
Translocating Peptides of Biomedical Interest Obtained from the Spike (S) Glycoprotein of the SARS-CoV-2


 , , ,
, , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Peptides and Structural Analysis
2.2. Prediction of Peptides Structure
2.3. Molecular Dynamics Simulations
2.3.1. Behavior of Peptides Inside the Membrane
2.3.2. Non-Equilibrium Pulling
2.4. Synthesis of Low Molecular Weight Chitosan Nanoparticles (CNPs)
2.5. Functionalization of CNPs with the Peptides and Rhodamine B
2.6. Hemolysis
2.7. Platelet Aggregation
2.8. Cytotoxicity and Cell Viability
2.9. Plasma Membrane Translocation and Endosomal Escape
3. Results and Discussion
3.1. Peptides’ Structure Prediction
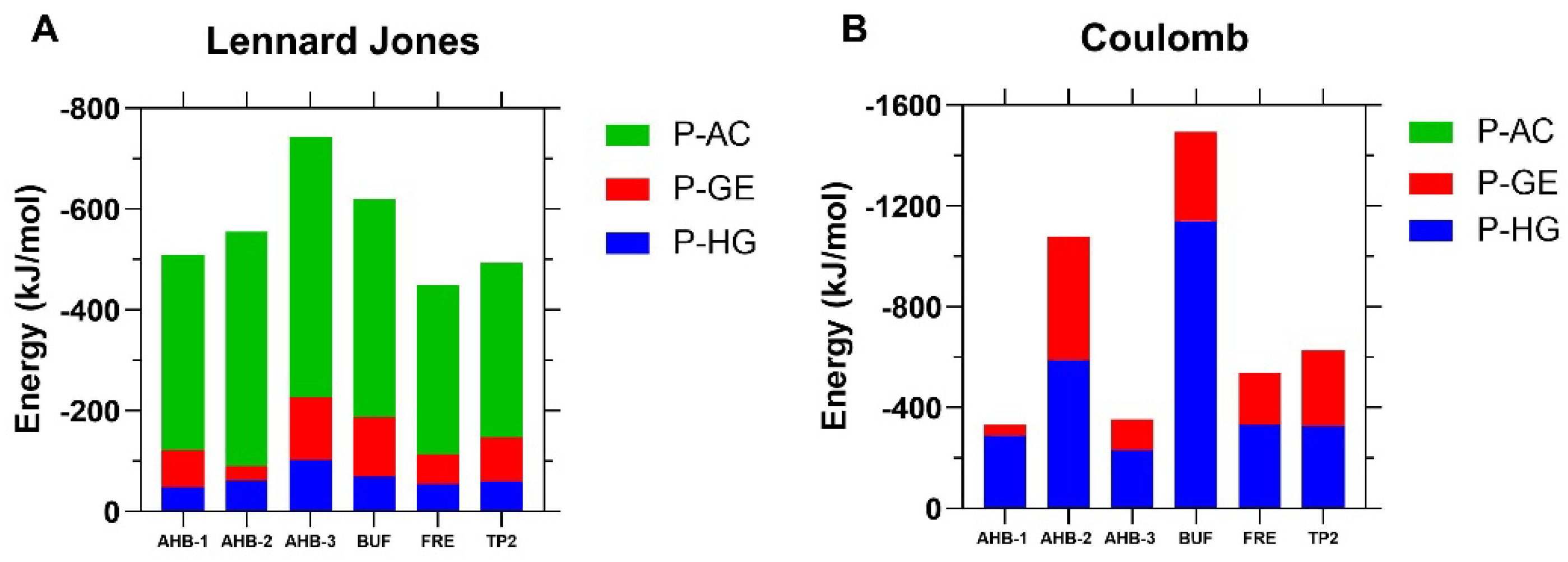
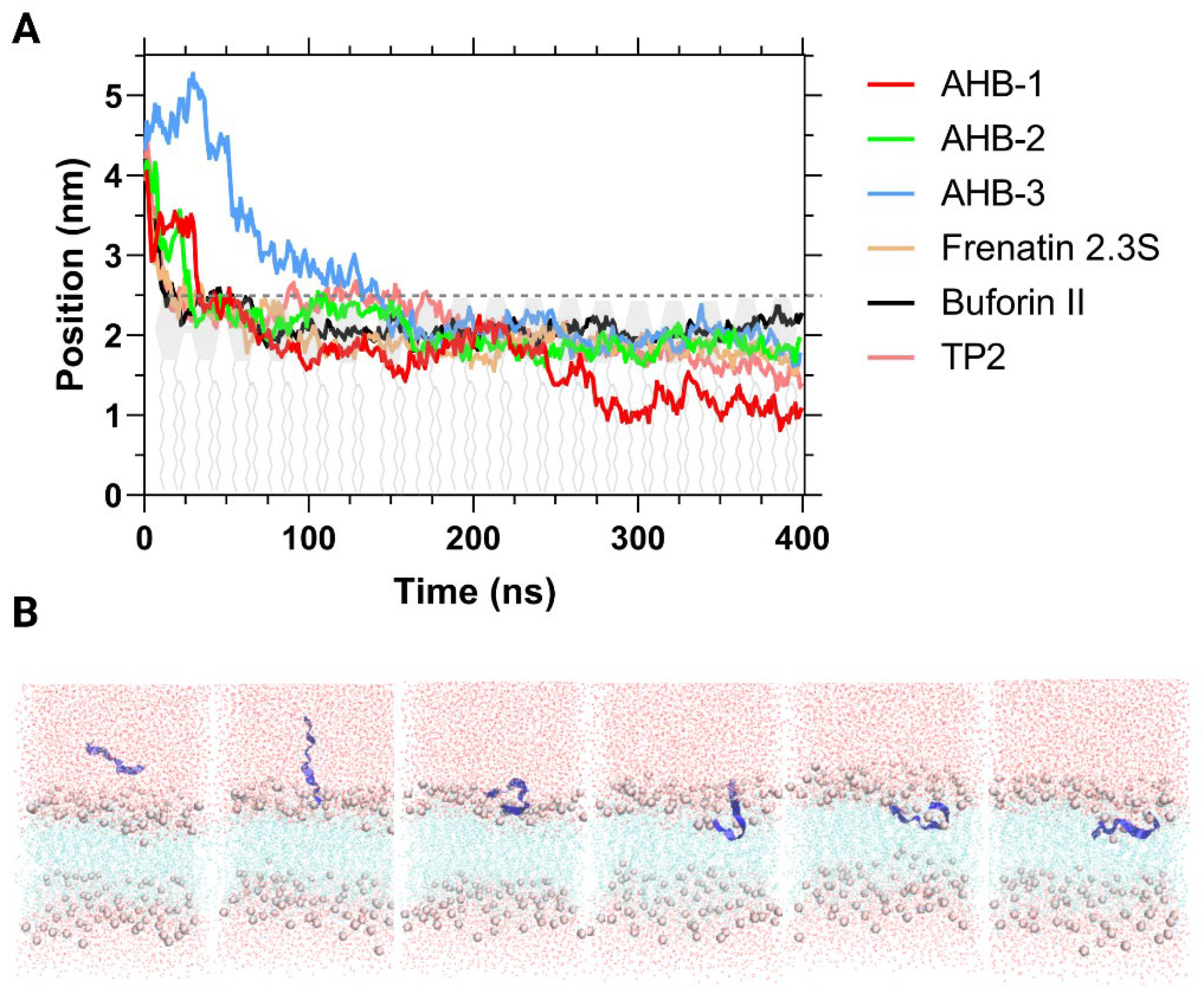
3.2. Molecular Dynamics Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boes, D.; Godoy-Hernandez, A.; McMillan, D. Peripheral membrane proteins: Promising therapeutic targets across domains of life. Membranes 2021, 11, 346. [Google Scholar] [CrossRef] [PubMed]
- Lombard, J. Once upon a time the cell membranes: 175 years of cell boundary research. Biol. Direct 2014, 9, 1–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loschwitz, J.; Olubiyi, O.O.; Hub, J.S.; Strodel, B.; Poojari, C.S. Computer simulations of protein–membrane systems. Prog. Mol. Biol. Transl. Sci. 2020, 170, 273–403. [Google Scholar] [PubMed]
- Zhang, R.; Qin, X.; Kong, F.; Chen, P.; Pan, G. Improving cellular uptake of therapeutic entities through interaction with components of cell membrane. Drug Deliv. 2019, 26, 328–342. [Google Scholar] [CrossRef] [Green Version]
- Bernacki, J.; Dobrowolska, A.; Nierwinska, K.; Małecki, A. Pharmacological reports PR. physiology and pharmacological role of the blood-brain barrier. Pharmacol. Rep. 2008, 60, 600–622. [Google Scholar]
- Miersch, S.; Sidhu, S.S. Intracellular targeting with engineered proteins. F1000Research 2016, 5, 1947. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sud, N.; Hettinghouse, A.; Liu, C.-J. Molecular regulations and therapeutic targets of gaucher disease. Cytokine Growth Factor Rev. 2018, 41, 65–74. [Google Scholar] [CrossRef]
- Galiullina, L.F.; Scheidt, H.A.; Huster, D.; Aganov, A.; Klochkov, V. Interaction of statins with phospholipid bilayers studied by solid-state NMR spectroscopy. Biochim. Biophys. Acta BBA Biomembr. 2018, 1861, 584–593. [Google Scholar] [CrossRef]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: An overview for small molecules, peptides, and proteins. In Site-Specific Protein Labeling: Methods and Protocols; Springer: New York, NY, USA, 2015; Volume 1266, pp. 29–53. [Google Scholar] [CrossRef] [Green Version]
