
Abstract
Introduction
In recent times, myriads of public have been infected with a novel SARS-CoV-2, and the fatality toll has reached thousands and been mounting step by step, which is a major crisis in the world. The challenge for this burning issue pertinent to repurposed medicines which prevent novel coronavirus is of immense concern for all scientists around the globe until the arrival of the vaccine.
Methods
Because of the global high priority rating on the search for the repurposed drugs which outfits clinical suitability to SARS-CoV-2, a unique theoretical methodology is proposed. The approach is based on explorations of biothermodynamics computed via molecular dynamics, root-mean-square deviation (RMSD), radius of gyration (Rg) and interactions. This unique methodology is tested for umifenovir/dexamethasone drugs on (SARS-CoV-2) main protease.
Results
This theoretical exploration not only suggested the presence of strong interactions between (SARS-CoV-2 + umifenovir/dexamethasone) but also emphasized the clinical suitability of dexamethasone over umifenovir to treat SARS-CoV-2. This supremacy of dexamethasone is well supported by the results of global clinical trials and COVID-19 therapeutic approved management guidelines of countries.
Conclusions
Thus, this work will pave a way for incremental advancement towards future design and development of more specific inhibitors for the treatment of SARS-CoV-2 infection in humans.
Similar content being viewed by others

Introduction
Coronavirus disease (COVID-19) is an infection causing the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a recently revealed novel coronavirus. SARS-CoV-2 is genetically different from viruses that trigger influenza. These are encased, single-stranded RNA viruses whose exterior is enclosed by a halo of protein spikes (corona) (Fig. 1). The SARS-CoV-2 fits into the cysteine protease family, and the fatality due to this has reached thousands and been mounting step by step, which is a major crisis in the world (Chen 2020; Roberts et al. 2007; Chen et al. 2020). Since SARS-CoV-2 is rapidly spreading worldwide, the World Health Organization (WHO) has declared it as a pandemic disease (Coronavirus disease 2019). Consequently, the challenge to search for medicines to prevent novel coronavirus is of immense concern for all scientists around the globe.

Diagram of coronavirus virion structure showing spikes that form a “crown” like the solar corona, hence the name. [Source: https://commons.wikimedia.org/wiki/File:3D_medical_animation_corona_virus.jpg]
Governments and pharmaceutical companies are paying attention to probing and developing the unambiguous vaccine or anti-viral drug to avert or manage budding infection of SARS-CoV-2. On the other hand, such selections need much time for the developing procedure. Drug repurposing permits to quickly examine medical management, at lower costs and with diminished danger of disappointment as the wellbeing profile of the medication is commonly entrenched. Growing new medications is a protracted procedure, in this way unfeasible to confront the prompt worldwide crisis. At present, anti-retroviral drugs are under clinical advancement based on earlier achievement of the therapeutic management pertinent to viruses of SARS-CoV and MERS-CoV (Huang et al. 2020; De Clercq and Li 2016; Liu and Wang 2020). Thus, this investigation aims to explore and distinguish the binding affinities and interactions of certain drugs against the main protease of SARS-CoV-2 utilizing computational and measurable tools.
It is to be noted that the viruses need host-cell functional receptors in humans to accumulate and attack the immune system. As per the studies (Gralinski and Menachery 2020; Paraskevis et al. 2020), the spike protein SARS-CoV-2 attacked the angiotensin-converting enzyme 2 (ACE2) target protein on the surface of pulmonary epithelial cells of human (Tipnis et al. 2000; Tikellis and Thomas 2012).
In the present study, 6M03 main protease pertinent to SARS-CoV-2 is considered the attractive drug target for the healing of SARS-CoV-2 infection, and hence, we are going to explore the binding capacity of dexamethasone and umifenovir drugs as ligands directly to the PDB6M03 pertinent to the structure of SARS-CoV-2 main protease via molecular dynamic studies using Gromacs with OPLS-AA force field. The sources from which SARS-CoV-2 main protease in apo form, drugs of dexamethasone/umifenovir taken with their structures, are shown in Table 1.
