, 2906. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, 293–296. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Bastit, L.; Rodon, J.; Pradenas, E.; Marfil, S.; Trinité, B.; Parera, M.; Roca, N.; Pou, A.; Cantero, G.; Lorca-Oró, C.; et al. First Detection of SARS-CoV-2 Delta (B.1.617.2) Variant of Concern in a dog with clinical signs in Spain. Viruses 2021, 13, 2526. [Google Scholar] [CrossRef]
- Barroso-Arévalo, S.; Barneto, A.; Ramos, Á.M.; Rivera, B.; Sánchez, R.; Sánchez-Morales, L.; Pérez-Sancho, M.; Buendía, A.; Ferreras, E.; Ortiz-Menéndez, J.C.; et al. Large-scale study on virological and serological prevalence of SARS-CoV-2 in cats and dogs in Spain. Transbound. Emerg. Dis. 2022, 69, e759–e774. [Google Scholar] [CrossRef] [PubMed]
- Calvet, G.A.; Pereira, S.A.; Ogrzewalska, M.; Pauvolid-Corrêa, A.; Resende, P.C.; Tassinari, W.S.; Costa, A.P.; Keidel, L.O.; da Rocha, A.S.B.; da Silva, M.F.B.; et al. Investigation of SARS-CoV-2 infection in dogs and cats of humans diagnosed with COVID-19 in Rio de Janeiro, Brazil. PLoS ONE 2021, 16, e0250853. [Google Scholar] [CrossRef]
- Cerino, P.; Buonerba, C.; Brambilla, G.; Atripaldi, L.; Tafuro, M.; Concilio, D.D.; Vassallo, L.; Conte, G.L.; Cuomo, M.C.; Maiello, I.; et al. No detection of SARS-CoV-2 in animals exposed to infected keepers: Results of a COVID-19 surveillance program. Future Sci. OA 2021, 7, FSO711. [Google Scholar] [CrossRef] [PubMed]
- Lawton, K.O.; Arthur, R.M.; Moeller, B.C.; Barnum, S.; Pusterla, N. Investigation of the role of healthy and sick equids in the COVID-19 pandemic through serological and molecular testing. Animals 2022, 12, 614. [Google Scholar] [CrossRef] [PubMed]
- Sit, T.H.; Brackman, C.J.; Ip, S.M.; Tam, K.W.; Law, P.Y.; To, E.M.; Yu, V.Y.; Sims, L.D.; Tsang, D.N.; Chu, D.K.; et al. Infection of dogs with SARS-CoV-2. Nature 2020, 586, 776–778. [Google Scholar] [CrossRef]
- Hammer, A.S.; Quaade, M.L.; Rasmussen, T.B.; Fonager, J.; Rasmussen, M.; Mundbjerg, K.; Lohse, L.; Strandbygaard, B.; Jørgensen, C.S.; AlfaroNúñez, A.; et al. SARS-CoV-2 transmission between mink (Neovison vison) and humans, Denmark. Emerg. Infect. Dis. 2021, 27, 547–551. [Google Scholar] [CrossRef]
- Kumar, M.; Patel, A.K.; Shah, A.V.; Raval, J.; Rajpara, N.; Joshi, M.; Joshi, C.G. First proof of the capability of wastewater surveillance for COVID-19 in India through detection of genetic material of SARS-CoV-2. Sci. Total Environ. 2020, 746, 141326. [Google Scholar] [CrossRef]
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.; Patil, D.Y.; Nyayanit, D.A.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. Neutralization of variant under investigation B.1.617.1 with Sera of BBV152 Vaccines. Clin. Infect. Dis. 2022, 74, 366–368. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name of District | Dog | Cattle | Buffalo | Goat | Sheep | Horse | Cat | Camel | Monkey | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| Ahmadabad | 114 | - | - | - | - | - | - | - | - | 114 |
| Anand | 17 | - | - | - | - | 1 | - | - | - | 18 |
| Gandhinagar | 39 | 26 | 13 | - | - | - | - | - | - | 78 |
| Banaskantha | 9 | 34 | 17 | 38 | 19 | 36 | 6 | 6 | 1 | 166 |