- Waller, D.G.; Sampson, A.P. Chemotherapy of infections. In Medical Pharmacology and Therapeutics; Elsevier: Amsterdam, The Netherlands, 2018; pp. 581–629. [Google Scholar] [CrossRef]
- Huang, Y.-W.; Lee, H.-J. Cell-penetrating peptides for medical theranostics and targeted drug delivery. In Peptide Applications in Biomedicine, Biotechnology and Bioengineering; Elsevier Inc.: Philadelphia, PA, USA, 2018; pp. 359–370. [Google Scholar] [CrossRef]
- Pan, X.; Veroniaina, H.; Su, N.; Sha, K.; Jiang, F.; Wu, Z.; Qi, X. Applications and developments of gene therapy drug delivery systems for genetic diseases. Asian J. Pharm. Sci. 2021, 16, 687–703. [Google Scholar] [CrossRef]
- Derakhshankhah, H.; Jafari, S. Cell penetrating peptides: A concise review with emphasis on biomedical applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef]
- Huang, Y.-W.; Lee, H.-J.; Tolliver, L.M.; Aronstam, R.S. Delivery of nucleic acids and nanomaterials by cell-penetrating peptides: Opportunities and challenges. BioMed Res. Int. 2015, 2015, 452603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghian, I.; Heidari, R.; Sadeghian, S.; Raee, M.J.; Negahdaripour, M. Potential of cell-penetrating peptides (CPPs) in delivery of antiviral therapeutics and vaccines. Eur. J. Pharm. Sci. 2021, 169, 106094. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.-H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep. 2019, 9, 6298. [Google Scholar] [CrossRef] [PubMed]
- El-Baky, N.A.; Uversky, V.N.; Redwan, E.M. Virucidal activity of cell-penetrating peptides of viral origin. J. Biomol. Struct. Dyn. 2017, 36, 1739–1746. [Google Scholar] [CrossRef]
- Pärn, K.; Eriste, E.; Langel, Ü. The antimicrobial and antiviral applications of cell-penetrating peptides. Cell-Penetrating Pept. 2015, 1324, 223–245. [Google Scholar] [CrossRef]
- Dong, E.; Hongru, D.; Lauren, G. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Jaimes, J.A.; André, N.M.; Millet, J.K.; Whittaker, G.R. Phylogenetic analysis and structural modeling of SARS-CoV-2 spike protein reveals an evolutionary distinct and proteolytically sensitive activation loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Hulswit, R.J.G.; De Haan, C.A.M.; Bosch, B.J. Coronavirus spike protein and tropism changes. In Advances in Virus Research; Academic Press Inc.: New York, NY, USA, 2016; Volume 96, pp. 29–57. [Google Scholar]