The information concerning interaction mechanisms of a drug with SARS-CoV-2 protein is requisite to know the drug’s pharmacodynamics and pharmacokinetics (Cui et al. 2008). To the best of our knowledge, this is the first report for estimation of the Gibb’s free energy of the dexamethasone and umifenovir drugs on the main protease (PDB6M03) of SARS-CoV-2 using molecular dynamic simulations by Gromacs with OPLS-AA force field. The results of this study show that the dexamethasone and umifenovir are considered a valuable resource recommended for attacking SARS-CoV-2 protein. The inhibiting action of dexamethasone and umifenovir drugs on ACE2 is already proved (Ghadhanfar et al. 2017; McKee et al. 2020).
Drug umifenovir is considered a vital drug for the treatment of HIV. Dexamethasone is a corticosteroid used in an extensive variety of conditions for its anti-inflammatory and immunosuppressant results. It became tested in hospitalized sufferers with COVID-19 inside the UK clinical trial recuperation and became observed to have benefits for significantly unwell sufferers. Consistent with preliminary findings shared with WHO, for sufferers on ventilators, the remedy was proven to lessen mortality by about one third, and for patients requiring only oxygen, mortality was cut by about one fifth, respectively (https://www.who.int/news-room/q-a-detail/q-a-dexamethasone-and-covid-19).
Theory
Biothermodynamics of the protein-drug complex
Application of thermodynamics in biochemical engineering is to rationalize bioprocess development and obviate a substantial fraction of this need for tedious experimental work. The understanding over thermodynamics is employed in choosing a drug (ligand) as it interacts, binds, and controls the function of biological receptors (protein) that helps to cure a disease. The main point in this is to evaluate the change in Gibb’s free energy (ΔGbind). This change in Gibb’s free energy includes various interactions, such as van der Waals, hydrogen bonds, and electrostatic and hydrophobic interactions. According to thermodynamics, ΔGbind determines the stability of any given protein-ligand complex, or the binding affinity of a ligand to a given acceptor (Bronowska 2011; Nayeem et al. 2014). Moreover, the protein-ligand association extent is determined by the magnitude of the negativeΔGbind. The negative values with high magnitude show a strong binding between ligand and protein. In this study, we adopted g_mmpbsa tool for the computation of ΔGbind. The evaluation of different energies by g_mmpbsa tool with formulae is available in the references (Kumari and Kumar 2014; Yang et al. 2013; Yang et al. 2016; Ishima and Torchia 2000; Adcock and McCammon 2006). Precisely, in MM/PBSA method, the enthalpy of the system is calculated using the molecular mechanics (MM) method; the contribution of the polar part and non-polar part of the solvent effect to the free energy is determined by solving the Poisson-Boltzmann (PB) equation and calculating the molecular surface area (SA), respectively. To explore binding affinity (ΔGbind), MM/PBSA method is outlined below:
The Gibb’s free energy (ΔGbind) is given by
where Gcomplex is the whole free energy of the protein-ligand system, and GproteinGligand are the gross free energies of the protein and ligand. The value of Gprotein or Gligand is evaluated by
where EMM is the average molecular mechanics potential energy in the vacuum. (TS) denotes the product of the temperature and the entropic contribution, and (Gsolvation) is the free energy of solvation. Further,
where Ebonded is bonded interactions, and Enon − bondedincludes (h-bond, hydrophobic) van der Waals’ (EvdW) and electrostatic (Eelect) interactions and are modeled using a Lennard-Jones (LJ) and Coulomb potential function, respectively.
Gsolvation (solvation free energy) is given by:
here, Gpolar and Gnon − polar are the electrostatic and non-electrostatic contributions to the solvation free energy, respectively. Gnon − polar includes attractive and repulsive forces between solvent and solute that are generated by van der Waals’ interactions and cavity formation, respectively. Furthermore,
We used solvent accessible surface area (SASA) for computation of free energy of solvation pertinent to non-polar model. In the end, average binding energy, average van der Waals, and electrostatic energies, as well as polar solvation (PS) and SASA non-polar energies, were calculated for each system.
