
Potential Resistance of SARS-CoV-2 Main Protease (Mpro) against Protease Inhibitors: Lessons Learned from HIV-1 Protease



Abstract
:1. Introduction
2. RNA Viruses
3. Proteases Inhibitors and Mutations
3.1. HIV-1 Protease and Protease Inhibitors
3.2. HIV-1 Protease Inhibitor-Associated Mutations and the Mechanism of Resistance
3.3. SARS-CoV-2 Mpro

3.4. Inhibitors of SARS-CoV-2 Mpro as (Potential) Therapeutic Drugs
3.5. Binding of Nirmatrelvir to SARS-CoV-2 Mpro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | 7MGR | 7MGS | 7N89 | 7VH8 | 7RFW | 7RFS | 7SI9 | 7TE0 | 7TLL | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | [82] | [82] | [83] | [79] | [70] | [70] | [84] | Unpublished Study | [80] | |||||||||
| Ligand | AVKLQ*NNE | SAVLQ*SGF | SAVLQ*SGF | nirmatrelvir | nirmatrelvir | nirmatrelvir | nirmatrelvir | nirmatrelvir | nirmatrelvir | |||||||||
| Interaction | H | NP | H | NP | H | NP | H | NP | H | NP | H | NP | H | NP | H | NP | H | NP |
| S5 site | P168 | P168 Q189 | P168 | |||||||||||||||
| S4 site | T190 | M165 L167 R188 Q189 Q192 | T190 | R188 Q189 Q192 | T190 | R188 Q192 | Q189 | M165 Q189 T190 | M165 Q189 T190 | Q189 | Q192 | Q189 | M165 Q189 Y190 | |||||
| S3 site | E166 N142 | M165 Q189 | E166 | M165 | E166 | M165 | E166 | M165 | E166 | M165 | E166 | M165 | E166 | M165 | E166 | M165 | E166 | M165 |
| S2 site | H41 M165 Q189 | H41 M49 H164 M165 | Q189 | H41 H164 M165 D187 | H41 M49 Y54 D187 R188 Q189 | H41 M49 Y54 H164 M165 D187 R188 Q189 | H41 M49 H164 R188 Q189 | H41 M49 Q189 | H41 M49 M165 Q189 | H41 M49 Y54 D187 R188 Q189 | ||||||||
| S1 site | F140 G143 A145* H163 H164 E166 | L141 S144 M165 H172 | F140 G143 A145 * H163 H164 | L141 H172 | F140 G143 A145 * H163 H164 E166 | L141 N142 S144 H172 | F140 G143 C145 H163 H164 E166 | H41 M49 Y54 L141 N142 H172 D187 | G143 C145 H163 | H41 M49 Y54 F140 N142 H164 M165 H172 D187 T190 | C145 H163 | H41 M49 F140 L141 N142 G143 H164 M165 H172 T190 | F140 H163 H164 E166 | H41 M49 N142 M165 H172 | G143 C145 H163 H164 E166 | F140 N142 G143 H172 | C145 H163 H164 E166 | H41 F140 L141 N142 G143 S144 H172 |
| S1’ site | G143 | T25 H41 M49 | N142 | T25 H41 M49 | T25 H41 M49 N142 | |||||||||||||
| S2’ site | T26 | T25 | T26 | T25 | T26 | T25 | ||||||||||||
| S3’ site | T24 | T24 | T24 S46 | |||||||||||||||