| Patan | 1 | 4 | 9 | - | - | 1 | - | - | - | 15 |
| Kutch | 15 | - | - | - | - | 1 | - | - | - | 16 |
| Mehasana | - | - | - | 3 | - | 1 | - | - | - | 4 |
| Others * | - | - | - | - | - | 2 | - | - | - | 2 |
| Total | 195 | 64 | 39 | 41 | 19 | 42 | 6 | 6 | 1 | 413 |
| Species | No of Animal Sampled | Nasal Swab Collected | Rectal Swab Collected | No. of Positive Samples in COVID-19 qPCR | |||
|---|---|---|---|---|---|---|---|
| Only Nasal Swab | Only Rectal Swab | Both Samples | Total | ||||
| Dog | 195 | 195 | 195 | 16 | 10 | 41 | 67 |
| Cattle | 64 | 64 | 63 | 5 | 2 | 8 | 15 |
| Buffalo | 39 | 39 | 39 | 4 | 3 | 6 | 13 |
| Goat | 41 | 40 | 37 | - | - | - | - |
| Sheep | 19 | 19 | 19 | - | - | - | - |
| Horse | 42 | 42 | 42 | - | - | - | - |
| Monkey | 1 | 1 | 1 | - | - | - | - |
| Camel | 6 | 6 | 5 | - | - | - | - |
| Cat | 6 | 6 | 6 | - | - | - | - |
| Total | 413 | 412 | 407 | 25 | 15 | 55 | 95 |
| Species (n = No. of Samples) | Type of Sample | N Gene (Cт) | ORF1ab (Cт) | S Gene (Cт) | |||
|---|---|---|---|---|---|---|---|
| Max–Min (± SD) | Mean | Max–Min (± SD) | Mean | Max–Min (± SD) | Mean | ||
| Dogs (n = 67) | Nasal | 27.63–34.98 ± 2.25 | 32.05 | 28.40–34.96 ± 2.03 | 31.88 | 29.60–34.96 ± 1.39 | 32.9 |
| Rectal | 27.89–34.97 ± 2.16 | 32.12 | 28.97–34.93 ± 1.64 | 32.51 | 30.68–34.96 ± 1.19 | 33.37 | |
| Cattle (n = 15) | Nasal | 32.53–34.76 ± 0.72 | 34.13 | 32.26–34.94 ± 0.97 | 33.67 | 30.87–33.69 ± 0.92 | 32.40 |
| Rectal | 33.78–35.00 ± 0.54 | 34.46 | 32.91–34.48 ± 0.57 | 33.8 | 30.30–34.95 ± 1.28 | 32.74 | |
| Buffaloes (n = 13) | Nasal | 31.63–34.61 ± 1.05 | 33.16 | 31.74–34.84 ± 0.94 | 33.36 | 29.88–34.85 ± 1.59 | 32.78 |
| Rectal | 29.00–34.93 ± 1.65 | 32.37 | 31.00–34.11 ± 2.16 | 32.92 | 29.79–34.35 ± 1.45 | 32.12 | |
| Reference Position | Reference | Allele | Count | Coverage | Frequency | Forward/Reverse Balance | Average Quality | Amino Acid Change |
|---|---|---|---|---|---|---|---|---|
| 210 | G | T | 16,195 | 16,339 | 99.11 | 0.48 | 29.90 | Synonymous |
| 241 | C | T | 5204 | 5233 | 99.44 | 0.45 | 23.23 | Synonymous |
| 3037 | C | T | 11,467 | 12,330 | 93.00 | 0.38 | 28.40 | Synonymous |
| 5184 | C | T | 2593 | 2595 | 99.92 | 0.48 | 29.64 | Pro1640Leu |
| 5584 | A | G | 712 | 1809 | 39.35 | 0.50 | 31.35 | Synonymous |
| 9891 | C | T | 6039 | 6047 | 99.86 | 0.49 | 30.39 | Ala3209Val |
| 11,418 | T | C | 7924 | 7980 | 99.29 | 0.48 | 29.17 | Val3718Ala |
| 11,514 | C | T | 4237 | 4281 | 98.97 | 0.45 | 30.47 | Thr3750Ile |
| 13,019 | C | T | 2598 | 2607 | 99.65 | 0.47 | 31.25 | Synonymous |
| 14,408 | C | T | 2845 | 3822 | 74.43 | 0.49 | 30.01 | Pro4715Leu |
| 15,451 | G | A | 16,545 | 16,645 | 99.39 | 0.47 | 31.40 | Gly5063Ser |
| 15,919 | G | T | 6848 | 6868 | 99.70 | 0.45 | 32.26 | Val5219Leu |
| 16,466 | C | T | 10,720 | 10,735 | 99.86 | 0.21 | 31.83 | Pro5401Leu |
| 21,618 | C | G | 6431 | 6438 | 99.89 | 0.49 | 30.68 | Thr19Arg |
| 21,987 | G | A | 3751 | 3946 | 95.05 | 0.47 | 30.27 | Gly142Asp |
| 22,029 | AGTTCA | - | 3379 | 3685 | 91.70 | 0.50 | 28.26 | Synonymous |
| 22,227 | C | T | 1562 | 1591 | 98.18 | 0.49 | 25.47 | Ala222Val |