Materials and methods
MD simulation in biothermodynamics
Molecular dynamics (MD) simulation is a computational method to enable us to calculate movements of atoms in a molecular system by numerically solving Newton equations of motions (Perozzo et al. 2004). In MD, one usually looks for a ligand which can bind easily with desired protein leading to the negative Gibb’s free energy. Parameters of mathematical functions describing the potential energy of a system, termed the force field, are set to simulate the movements of atoms and molecules (Ren et al. 2019). OPLS-AA (optimize potential for liquid systems–all atom) is the most widely used biomolecular force field and is the latest and better force fields over others when ligands are to be included (Robertson et al. 2015). Hence, we used the OPLS-AA force fields on (SARS-CoV-2 + dexamethasone) and (SARS-CoV-2 + umifenovir).
The method fabrication in an understanding of binding energetics between protein-ligand interactions with the aid of thermodynamics and is recently used to assist in the discovery, development of antiviral drugs (Jiayi et al. 2017) and computer-aided design of drug molecules (CADD).
In the present context, molecular dynamics study is performed with Gromacs-2020.1 on Ubuntu platform. The molecular mechanics Poisson-Boltzmann surface area (MM/PBSA) method needs the trajectories generated by GROMACS (g_mmpbsa) (Sun et al. 2014; Sharp and Honig 1990; Tsui and Case 2000) to calculate the ΔGbind between the SARS-CoV-2 receptor and dexamethasone and umifenovir.
Preparation of SARS-CoV-2 + dexamethasone/umifenovir
The molecular dynamic simulation is followed as per the usual standard operating procedure (SOP) (Lemkul 2018). The SARS-CoV-2 main protease biological target is the PDB6M03 protein presented at RCSB Protein Data Bank (Liu et al. 2020). The structure of the protease was optimized and checked by Swiss-PDB viewer software packages (version 4.1.0) in light of their minimum energy. Some noteworthy components, for example, bond order, side-chain geometry, and missing H-bonds, were seen in the structure of the protease. PyMol (version1.1) software package was utilized to eradicate all the heteroatoms, H2O, and inhibitors present in the structure (DeLano 2002). This protein at pH = 7.4 is optimized by using Avogadro software with the purpose to correct the mismatch values of bond lengths, bond angles, bending angles, and unusual non-bonding interactions due to the atoms in different parts of the compounds occupying the same space. The OPLS-AA force field was utilized for this molecular dynamics to depict the macromolecular framework. Later, protein-energy is minimized with Gromacs. This structure is finally used for simulation. The chemical structures of dexamethasone and umifenovir were downloaded from the DrugBank (https://www.drugbank.ca/) database. Later, dexamethasone and umifenovir drug files were prepared and optimized using Avogadro software and converted to the executable OPLS-AA topology files (Dodda et al. 2017). Later, their energy is minimized with Gromacs tool. The minimized structure is finally used for simulation. With Gromacs-2020.1, executable files of SARS-CoV-2 protein were created with OPLS-AA force field. In a cubical box (d =1 nm), the executable files of protein-ligands are combined, solvated, and neutralized respectively. The initial MD simulation is energy minimized by using steepest descent. This is immediately followed by the equilibration of the simulation box in NVT ensemble. The temperature of the system was relaxed for 20 ns using modified Berendsen thermostat with a relaxation time of 2 fs and a reference temperature of 300 K. Temperature relaxation was followed by pressure relaxation of the simulation box in NPT ensemble for 40 ns. Pressure equilibration was achieved using Berendsen barostat. A relaxation time of 2 fs and reference pressure of 1 atm was used in pressure relaxation of the simulation box. Leapfrog algorithm was used during simulation to integrate the equation of motion. Long-range electrostatic interactions were accounted during simulation by particle mesh Ewald method. During the simulation, a spherical cut-off of 1.2 nm was used for both electrostatic and van der Waals forces. Upon completion of NVT and NPT equilibrations, final MD simulations have been carried for 100 ns and observed that the complexes are equilibrated and stabilized well below 10 ns and a relaxation time of 2 fs with reference temperature at 300 K and pressure of 1 atm using modified Berendsen thermostat and Parrinello-Rahman barostat, respectively. All the bond lengths were limited by the LINCS algorithm. The electrostatic interactions were calculated using the particle Mesh Ewald (PME) summation scheme (Essman et al. 1995), and the conformations were stored every 20 ps. In this study, three multisimulations for each are systems performed to check the reproducibility in computational values.
