
Targeted Virome Sequencing Enhances Unbiased Detection and Genome Assembly of Known and Emerging Viruses—The Example of SARS-CoV-2

 , ,
, ,  ,
,
Abstract
:1. Introduction
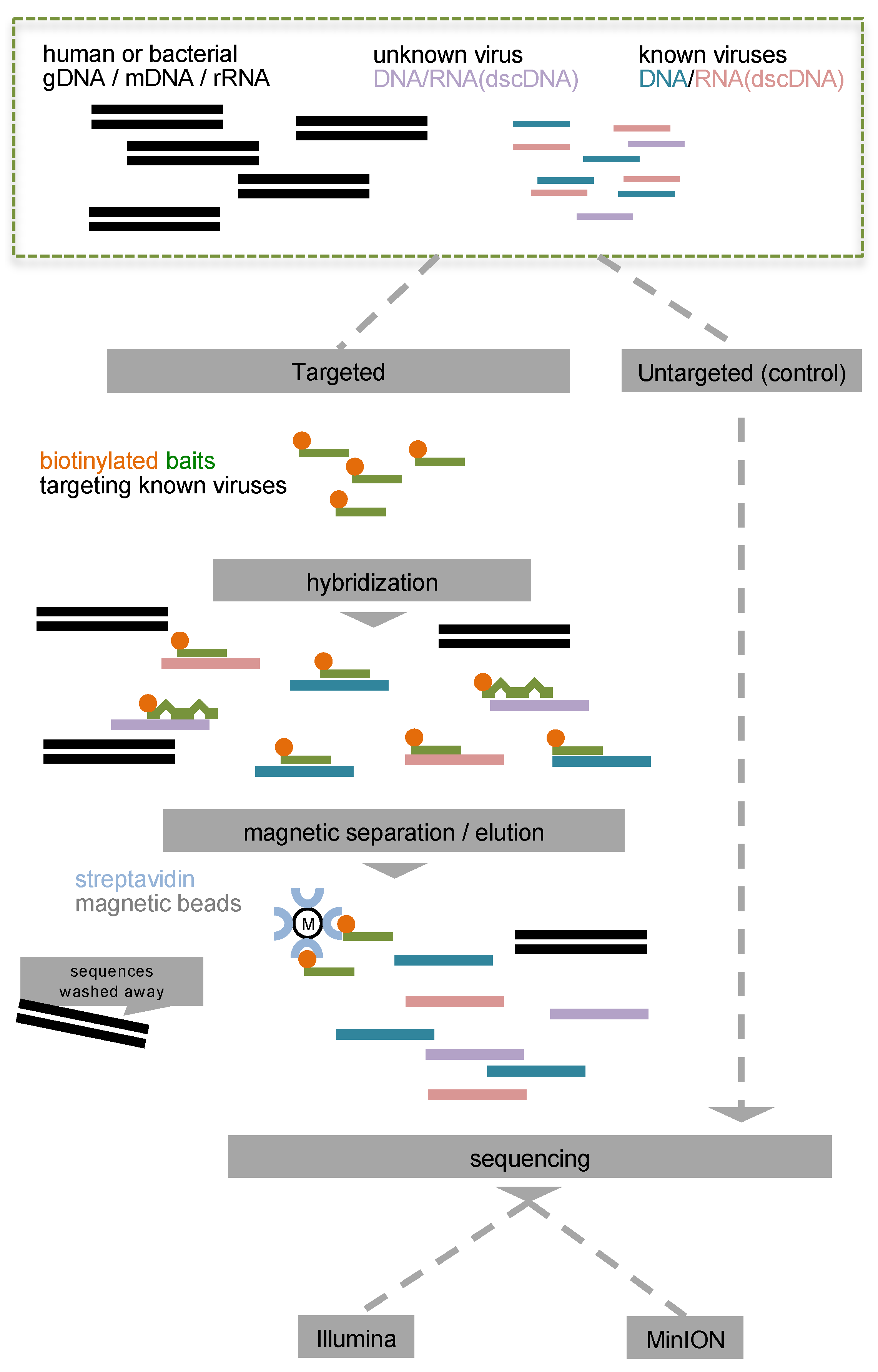
2. Materials and Methods
2.1. Sample Preparation
2.2. Bioinformatics
3. Results
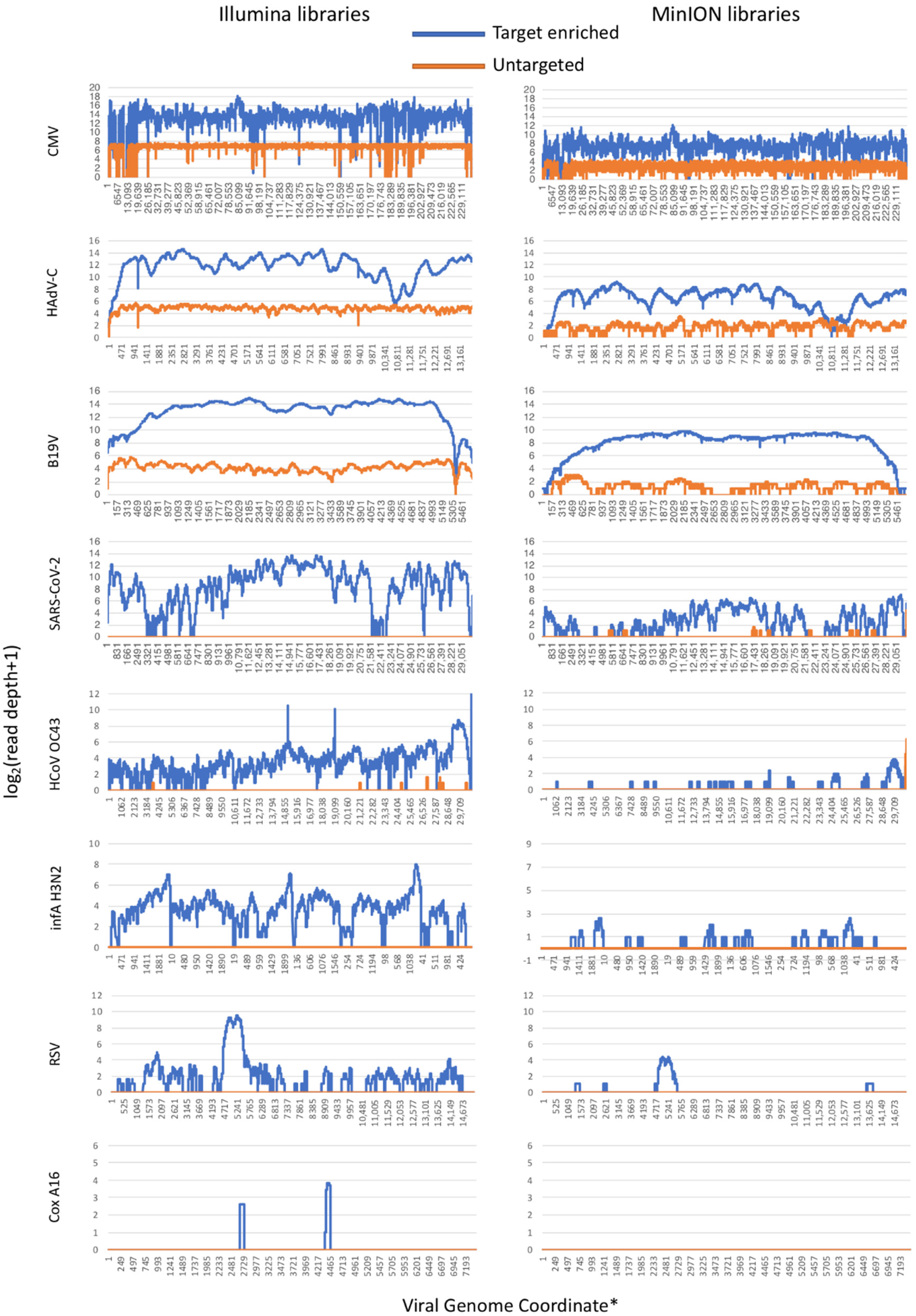
3.1. Efficiency of Total Virome Target Enrichment
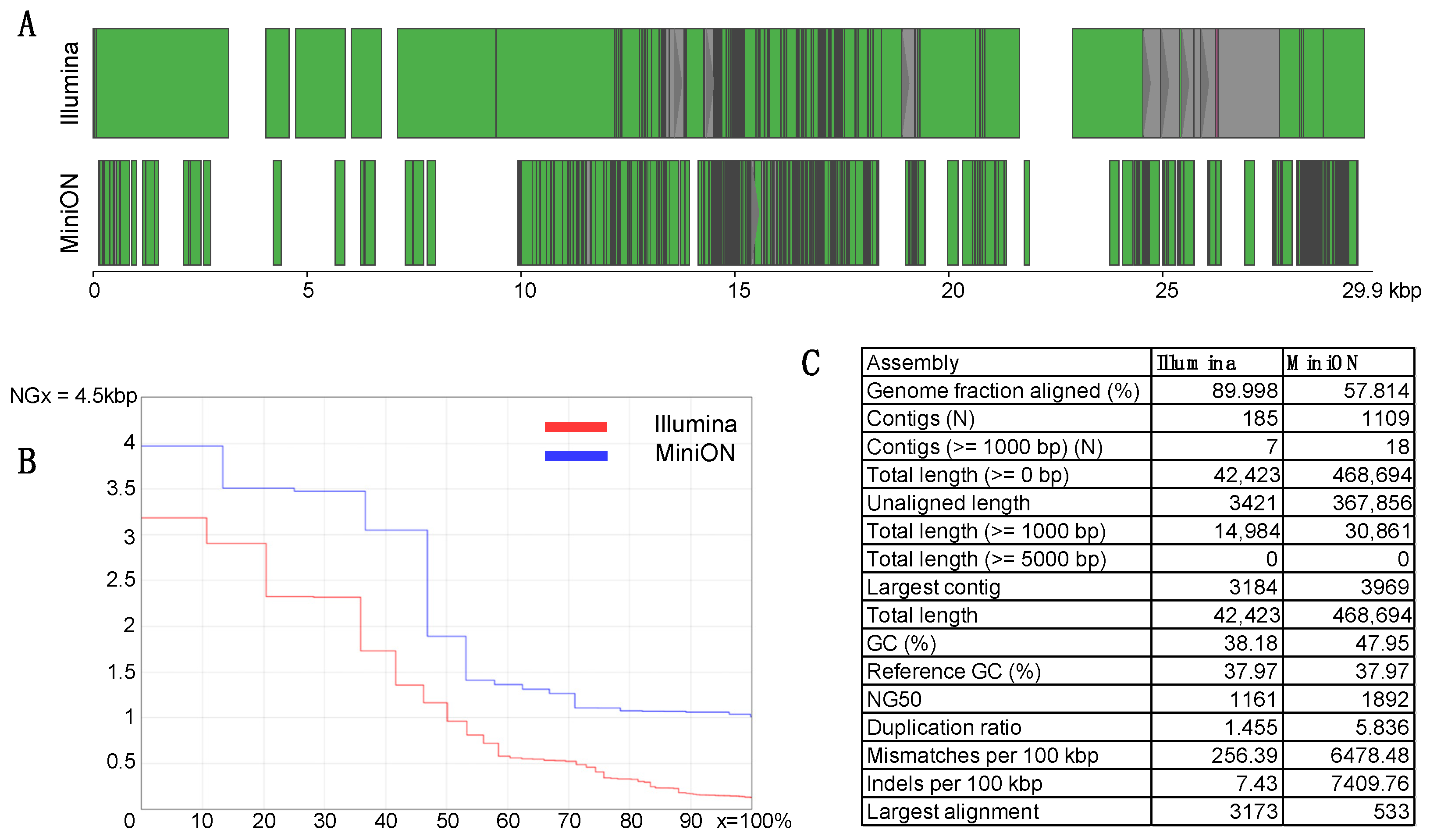
3.2. De Novo Assembly—Reconstruction of SARS-CoV-2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding

Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Regional Office for South-East Asia. In A Brief Guide to Emerging Infectious Diseases and Zoonoses; WHO Regional Office for South-East Asia: New Delhi, India, 2014; ISBN 9789290224587. [Google Scholar]
- Chabas, H.; Lion, S.; Nicot, A.; Meaden, S.; van Houte, S.; Moineau, S.; Wahl, L.M.; Westra, E.R.; Gandon, S. Evolutionary Emergence of Infectious Diseases in Heterogeneous Host Populations. PLoS Biol. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Falcinelli, S.D.; Chertow, D.S.; Kindrachuk, J. Integration of Global Analyses of Host Molecular Responses with Clinical Data to Evaluate Pathogenesis and Advance Therapies for Emerging and Re-Emerging Viral Infections. ACS Infect. Dis. 2016, 2, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.-C.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-Species Virus Transmission and the Emergence of New Epidemic Diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artika, I.M.; Wiyatno, A.; Ma’roef, C.N. Pathogenic Viruses: Molecular Detection and Characterization. Infect. Genet. Evol. 2020. [Google Scholar] [CrossRef]
- Parvez, M.K.; Parveen, S. Evolution and Emergence of Pathogenic Viruses: Past, Present, and Future. Intervirology 2017, 60, 1–7. [Google Scholar] [CrossRef]