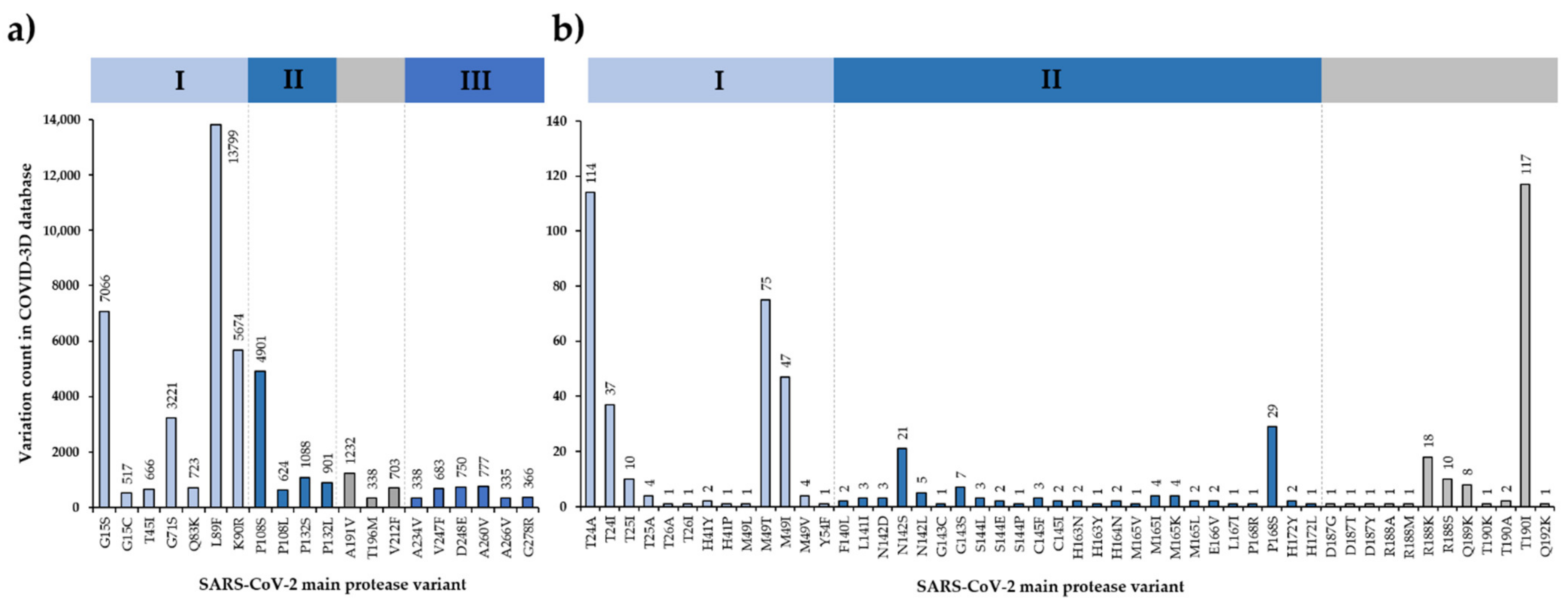
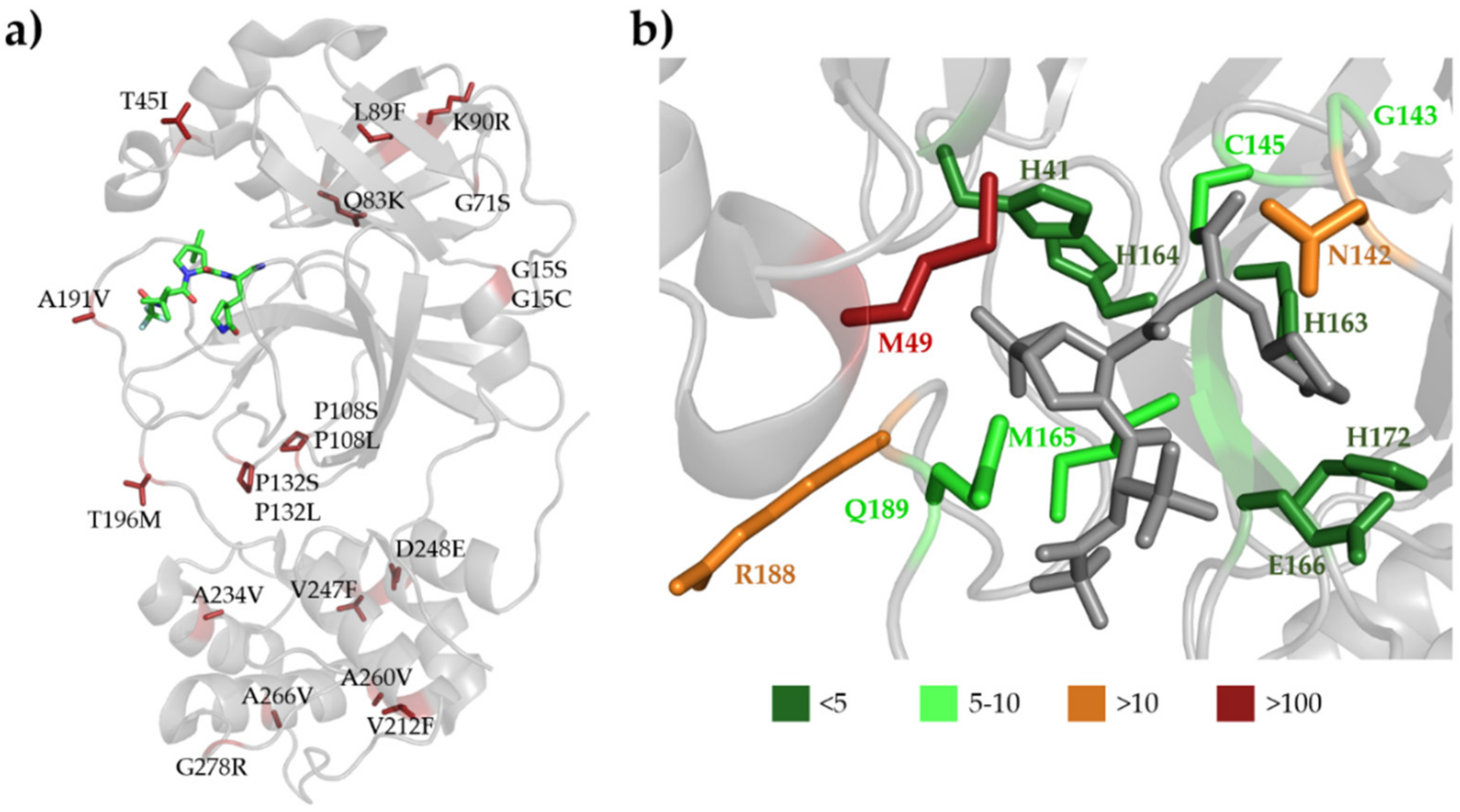
3.6. SARS-CoV-2 Mpro Variants
3.7. Efficacy of Nirmatrelvir against SARS-CoV-2 Variants
3.8. SARS-CoV-2 Cleavage-Site Variants
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahdi, M.; Mótyán, J.A.; Szojka, Z.I.; Golda, M.; Miczi, M.; Tőzsér, J. Analysis of the efficacy of HIV protease inhibitors against SARS-CoV-2’s main protease. Virol. J. 2020, 17, 190. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A trial of Lopinavir-Ritonavir in adults hospitalized with severe COVID-19. N. Engl. J. Med. 2020, 382, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- von Hentig, N. Repositioning HIV protease inhibitors and nucleos(t)ide RNA polymerase inhibitors for the treatment of SARS-CoV-2 infection and COVID-19. Eur. J. Clin. Pharmacol. 2021, 77, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 quasispecies provides an advantage mutation pool for the epidemic variants. Microbiol. Spectr. 2021, 9, e0026121. [Google Scholar] [CrossRef]
- Karakasiliotis, I.; Lagopati, N.; Evangelou, K.; Gorgoulis, V.G. Cellular senescence as a source of SARS-CoV-2 quasispecies. FEBS J. 2021. [Google Scholar] [CrossRef]
- Cann, A.J. Principles of Molecular Virology, 6th ed.; Academic Press: London, UK, 2015; pp. 128–130. [Google Scholar]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers. 2015, 1, 15035. [Google Scholar] [CrossRef]
- Bell, N.M.; Lever, A.M. HIV Gag polyprotein: Processing and early viral particle assembly. Trends Microbiol. 2013, 21, 136–144. [Google Scholar] [CrossRef]
- Smyth, R.P.; Davenport, M.P.; Mak, J. The origin of genetic diversity in HIV-1. Virus Res. 2012, 169, 415–429. [Google Scholar] [CrossRef]
- Craigie, R.; Bushman, F.D. HIV DNA integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef] [Green Version]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Coffin, J.M. HIV population dynamics in vivo: Implications for genetic variation, pathogenesis, and therapy. Science 1995, 267, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Genet. 2015, 13, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Cherepanov, P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol. 2012, 10, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Zhou, H.; Yang, J.; Zhou, C.; Chen, B.; Fang, H.; Chen, S.; Zhang, X.; Wang, L.; Zhang, L. A review of SARS-CoV-2: Compared with SARS-CoV and MERS-CoV. Front Med. 2021, 8, 628370. [Google Scholar] [CrossRef]