Result analysis
Analysis of MD trajectories
Root-mean-square deviation (RMSD) and radius of gyration (Rg), from the MD simulations, using the GROMACS routines. The graphs related to RMSD and Rg properties are widely used in predicting the structural activity of a protein molecule (Falsafi-Zadeh et al. 2012).
(i) RMSD of the trajectory of the SARS-CoV-2 backbone: the stabilities of the trajectories for (SARS-CoV-2 + dexamethasone) and (SARS-CoV-2 + umifenovir) were examined (Graph 1) using the RMSD for the backbone of SARS-CoV-2. Between ranges of 0–2 ns, the structure of (SARS-CoV-2 + dexamethasone) attained a maximum RMSD value of 0.31 nm and later remained constant about 0.12 nm throughout the rest of the period. Similarly, (SARS-CoV-2 + umifenovir) attained the highest of 6.50 nm between 0 and 2 ns and later decreased to 6.41 nm and maintained constant throughout the rest of the time. The backbone RMSD graph suggests that the (SARS-CoV-2 + dexamethasone) is more stable than (SARS-CoV-2 + umifenovir).

RMSD graph for the backbone of (SARS-CoV-2 + dexamethasone) and (SARS-CoV-2 + umifenovir) systems
The Graph 1 shows that (SARS-CoV-2 + dexamethasone)/(SARS-CoV-2 + umifenovir) systems reached equilibrium from initial stages of md run.
ii) Radius of gyration of the trajectory of the SARS-CoV-2 backbone: the radius of gyration (Rg) speaks about the structure compactness. The lower level of vacillation with its consistency all through the recreation demonstrates the more noteworthy compactness and unbending nature of a framework. Thus, if a protein is stably folded, it will likely maintain a relatively steady value of the radius of gyration. The radius of gyration for the systems was computed and drawn as Graph 2 against simulation time to study the protein compactness at 300 K. The study values in graph clearly showed that the systems stabilized properly and achieved compact structures (Seeliger and de Groot 2010; Daidone et al. 2003). The radius of gyration (Rg) of (SARS-CoV-2 + dexamethasone)/(SARS-CoV-2 + umifenovir) systems is raised slightly to 2.35 nm/3.98 nm at the initial stages of MD run. Later, both the systems soon reached a slightly lower steady value of 2.22 nm and 2.44 nm until 100 ns. This study value for the systems indicates that the drugs dexamethasone/umifenovir are not only stabilized properly but also achieved compact structures. Furthermore, the change in Rg magnitude revealed that the dexamethasone binds well to SARS-CoV-2 protein than umifenovir.

The radius of gyration graph for the backbone of (SARS-CoV-2 + dexamethasone) and (SARS-CoV-2 + umifenovir) systems
Predicted inhibitory efficiency
The interaction strengths of bonded and non-bonded for the whole system (with solvent) under study are evaluated under certain heads and finally summed up to the total binding energy in MM/PBSA. The susceptibility of dexamethasone and umifenovir towards the SARS-CoV-2 protein is estimated using the MM/PBSA approach on 100 snapshots extracted from the last 20 ns of simulation. The MM/PBSA results are tabulated in Table 2. The susceptibility of dexamethasone/umifenovir towards the SARS-CoV-2 protein is estimated using the MM/PBSA approach to the whole 100 ns of multiple simulations and the reproducibility pertinent to ΔGbind is found to be < 1.7%. From Table 2, the magnitude of different energy terms clearly shows that energy for (SARS-CoV-2 + dexamethasone) is greater than (SARS-CoV-2 + umifenovir). Finally, the binding energy (ΔGbind) is as follows: (SARS-CoV-2 + dexamethasone) > (SARS-CoV-2 + umifenovir).
Therefore, ΔGbind of dexamethasone is approximately 1.5 times of ΔGbind umifenovir. This result shows that dexamethasone drug can clinically boost efficacy in the fight against SARS-CoV-2 infection in humans over umifenovir via its strong binding affinity. Thus, this result paves a way for the usage of these drugs (dexamethasone and umifenovir) as a clinical trial on SARS-CoV-2 infection.