- Longini, I.M.; Nizam, A.; Xu, S.; Ungchusak, K.; Hanshaoworakul, W.; Cummings, D.A.T.; Halloran, M.E. Containing Pandemic Influenza at the Source. Science (1979) 2005, 309, 1083–1087. [Google Scholar] [CrossRef] [Green Version]
- Bauch, C.T.; Lloyd-Smith, J.O.; Coffee, M.P.; Galvani, A.P. Dynamically_Modeling_SARS_and_Other_Newly_Emerging.13. Epidemiology 2005, 16, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Litvinova, M.; Liang, Y.; Wang, Y.; Wang, W.; Zhao, S.; Wu, Q.; Merler, S.; Viboud, C.; Vespignani, A.; et al. Changes in Contact Patterns Shape the Dynamics of the COVID-19 Outbreak in China. Science 2020, 368, 1481–1486. [Google Scholar] [CrossRef]
- de Salazar, P.M.; Gómez-Barroso, D.; Pampaka, D.; Gil, J.M.; Peñalver, B.; Fernández-Escobar, C.; Lipsitch, M.; Larrauri, A.; Goldstein, E.; Hernán, M.A. Lockdown Measures and Relative Changes in the Age-Specific Incidence of SARS-CoV-2 in Spain. Epidemiol. Infect. 2020. [Google Scholar] [CrossRef]
- Bousali, M.; Dimadi, A.; Kostaki, E.G.; Tsiodras, S.; Nikolopoulos, G.K.; Sgouras, D.N.; Magiorkinis, G.; Papatheodoridis, G.; Pogka, V.; Lourida, G.; et al. Sars-cov-2 Molecular Transmission Clusters and Containment Measures in Ten European Regions during the First Pandemic Wave. Life 2021, 11, 219. [Google Scholar] [CrossRef]
- Fang, G.; Munera, D.; Friedman, D.I.; Mandlik, A.; Chao, M.C.; Banerjee, O.; Feng, Z.; Losic, B.; Mahajan, M.C.; Jabado, O.J.; et al. Genome-Wide Mapping of Methylated Adenine Residues in Pathogenic Escherichia Coli Using Single-Molecule Real-Time Sequencing. Nat. Biotechnol. 2012, 30, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Albert, T.J.; Molla, M.N.; Muzny, D.M.; Nazareth, L.; Wheeler, D.; Song, X.; Richmond, T.A.; Middle, C.M.; Rodesch, M.J.; Packard, C.J.; et al. Direct Selection of Human Genomic Loci by Microarray Hybridization. Nat. Methods 2007, 4, 903–905. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, U.S.; Tan, J.H.; Mei, O.X.; Pan, L.; Chawla, T.; Uehara, A.; Wang, L.F.; Ooi, E.E.; Gubler, D.J.; Tissera, H.; et al. Application of a Targeted-Enrichment Methodology for Full-Genome Sequencing of Dengue 1-4, Chikungunya and Zika Viruses Directly from Patient Samples. PLoS Negl. Trop. Dis. 2019, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkina, W.I. Virome Capture Sequencing Enables Sensitive Viral Diagnosis and Comprehensive Virome Analysis. mBio 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-Time, Portable Genome Sequencing for Ebola Surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [Green Version]
- Faria, N.R.; Sabino, E.C.; Nunes, M.R.T.; Alcantara, L.C.J.; Loman, N.J.; Pybus, O.G. Mobile Real-Time Surveillance of Zika Virus in Brazil. Genome Med. 2016, 8. [Google Scholar] [CrossRef] [Green Version]
- Karamitros, T.; Magiorkinis, G. A Novel Method for the Multiplexed Target Enrichment of MinION next Generation Sequencing Libraries Using PCR-Generated Baits. Nucleic Acids Res. 2015, 43, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Karamitros, T.; Magiorkinis, G. Multiplexed Targeted Sequencing for Oxford Nanopore MinION: A Detailed Library Preparation Procedure. Methods Mol. Biol. 2018, 1712, 43–51. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; Desantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An Improved Greengenes Taxonomy with Explicit Ranks for Ecological and Evolutionary Analyses of Bacteria and Archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wu, S.; Li, A.; Ruan, J. SMARTdenovo: A de Novo Assembler Using Long Noisy Reads. Gigabyte 2021, 2021, 1–9. [Google Scholar] [CrossRef]
- Altschup, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kozarewa, I.; Armisen, J.; Gardner, A.F.; Slatko, B.E.; Hendrickson, C.L. Overview of Target Enrichment Strategies. Curr. Protoc. Mol. Biol. 2015, 2015. [Google Scholar] [CrossRef]