- Hardenbrook, N.J.; Zhang, P. A structural view of the SARS-CoV-2 virus and its assembly. Curr. Opin. Virol. 2022, 52, 123–134. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication-transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef]
- Baggen, J.; Vanstreels, E.; Jansen, S.; Daelemans, D. Cellular host factors for SARS-CoV-2 infection. Nat. Microbiol. 2021, 6, 1219–1232. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. Coronavirus infection, ER stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Wang, L.; Cheng, G. The battle between host and SARS-CoV-2: Innate immunity and viral evasion strategies. Mol. Ther. 2022, S1525-0016(22)00097-1. [Google Scholar] [CrossRef] [PubMed]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. (Eds.) Retroviruses; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Tözsér, J.; Gustchina, A.; Weber, I.T.; Blaha, I.; Wondrak, E.M.; Oroszlan, S. Studies on the role of the S4 substrate binding site of HIV proteinases. FEBS Lett. 1991, 279, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Pokorná, J.; Machala, L.; Rezáčová, P.; Konvalinka, J. Current and novel inhibitors of HIV protease. Viruses 2009, 1, 1209–1239. [Google Scholar] [CrossRef] [Green Version]
- Tözsér, J. Comparative studies on retroviral proteases: Substrate specificity. Viruses 2010, 2, 147–165. [Google Scholar] [CrossRef] [Green Version]
- Tözsér, J.; Bagossi, P.; Weber, I.T.; Louis, J.M.; Copeland, T.D.; Oroszlan, S. Studies on the symmetry and sequence context dependence of the HIV-1 proteinase specificity. J. Biol. Chem. 1997, 272, 16807–16814. [Google Scholar] [CrossRef] [Green Version]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef]
- King, N.M.; Prabu-Jeyabalan, M.; Nalivaika, E.A.; Schiffer, C.A. Combating susceptibility to drug resistance: Lessons from HIV-1 protease. Chem. Biol. 2004, 11, 1333–1338. [Google Scholar]
- Ali, A.; Bandaranayake, R.M.; Cai, Y.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.; Nalivaika, E.A.; Ozen, A.; et al. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses 2010, 2, 2509–2535. [Google Scholar] [CrossRef]