The changes in magnitude and sign of the thermodynamic parameters can be predicted from the main interactions computed between a protein and a ligand (Moradi et al. 2018). If changes in enthalpy and entropy are represented by ΔH and ΔS, respectively, then for ΔH > 0 and ΔS > 0, the main dominating interaction forces are hydrophobic; a combination of ΔH < 0 and ΔS < 0 implies the existence and dominance of van der Waals interactions (with hydrogen bonds); and the combination ΔH < 0 and ΔS > 0 indicates the dominance of electrostatic interactions. In our work, the negative ΔG implies a spontaneous interaction process. From Table 2 pertinent to (SARS-CoV-2 + dexamehasone) and (SARS-CoV-2 + umifenovir) systems, we can infer that the dominance of van der Waals interactions (high value) suggest the presence of ΔH < 0 and ΔS < 0 between SARS-CoV-2 and dexamethasone/umifenovir over the entire simulation run of 100 ns at 300 K /1 atm pressure.
Interactions present in SARS-CoV-2 + dexamethasone/umifenovir
If the computed free energy is positive, bonded interactions > non-bonded terms. If the computed free energy is negative, non-bonded interactions > bonded terms. For the present case, Gibb’s free energy is negative indicating the dominance of favorable non-bonded interactions over unfavorable bonded interactions. The supremacy of non-bonded interactions stabilizes the three-dimensional structure of the protein-ligand complex and consists of electrostatic, π effects, van der Waals forces, H-bonds, and hydrophobic effects. In these circumstances, we restrict to the non-bonded interactions (Atkins et al. 2018; Chang 2005; Schauperl et al. 2016; Kamps et al. 2015) between SARS-CoV-2 + dexamethasone/umifenovir only. Certain non-bonded interactions between the protein and drugs are shown (at 10 ns) in Figs. 2 and 3 (Biovia 2017; Laskowski and Swindells 2011).

SARS-CoV-2 + dexamethasone interactions

SARS-CoV-2 + umifenovir interactions
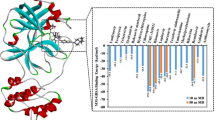
The importance of these drugs is evident in Graph 3. The obtained values ofΔGbind in the units of kJ/mol for SARS-CoV-2 main protease with dexamethasone/umifenovir and other drugs (Tachoua and Kabrine 2020) are compared in Graph 3. It is clear from the graph that dexamethasone has the highest value of ΔGbind when compared with other drugs. This emphasizes the presence of strong interactions between (SARS-CoV-2 + dexamethasone). The output of literature survey over clinical trials of umifenovir (Lian et al. 2020; Vankadari 2020; Choudhary and Silakari 2020; Costanzo et al. 2020; Zhang et al. 2020; Kadam and Wilson 2017; Valle et al. 2020)/dexamethasone (Ortolani and Pastorello 2020; Li and De Clercq 2020; Mittal et al. 2020; Ledford 2020; Low-cost dexamethasone 2020; WHO welcomes preliminary 2020; Alessi et al. 2020; Cain and Cidlowski 2020; Lammers et al. 2020; Stauffer et al. 2020) on SARS-CoV-2 infected patients also suggests that dexamethasone is the best and the first approved referral drug in UK, Australia, and South Africa countries for the treatment of COVID-19 infection. Thus, dexamethasone is one of the best-repurposed drugs to bind SARS-CoV-2 over umifenovir.