- Mori, T.; Miyashita, N.; Im, W.; Feig, M.; Sugita, Y. Molecular dynamics simulations of biological membranes and membrane proteins using enhanced conformational sampling algorithms. Biochim. Biophys. Acta BBA Biomembr. 2016, 1858, 1635–1651. [Google Scholar] [CrossRef] [Green Version]
- Azad, T.; Singaravelu, R.; Crupi, M.J.; Jamieson, T.; Dave, J.; Brown, E.E.; Rezaei, R.; Taha, Z.; Boulton, S.; Martin, N.T.; et al. Implications for SARS-CoV-2 vaccine design: Fusion of spike glycoprotein transmembrane domain to receptor-binding domain induces trimerization. Membranes 2020, 10, 215. [Google Scholar] [CrossRef]
- Moraes, I.; Evans, G.; Sanchez-Weatherby, J.; Newstead, S.; Stewart, P.D.S. Membrane protein structure determination—The next generation. Biochim. Biophys. Acta BBA Biomembr. 2014, 1838, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, E.; Sansom, M.S.P. Membrane proteins: Molecular dynamics simulations. Curr. Opin. Struct. Biol. 2008, 18, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Corradi, V.; Souza, P.C.; Ingólfsson, H.I.; Tieleman, D.P.; Sansom, M.S. Computational modeling of realistic cell membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puentes, P.R.; Henao, M.C.; Torres, C.E.; Gómez, S.C.; Gómez, L.A.; Burgos, J.C.; Arbeláez, P.; Osma, J.F.; Muñoz-Camargo, C.; Reyes, L.H.; et al. Design, screening, and testing of non-rational peptide libraries with antimicrobial activity: In silico and experimental approaches. Antibiotics 2020, 9, 854. [Google Scholar] [CrossRef] [PubMed]
- González, M. Force fields and molecular dynamics simulations. École Thématique Société Française Neutron. 2011, 12, 169–200. [Google Scholar] [CrossRef]
- Monticelli, L.; Tieleman, D.P. Force fields for classical molecular dynamics. In Biomolecular Simulations; Springer: Berlin/Heidelberg, Germany, 2012; pp. 197–213. [Google Scholar]
- Goossens, K.; De Winter, H. Molecular dynamics simulations of membrane proteins: An overview. J. Chem. Inf. Model. 2018, 58, 2193–2202. [Google Scholar] [CrossRef] [PubMed]
- Feig, M.; Nawrocki, G.; Yu, I.; Wang, P.-H.; Sugita, Y. Challenges and opportunities in connecting simulations with experiments via molecular dynamics of cellular environments. J. Phys. Conf. Ser. 2018, 1036, 012010. [Google Scholar] [CrossRef]