- Wei, X.; Ju, X.; Yi, X.; Zhu, Q.; Qu, N.; Liu, T.; Chen, Y.; Jiang, H.; Yang, G.; Zhen, R.; et al. Identification of Sequence Variants in Genetic Disease-Causing Genes Using Targeted next-Generation Sequencing. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Salmon, A.; Udall, J.A.; Jeddeloh, J.A.; Wendel, J. Targeted Capture of Homoeologous Coding and Noncoding Sequence in Polyploid Cotton. G3 Genes Genomes Genet. 2012, 2, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Paskey, A.C.; Frey, K.G.; Schroth, G.; Gross, S.; Hamilton, T.; Bishop-Lilly, K.A. Enrichment Post-Library Preparation Enhances the Sensitivity of High-Throughput Sequencing-Based Detection and Characterization of Viruses from Complex Samples. BMC Genom. 2019, 20, 1–14. [Google Scholar] [CrossRef]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-Enrichment Strategies for next-Generation Sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef]
- Wang, M.; Fu, A.; Hu, B.; Tong, Y.; Liu, R.; Liu, Z.; Gu, J.; Xiang, B.; Liu, J.; Jiang, W.; et al. Nanopore Targeted Sequencing for the Accurate and Comprehensive Detection of SARS-CoV-2 and Other Respiratory Viruses. Small 2020, 16. [Google Scholar] [CrossRef]
- Furtwängler, A.; Neukamm, J.; Böhme, L.; Reiter, E.; Vollstedt, M.; Arora, N.; Singh, P.; Cole, S.T.; Knauf, S.; Calvignac-Spencer, S.; et al. Comparison of Target Enrichment Strategies for Ancient Pathogen DNA. Biotechniques 2020, 69, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Karamitros, T.; Pogka, V.; Papadopoulou, G.; Tsitsilonis, O.; Evangelidou, M.; Sympardi, S.; Mentis, A. Dual Rna-Seq Enables Full-Genome Assembly of Measles Virus and Characterization of Host–Pathogen Interactions. Microorganisms 2021, 9, 1538. [Google Scholar] [CrossRef] [PubMed]
- Marston, D.A.; McElhinney, L.M.; Ellis, R.J.; Horton, D.L.; Wise, E.L.; Leech, S.L.; David, D.; de Lamballerie, X.; Fooks, A.R. Next Generation Sequencing of Viral RNA Genomes. BMC Genom. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamitros, T.; Papadopoulou, G.; Bousali, M.; Mexias, A.; Tsiodras, S.; Mentis, A. SARS-CoV-2 Exhibits Intra-Host Genomic Plasticity and Low-Frequency Polymorphic Quasispecies. J. Clin. Virol. 2020, 131, 104585. [Google Scholar] [CrossRef]