Comparative free energies of SARS-CoV-2 main protease with different drugs
Conclusion
This study proposes a potential approach to the use of dexamethasone/umifenovir, to tackle the current pandemic SARS-CoV-2. The MD simulation studies revealed that the RMSD/radius of gyration of the (SARS-CoV-2 + dexamethasone) and (SARS-CoV-2 + umifenovir) systems reached equilibrium, and the binding site remained compact/rigid during the simulation. According to ΔGbind bind prediction, the vulnerabilities against the SARS-CoV-2 with dexamethasone and umifenovir are − 79.032 ± 0.673 and − 54.297 ± 2.013) kJ mol−1, respectively. This statistics indicates that that the strength of binding energy of dexamethasone is 1.5 times stronger than umifenovir over SARS-CoV-2. Since there are already experimental studies using these drugs to treat COVID-19, the result of the article made it possible to verify from simulations and the effectiveness of each of these drugs.Further, in this work, the binding pattern and susceptibility of the inhibitors dexamethasone/umifenovir in complex with the SARS-CoV-2 were fully revealed by MD simulations with Gromacs using OPLS-AA force field in conjunction with binding free energy estimation based on the MM/PBSA calculations. Basing on the biothermodynamics and reported clinical trials, dexamethasone is the efficient drug when compared with umifenovir for the treatment of SARS-CoV-2 infection. We hope that, as a minimum, this work can be helpful for future design or development of more specific inhibitors for the treatment of SARS-CoV-2 infection. This study at the atomic level could also be helpful for the design or development of more specific inhibitors in treating human SARS-CoV-2 infection.
References
Adcock SA, McCammon JA. Molecular dynamics: survey of methods for simulating the activity of proteins. Chem Rev. 2006;106:1589–615.
Alessi J, de Oliveira GB, Schaan BD, Telo GH. Dexamethasone in the era of COVID-19: friend or foe? An essay on the effects of dexamethasone and the potential risks of its inadvertent use in patients with diabetes. Diabetol Metab Syndr. 2020;12:80. https://doi.org/10.1186/s13098-020-00583-7.
Atkins PW, De Paula J, Keeler J (2018) Atkins’ physical chemistry. Oxford university press.
Biovia DS (2017) Discovery studio visualizer. San Diego.
Bronowska AK (2011) Thermodynamics of ligand-protein interactions: implications for molecular design. In: Thermodynamics interaction studies-solids, liquids and gases. IntechOpen.
Cain DW, Cidlowski JA. After 62 years of regulating immunity, dexamethasone meets COVID-19. Nat Rev Immunol. 2020;20:587–8. https://doi.org/10.1038/s41577-020-00421-x.
Chang R (2005) Physical chemistry for the biosciences. University Science Books.
Chen J. Pathogenicity and transmissibility of 2019-nCoV—a quick overview and comparison with other emerging viruses. Microbes Infect. 2020;22:69–71.
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395:507–13.
Choudhary S, Silakari O. Scaffold morphing of arbidol (umifenovir) in search of multi-targeting therapy halting the interaction of SARS-CoV-2 with ACE2 and other proteases involved in COVID-19. Virus Res. 2020;289:198146. https://doi.org/10.1016/j.virusres.2020.198146.
Coronavirus disease 2019 https://www.who.int/emergencies/diseases/novel-coronavirus-2019
Costanzo M, Giglio MAR d, Roviello GN. SARS-CoV-2: recent reports on antiviral therapies based on lopinavir/ritonavir, darunavir/umifenovir, hydroxychloroquine, remdesivir, favipiravir and other drugs for the treatment of the new coronavirus. Curr Med Chem. 2020;27(27):4536–41. https://doi.org/10.2174/0929867327666200416131117.
Cui F, Qin L, Zhang G, Liu Q, Yao X, Lei B. Interaction of anthracycline disaccharide with human serum albumin: investigation by fluorescence spectroscopic technique and modeling studies. J Pharm Biomed Anal. 2008;48:1029–36.
Daidone I, Amadei A, Roccatano D, Di Nola A. Molecular dynamics simulation of protein folding by essential dynamics sampling: folding landscape of horse heart cytochrome c. Biophys J. 2003;85:2865–71.
De Clercq E, Li G. Approved antiviral drugs over the past 50 years. Clin Microbiol Rev. 2016;29:695–747.
DeLano WL. The PyMOL molecular graphics system. San Carlos: DeLano Scientific LLC; 2002.
Dodda LS, Cabeza de Vaca I, Tirado-Rives J, Jorgensen WL. LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017;45:W331–6.
Essman U, Perera L, Berkowitz ML, et al. A smooth particle mesh Ewald potential. J Chem Phys. 1995;103:8577–92.
Falsafi-Zadeh S, Karimi Z, Galehdari H. VMD DisRg: new user-friendly implement for calculation distance and radius of gyration in VMD program. Bioinformation. 2012;8:341–3.