- Aponte-Santamaría, C.; Camilo, R.B.; Andreas, D.S.; Thomas, W.; Bert, L.D.G. Molecular driving forces defining lipid positions around aquaporin-0. Proc. Natl. Acad. Sci. USA 2012, 109, 9887–9892. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, A.; Srikanth, B.; Kumar, A.; DasMahapatra, A.K. Coarse-grain molecular dynamics study of fullerene transport across a cell membrane. J. Chem. Phys. 2015, 143, 24907. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Uppal, S.; Kaur, K.; Kumar, R.; Kaur, N.D.; Shukla, G.; Mehta, S. Chitosan nanoparticles as a biocompatible and efficient nanowagon for benzyl isothiocyanate. Int. J. Biol. Macromol. 2018, 115, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Melo, C.; Garcia-Brand, A.J.; Quezada, V.; Reyes, L.H.; Muñoz-Camargo, C.; Cruz, J.C. Highly efficient synthesis of type B gelatin and low molecular weight chitosan nanoparticles: Potential applications as bioactive molecule carriers and cell-penetrating agents. Polymers 2021, 13, 4078. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadaf, A.; Cho, K.H.; Byrne, B.; Chae, P.S. Amphipathic agents for membrane protein study. Methods Enzymol. 2015, 557, 57–94. [Google Scholar] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M. The Cell: A Molecular Approach; Sinauer Associates: Sunderland, MA, USA, 2000; Volume 8, pp. 103–108. [Google Scholar]
- Soundrarajan, N.; Cho, H.-S.; Ahn, B.; Choi, M.; Thong, L.M.; Choi, H.; Cha, S.-Y.; Kim, J.-H.; Park, C.-K.; Seo, K.; et al. Green fluorescent protein as a scaffold for high efficiency production of functional bacteriotoxic proteins in escherichia Coli. Sci. Rep. 2016, 6, 20661. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Camargo, C.; Salazar, V.A.; Barrero-Guevara, L.; Camargo, S.; Mosquera, A.; Groot, H.; Boix, E. Unveiling the multifaceted mechanisms of antibacterial activity of buforin II and frenatin 2.3S peptides from skin micro-organs of the orinoco lime treefrog (sphaenorhynchus lacteus). Int. J. Mol. Sci. 2018, 19, 2170. [Google Scholar] [CrossRef] [Green Version]
- Cruz, J.C.; Mihailescu, M.; Wiedman, G.; Herman, K.; Searson, P.; Wimley, W.C.; Hristova, K. A membrane-translocating peptide penetrates into bilayers without significant bilayer perturbations. Biophys. J. 2013, 104, 2419–2428. [Google Scholar] [CrossRef] [Green Version]
- Berger, O.; Edholm, O.; Jähnig, F.; Berger, O.; Edholm, O.; Jähnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [Google Scholar] [CrossRef] [Green Version]
- Lemkul, J. From proteins to perturbed hamiltonians: A suite of tutorials for the gromacs-2018 molecular simulation package [Article v1.0]. Living J. Comput. Mol. Sci. 2019, 1, 5068. [Google Scholar] [CrossRef]
- Mark, P.; Lennart, N. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Roux, B. The calculation of the potential of mean force using computer simulations. Comput. Phys. Commun. 1995, 91, 275–282. [Google Scholar] [CrossRef]