- Karamitros, T.; Harrison, I.; Piorkowska, R.; Katzourakis, A.; Magiorkinis, G.; Mbisa, J.L. De Novo Assembly of Human Herpes Virus Type 1 (HHV-1) Genome, Mining of Non-Canonical Structures and Detection of Novel Drug-Resistance Mutations Using Short- and Long-Read next Generation Sequencing Technologies. PLoS ONE 2016, 11, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Karamitros, T.; van Wilgenburg, B.; Wills, M.; Klenerman, P.; Magiorkinis, G. Nanopore Sequencing and Full Genome de Novo Assembly of Human Cytomegalovirus TB40/E Reveals Clonal Diversity and Structural Variations. BMC Genom. 2018, 19, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cutler, S.J.; Fooks, A.R.; van der Poel, W.H.M. Public Health Threat of New, Reemerging, and Neglected Zoonoses in the Industrialized World. Emerg. Infect. Dis. 2010, 16, 1–7. [Google Scholar] [CrossRef]
- Naicker, P.R. The Impact of Climate Change and Other Factors on Zoonotic Diseases. Arch. Clin. Microbiol. 2011, 2, 1–6. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Illumina | MinION(ONT) | |||||||
|---|---|---|---|---|---|---|---|---|
| Untargeted/Targeted | Untargeted/Targeted | |||||||
| Total Reads(N) | 32,235,720/63,664,520 | 1,866,680/1,129,865 | ||||||
| Mapped Reads (N) | Mapped Reads (%) | Genome Coverage (%) | Read Depth (X, Mean) | Mapped Reads, N | Mapped Reads (%) | Genome Coverage (%) | Read Depth (X, Mean) | |
| Viral Genome | ||||||||
| CMV | 295,623/48,693,256 | 0.9171/76.4841 | 96.71/97.38 | 17,873.30/20,176.00 | 9522/367,035 | 0.5101/32.4849 | 95.58/96.81 | 8.09/310.59 |
| HAdV-C | 8549/2,438,953 | 0.0265/3.8309 | 88.46/92.83 | 23.42/6617.77 | 348/22,324 | 0.0186/1.9758 | 69.58/97.48 | 2.51/154.86 |
| B19V | 1129/726,199 | 0.0035/1.1405 | 99.76/99.98 | 19.88/12,782.60 | 38/7620 | 0.0020/0.6744 | 63.44/98.86 | 1.84/433.58 |