Ghadhanfar E, Alsalem A, Al-Kandari S, Naser J, Babiker F, Al-Bader M. The role of ACE2, angiotensin-(1–7) and Mas1 receptor axis in glucocorticoid-induced intrauterine growth restriction. Reprod Biol Endocrinol. 2017;15:97. https://doi.org/10.1186/s12958-017-0316-8.
Gralinski LE, Menachery VD. Return of the coronavirus: 2019-nCoV. Viruses. 2020;12:135 Google Sch.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506.
Ishima R, Torchia DA. Protein dynamics from NMR. Nat Struct Biol. 2000;7:740–3.
Jiayi R, Zhiwei Y, Nianjue Z, et al. Effect of force fields and water models on egfrviii-MR1 (scfv) complex by molecular dynamics simulation, MM-PBSA calculation, and ITC experiment. Chem J Chin Univ. 2017;38:2070–6.
Kadam RU, Wilson IA. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc Natl Acad Sci U S A. 2017;114:206–14.
Kamps JJAG, Huang J, Poater J, et al. Chemical basis for the recognition of trimethyllysine by epigenetic reader proteins. Nat Commun. 2015;6:1–12.
Kumari R, Kumar R. C. Open source drug discovery and A. Lynn. J Chem Inf Model. 2014;54:1951–62.
Lammers T, Sofias AM, van der Meel R, Schiffelers R, Storm G, Tacke F, et al. Dexamethasone nanomedicines for COVID-19. Nat Nanotechnol. 2020;15:622–4. https://doi.org/10.1038/s41565-020-0752-z.
Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–86.
Ledford, H., Coronavirus breakthrough: dexamethasone is first drug shown to save lives. 2020.
Lemkul J. From proteins to perturbed Hamiltonians: a suite of tutorials for the GROMACS-2018 molecular simulation package [article v1. 0]. Living J Comput Mol Sci. 2018;1:5068.
Li, G. and E. De Clercq, Therapeutic options for the 2019 novel coronavirus (2019-nCoV). 2020, Nat Publ Group.
Lian N, Xie H, Lin S, Huang J, Zhao J, Lin Q. Umifenovir treatment is not associated with improved outcomes in patients with coronavirus disease 2019: a retrospective study. Clin Microbiol Infect. 2020 Jul;26(7):917–21. https://doi.org/10.1016/j.cmi.2020.04.026.
Liu X, Wang X-J. Potential inhibitors against 2019-nCoV coronavirus M protease from clinically approved medicines. J Genet Genomics. 2020;47:119–21.
Liu X, Zhang B, Jin Z, et al (2020) The crystal structure of COVID-19 main protease in complex with an inhibitor N3. Protein DataBank
Low-cost dexamethasone reduces death by up to one third in hospitalised patients with severe respiratory complications of COVID-19. http://www.ox.ac.uk/news/2020-06-16-low-cost-dexamethasone-reduces-death-one-third-hospitalised-patients-severe, 2020.
McKee DL, Sternberg A, Stange U, Laufer S, Naujokat C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res. 2020;157:05. https://doi.org/10.1016/j.phrs.2020.104859.
Mittal, L., et al., Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. Journal of Biomolecular Structure and Dynamics, 2020(just-accepted): p. 1–26.
Moradi SZ , Nowroozi A , Sadrjavadi K , Moradi S , Mansouri K , Hosseinzadeh L, Shahlaei M (2018), Int. J. Biol. Macromol.114 , 40 —53.
Nayeem SM, Kondaiah M, Sreekanth K, Krishna Rao D. Thermoacoustic, volumetric, and viscometric investigations in binary liquid system of cyclohexanone with methyl benzoate at T = 30815, 31315, and 31815 K. J Thermodyn. 2014;2014:1–13. https://doi.org/10.1155/2014/487403.
Ortolani C, Pastorello EA. Hydroxychloroquine and dexamethasone in COVID-19: who won and who lost? Clin Mol Allergy. 2020;18:17. https://doi.org/10.1186/s12948-020-00132-7.