- Castro, A. Formulación, Síntesis, Optimización y Caracterización de Dos Tipos de Nanosistemas de Encapsulamiento Basados En Quitosano. Bachelor’s Dissertation, Universidad de la Republica, Montevideo, Uruguay, 2014. [Google Scholar]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: Implications for the design of spike-based vaccine immunogens. Front. Immunol. 2020, 11, 576622. [Google Scholar] [CrossRef]
- Karamyshev, A.L.; Tikhonova, E.B.; Karamysheva, Z.N. Translational control of secretory proteins in health and disease. Int. J. Mol. Sci. 2020, 21, 2538. [Google Scholar] [CrossRef] [Green Version]
- Zahid, M.; Robbins, P.D. Cell-type specific penetrating peptides: Therapeutic promises and challenges. Molecules 2015, 20, 13055–13070. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-NCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Giralt, E. Cell-penetrating peptides: Design strategies beyond primary structure and amphipathicity. Molecules 2017, 22, 1929. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Camargo, C.; Montoya, V.S.; Barrero-Guevara, L.A.; Groot, H.; Boix, E. Buforin II Bacteria agglutination activity as part of its antimicrobial action mechanism. In Proceedings of the 2018 9th International Seminar of Biomedical Engineering, SIB 2018—Conference Proceedings, Bogota, Colombia, 16–18 May 2018. [Google Scholar]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Zhang, Y. Protein structure prediction: When is it useful? Curr. Opin. Struct. Biol. 2009, 19, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure of biomolecules through molecular dynamics simulations. Procedia Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Nunes, J.P.F.; Ledbetter, K.; Lin, M.; Kozina, M.; DePonte, D.P.; Biasin, E.; Centurion, M.; Crissman, C.J.; Dunning, M.; Guillet, S.; et al. Liquid-phase mega-electron-volt ultrafast electron diffraction. Struct. Dyn. 2020, 7, 024301. [Google Scholar] [CrossRef] [PubMed]
- Galzitskaya, O.V.; Bogatyreva, N.S.; Ivankov, D.N. Compactness determines protein folding type. J. Bioinform. Comput. Biol. 2008, 6, 667–680. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Akimoto, T.; Mitsutake, A.; Metzler, R. Universal relation between instantaneous diffusivity and radius of gyration of proteins in aqueous solution. Phys. Rev. Lett. 2021, 126, 128101. [Google Scholar] [CrossRef] [PubMed]
- Braun, A.; Sachs, J.N. Determining structural and mechanical properties from molecular dynamics simulations of lipid vesicles. J. Chem. Theory Comput. 2014, 10, 4160–4168. [Google Scholar] [CrossRef] [Green Version]