| 22,917 | T | G | 7672 | 7719 | 99.39 | 0.47 | 30.04 | Leu452Arg |
| 22,995 | C | A | 2725 | 2779 | 98.06 | 0.45 | 32.79 | Thr478Lys |
| 23,403 | A | G | 10,686 | 10,759 | 99.32 | 0.49 | 27.51 | Asp614Gly |
| 23,604 | C | G | 19,277 | 19,301 | 99.87 | 0.47 | 32.46 | Pro681Arg |
| 25,139 | T | C | 9114 | 9125 | 99.88 | 0.43 | 32.48 | Synonymous |
| 25,469 | C | T | 8791 | 8844 | 99.40 | 0.48 | 27.93 | Ser26Leu |
| 26,767 | T | C | 5947 | 5949 | 99.97 | 0.47 | 29.88 | Ile82Thr |
| 27,638 | T | C | 8633 | 8692 | 99.32 | 0.47 | 30.96 | Val82Ala |
| 27,752 | C | T | 6990 | 6993 | 99.96 | 0.45 | 30.07 | Thr120Ile |
| 28,248 | GA | - | 7439 | 7614 | 97.70 | 0.48 | 21.41 | Synonymous |
| 28,271 | A | - | 14,715 | 14,950 | 98.43 | 0.46 | 24.47 | Synonymous |
| 28,461 | A | G | 8647 | 8651 | 99.95 | 0.46 | 29.38 | Asp63Gly |
| 28,881 | G | T | 4107 | 4127 | 99.51 | 0.45 | 30.01 | Arg203Met |
| 29,402 | G | T | 5132 | 5192 | 98.84 | 0.49 | 27.77 | Asp377Tyr |
| 29,742 | G | T | 889 | 889 | 100.0 | 0.45 | 31.86 | Synonymous |
| Nucleotide Position | Nucleotide in Test Strain | Nucleotide in Reference Strain | Type of Mutation | Amino Acid Change | Possible Outcome of Mutation |
|---|---|---|---|---|---|
| 21,618 | C | G | SNV | Thr19Arg | Removes a potential N-glycosylation site that might also affect antigenic and other properties of this strain |
| 21,987 | G | A | SNV | Gly142Asp | |
| 22,029 | AGTTCA | - | Deletion | Possible deletion of antibody recognition site at amino acid position 156–157 | |
| 22,227 | C | T | SNV | Ala222Val | - |
| 22,917 | T | G | SNV | Leu452Arg | Host and other changes; antigenic drift; antibody recognition sites |
| 22,995 | C | A | SNV | Thr478Lys | Host and other changes; antigenic drift; host surface receptor binding; antibody recognition sites; viral oligomerization interfaces |
| 23,403 | A | G | SNV | Asp614Gly | Antigenic drift; virulence and host change; ligand binding; viral oligomerization interfaces |
| 23,604 | C | G | SNV | Pro681Arg | Increased rate of membrane fusion, internalization, and thus better transmissibility |
| 25,139 | T | C | SNV | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, D.; Antiya, S.P.; Patel, S.S.; Pandit, R.; Joshi, M.; Mishra, A.K.; Joshi, C.G.; Patel, A.C. Surveillance and Molecular Characterization of SARS-CoV-2 Infection in Non-Human Hosts in Gujarat, India. Int. J. Environ. Res. Public Health 2022, 19, 14391. https://doi.org/10.3390/ijerph192114391
Kumar D, Antiya SP, Patel SS, Pandit R, Joshi M, Mishra AK, Joshi CG, Patel AC. Surveillance and Molecular Characterization of SARS-CoV-2 Infection in Non-Human Hosts in Gujarat, India. International Journal of Environmental Research and Public Health. 2022; 19(21):14391. https://doi.org/10.3390/ijerph192114391
Chicago/Turabian StyleKumar, Dinesh, Sejalben P. Antiya, Sandipkumar S. Patel, Ramesh Pandit, Madhvi Joshi, Abhinava K. Mishra, Chaitanya G. Joshi, and Arunkumar C. Patel. 2022. "Surveillance and Molecular Characterization of SARS-CoV-2 Infection in Non-Human Hosts in Gujarat, India" International Journal of Environmental Research and Public Health 19, no. 21: 14391. https://doi.org/10.3390/ijerph192114391