Paraskevis D, Kostaki EG, Magiorkinis G, Panayiotakopoulos G, Sourvinos G, Tsiodras S. Full-genome evolutionary analysis of the novel corona virus (2019-nCoV) rejects the hypothesis of emergence as a result of a recent recombination event. Infect Genet Evol. 2020;79:104212.
Perozzo R, Folkers G, Scapozza L. Thermodynamics of protein–ligand interactions: history, presence, and future aspects. J Recept Signal Transduct. 2004;24:1–52.
Ren J, Yuan X, Li J, et al (2019) Assessing the performance of the g_mmpbsa tools to simulate the inhibition of oseltamivir to influenza virus neuraminidase by molecular mechanics Poisson–Boltzmann surface area methods. J Chinese Chem Soc.
Roberts A, Deming D, Paddock CD, et al (2007) A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice PLoS Pathog 3:
Robertson MJ, Tirado-Rives J, Jorgensen WL. Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput. 2015;11(7):3499–509.
Schauperl M, Podewitz M, Waldner BJ, Liedl KR. Enthalpic and entropic contributions to hydrophobicity. J Chem Theory Comput. 2016;12:4600–10.
Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24:417–22.
Sharp KA, Honig B. Calculating total electrostatic energies with the nonlinear Poisson-Boltzmann equation. J Phys Chem. 1990;94:7684–92.
Stauffer WM, Alpern JD, Walker PF. COVID-19 and dexamethasone: a potential strategy to avoid steroid-related strongyloides hyperinfection. JAMA. 2020;324(7):623–4. https://doi.org/10.1001/jama.2020.13170.
Sun H, Li Y, Shen M, Tian S, Xu L, Pan P, et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys Chem Chem Phys. 2014;16:22035–45.
Tachoua W, Kabrine M. Molecular docking study of COVID-19 main protease with clinically approved drugs. ChemRxiv. 2020. https://doi.org/10.26434/chemrxiv.12318689.v1.
Tikellis C, Thomas MC. Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept. 2012;2012:1–8.
Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–43.
Tsui V, Case DA. Theory and applications of the generalized born solvation model in macromolecular simulations. Biopolym Orig Res Biomol. 2000;56:275–91.
Coralie Valle, Baptiste Martin, Franck Touret, Ashleigh Shannon, Bruno Canard, Jean-Claude Guillemot, Bruno Coutard, Etienne Decroly; Drugs against SARS-CoV-2: what do we know about their mode of action? Rev Med Virol. 2020;e2143. https://doi.org/10.1002/rmv.2143
Vankadari N. Arbidol: a potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int J Antimicrob Agents. 2020;56(2):105998. https://doi.org/10.1016/j.ijantimicag.2020.105998.
https://www.who.int/news-room/detail/16-06-2020-who-welcomes-preliminary-results-aboutdexamethasone-use-in-treating-critically-ill-covid-19-patients, WHO welcomes preliminary results about dexamethasone use in treating critically ill COVID-19 patients. 2020.
Yang Z, Yang G, Zhou L. Mutation effects of neuraminidases and their docking with ligands: a molecular dynamics and free energy calculation study. J Comput Aided Mol Des. 2013;27:935–50.
Yang Z, Wu F, Yuan X, Zhang L, Zhang S. Novel binding patterns between ganoderic acids and neuraminidase: insights from docking, molecular dynamics and MM/PBSA studies. J Mol Graph Model. 2016;65:27–34.
Zhang J, Zhou L, Yang Y, Peng W, Wang W, Chen X. Therapeutic and triage strategies for 2019 novel coronavirus disease in fever clinics. Lancet Respir Med. 2020;8:e11–2.
Acknowledgments
The authors are very thankful to the Government of Andhra Pradesh for taking all measures to control the widespread of SARS-CoV-2 virus and paying much attention to trace, test, and treatment of the COVID-19 infected patients.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

{kind=link}
Cite this article
Nayeem, S.M., Sohail, E.M., Ridhima, G. et al. Target SARS-CoV-2: computation of binding energies with drugs of dexamethasone/umifenovir by molecular dynamics using OPLS-AA force field. Res. Biomed. Eng. 38, 117–126 (2022). https://doi.org/10.1007/s42600-020-00119-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42600-020-00119-y