- Waidyasooriya, H.M.; Hariyama, M.; Kasahara, K. An FPGA accelerator for molecular dynamics simulation using openCL. Int. J. Netw. Distrib. Comput. 2017, 5, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Yesylevskyy, S.; Marrink, S.-J.; Mark, A.E. Alternative mechanisms for the interaction of the cell-penetrating peptides penetratin and the TAT peptide with lipid bilayers. Biophys. J. 2009, 97, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Chen, J.; Zhou, G.; Wang, Y.; Xu, C.; Wang, X. Molecular dynamics simulations of the permeation of bisphenol A and pore formation in a lipid membrane. Sci. Rep. 2016, 6, 33399. [Google Scholar] [CrossRef]
- Schaefer, S.L.; Hendrik, J.; Gerhard, H. Binding of SARS-CoV-2 fusion peptide to host endosome and plasma membrane. J. Phys. Chem. B 2021, 125, 7732–7741. [Google Scholar] [CrossRef]
- John, C.M.; Li, M.; Feng, D.; Jarvis, A.G. Cationic cell-penetrating peptide is bactericidal against neisseria gonorrhoeae. J. Antimicrob. Chemother. 2019, 74, 3245–3251. [Google Scholar] [CrossRef] [PubMed]
- Jobin, M.-L.; Bonnafous, P.; Temsamani, H.; Dole, F.; Grélard, A.; Dufourc, E.J.; Alves, I.D. The enhanced membrane interaction and perturbation of a cell penetrating peptide iAn the presence of anionic lipids: Toward an understanding of its selectivity for cancer cells. Biochim. Biophys. Acta BBA Biomembr. 2013, 1828, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Allolio, C.; Aniket, M.; Piotr, J.; Katarína, B.; Matti, J.; Philip, E.M.; Radek, Š.; Marek, C.; Martin, H.; Dominik, H.; et al. Arginine-rich cell-penetrating peptides induce membrane multilamellarity and subsequently enter via formation of a fusion pore. Proc. Natl. Acad. Sci. USA 2018, 115, 11923–11928. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.A.; Selby, L.I.; Johnston, A.P.R.; Such, G.K. The endosomal escape of nanoparticles: Toward more efficient Cellular delivery. Bioconjug. Chem. 2018, 30, 263–272. [Google Scholar] [CrossRef] [PubMed]
- González, C.; Reyes, L.H.; Muñoz-Camargo, C.; Cruz, J.C. Synthesis, characterization, and functionalization of chitosan and gelatin type B nanoparticles to develop novel highly biocompatible cell-penetrating agents. Mater. Proc. 2021, 4, 30. [Google Scholar]
- Weber, M.; Steinle, H.; Golombek, S.; Hann, L.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Blood-contacting biomaterials: In vitro evaluation of the hemocompatibility. Front. Bioeng. Biotechnol. 2018, 6, 99. [Google Scholar] [CrossRef]