| SARS-CoV-2 | 0/797,191 | 0.0000/1.2522 | 0.00/96.12 | 0.00/1800.46 | 1285/2974 | 0.0688/0.2632 | 4.18/74.46 | 1.53/14.39 |
| HCoV-OC43 | 9/141,470 | 0.0041/0.2222 | 2.44/96.07 | 0.03/152.40 | 76/102 | 0.0041/0.0090 | 0.14/20.09 | 0.08/0.54 |
| infA-H3N2 | 0/2733 | 0.0000/0.0043 | 0.00/93.87 | 0.00/19.40 | 0/32 | 0.0000/0.0028 | 0.00/25.72 | 0.00/0.45 |
| RSV | 0/4119 | 0.0000/0.0065 | 0.00/63.62 | 0.00/24.90 | 0/49 | 0.0000/0.0043 | 0.00/9.23 | 0.00/0.61 |
| Cox A16 | 0/18 | 0.0000/0.0000 | 0.00/2.64 | 0.00/0.21 | 0/0 | 0.0000/0.0000 | 0.00/0.00 | 0.00/0.00 |
| Background | ||||||||
| Human Genome (hg19) | 30,125,987/6,016,007 | 93.4553/9.4495 | 1,322,818/158,985 | 70.8647/14.0712 | ||||
| Human mtDNA | 62,049/12,196 | 0.1925/0.0192 | 3461/371 | 0.1854/0.0328 | ||||
| Human rRNA/DNA | 228,575/501,020 | 0.7091/0.7870 | 44,473/13,166 | 2.3825/1.1653 | ||||
| 16S DNA | 11,444/53,525 | 0.0355/0.0841 | 1456/1172 | 0.0780/0.1037 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pogka, V.; Papadopoulou, G.; Valiakou, V.; Sgouras, D.N.; Mentis, A.F.; Karamitros, T. Targeted Virome Sequencing Enhances Unbiased Detection and Genome Assembly of Known and Emerging Viruses—The Example of SARS-CoV-2. Viruses 2022, 14, 1272. https://doi.org/10.3390/v14061272
Pogka V, Papadopoulou G, Valiakou V, Sgouras DN, Mentis AF, Karamitros T. Targeted Virome Sequencing Enhances Unbiased Detection and Genome Assembly of Known and Emerging Viruses—The Example of SARS-CoV-2. Viruses. 2022; 14(6):1272. https://doi.org/10.3390/v14061272
Chicago/Turabian StylePogka, Vasiliki, Gethsimani Papadopoulou, Vaia Valiakou, Dionyssios N. Sgouras, Andreas F. Mentis, and Timokratis Karamitros. 2022. "Targeted Virome Sequencing Enhances Unbiased Detection and Genome Assembly of Known and Emerging Viruses—The Example of SARS-CoV-2" Viruses 14, no. 6: 1272. https://doi.org/10.3390/v14061272