- Beztsinna, N.; De Matos, M.B.C.; Walther, J.; Heyder, C.; Hildebrandt, E.; Leneweit, G.; Mastrobattista, E.; Kok, R.J. Quantitative analysis of receptor-mediated uptake and pro-apoptotic activity of mistletoe lectin-1 by high content imaging. Sci. Rep. 2018, 8, 2768. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.; Almeida, A.J.; Vale, N. Combination of cell-penetrating peptides with nanoparticles for therapeutic application: A review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef]
- Perez, J.; Rueda, J.; Cuellar, M.; Suarez-Arnedo, A.; Cruz, J.C.; Muñoz-Camargo, C. Cell-penetrating and antibacterial BUF-II nanobioconjugates: Enhanced potency via immobilization on polyetheramine-modified magnetite nanoparticles. Int. J. Nanomed. 2019, ume 14, 8483–8497. [Google Scholar] [CrossRef] [Green Version]
- Malatesta, M.; Grecchi, S.; Chiesa, E.; Cisterna, B.; Costanzo, M.; Zancanaro, C. Internalized chitosan nanoparticles persist for long time in cultured cells. Eur. J. Histochem. 2015, 59, 2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | AHB-1 | AHB-2 | AHB-3 | Frenatin 2.3s | Buforin II | TP2 |
|---|---|---|---|---|---|---|
| Sequence | MFVFLVLLPLVS | IYKTPPIKDFGGFNFSQIL | WYIWLGFIAGLIAIVMVTIMLCC | GLVGTLLGHIGKAILGG | TRSSRAGLQFPVGRVHRLLRK | PLIYLRLLRGQF |
| Residues | 12 | 19 | 23 | 17 | 21 | 12 |
| (Asp + Glu) | 0 | 1 | 0 | 0 | 0 | 0 |
| (Arg + Lys) | 0 | 2 | 0 | 1 | 6 | 2 |
| Net Charge | 0 | +1 | 0 | +1 | +6 | +2 |
| GRAVY | +2.74 | +0.03 | +2.3 | +1.18 | −0.64 | +0.56 |
| Theo. pI | 5.28 | 8.5 | 5.51 | 8.76 | 12.6 | 10.84 |
| mW | 1377.79 | 2185.55 | 2630.36 | 1575.91 | 2434.88 | 1488.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henao, M.C.; Ocasion, C.; Puentes, P.R.; González-Melo, C.; Quezada, V.; Cifuentes, J.; Yepes, A.; Burgos, J.C.; Cruz, J.C.; Reyes, L.H. Translocating Peptides of Biomedical Interest Obtained from the Spike (S) Glycoprotein of the SARS-CoV-2. Membranes 2022, 12, 600. https://doi.org/10.3390/membranes12060600
Henao MC, Ocasion C, Puentes PR, González-Melo C, Quezada V, Cifuentes J, Yepes A, Burgos JC, Cruz JC, Reyes LH. Translocating Peptides of Biomedical Interest Obtained from the Spike (S) Glycoprotein of the SARS-CoV-2. Membranes. 2022; 12(6):600. https://doi.org/10.3390/membranes12060600
Chicago/Turabian StyleHenao, Maria C., Camila Ocasion, Paola Ruiz Puentes, Cristina González-Melo, Valentina Quezada, Javier Cifuentes, Arnovis Yepes, Juan C. Burgos, Juan C. Cruz, and Luis H. Reyes. 2022. "Translocating Peptides of Biomedical Interest Obtained from the Spike (S) Glycoprotein of the SARS-CoV-2" Membranes 12, no. 6: 600. https://doi.org/10.3390/membranes12060600