- Nalam, M.N.; Ali, A.; Reddy, G.S.; Cao, H.; Anjum, S.G.; Altman, M.D.; Yilmaz, N.K.; Tidor, B.; Rana, T.M.; Schiffer, C.A. Substrate envelope-designed potent HIV-1 protease inhibitors to avoid drug resistance. Chem. Biol. 2013, 20, 1116–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sungkanuparph, S.; Sukasem, C.; Manosuthi, W.; Wiboonchutikul, S.; Piyavong, B.; Chantratita, W. Tipranavir resistance associated mutations in protease inhibitor-naïve patients with HIV-1 subtype A/E infection. J. Clin. Virol. 2008, 43, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid evolution of RNA genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Pillay, D.; Bhaskaran, K.; Jurriaans, S.; Prins, M.; Masquelier, B.; Dabis, F.; Gifford, R.; Nielsen, C.; Pedersen, C.; Balotta, C.; et al. The impact of transmitted drug resistance on the natural history of HIV infection and response to first-line therapy. AIDS 2006, 20, 21–28. [Google Scholar]
- Bennett, D.E.; Camacho, R.J.; Otelea, D.; Kuritzkes, D.R.; Fleury, H.; Kiuchi, M.; Heneine, W.; Kantor, R.; Jordan, M.R.; Schapiro, J.M.; et al. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS ONE 2009, 4, e4724. [Google Scholar] [CrossRef] [Green Version]
- Vercauteren, J.; Wensing, A.M.; van de Vijver, D.A.; Albert, J.; Balotta, C.; Hamouda, O.; Kücherer, C.; Struck, D.; Schmit, J.C.; Asjö, B.; et al. Transmission of drug-resistant HIV-1 is stabilizing in Europe. J. Infect Dis. 2009, 200, 1503–1508. [Google Scholar] [CrossRef]
- Grgic, I.; Lepej, S.Z.; Lunar, M.M.; Poljak, M.; Vince, A.; Vrakela, I.B.; Planinic, A.; Seme, K.; Begovac, J. The prevalence of transmitted drug resistance in newly diagnosed HIV-infected individuals in Croatia: The role of transmission clusters of men who have sex with men carrying the T215S surveillance drug resistance mutation. AIDS Res. Hum. Retroviruses 2013, 29, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Áy, É.; Müller, V.; Mezei, M.; Pocskay, Á.; Koroknai, A.; Müller, D.; Győri, Z.; Marschalkó, M.; Tóth, B.; Kárpáti, S.; et al. Transmitted drug resistance in newly diagnosed and treatment-naïve HIV type 1-infected patients in Hungary. J. Glob. Antimicrob. Resist. 2020, 20, 124–130. [Google Scholar] [CrossRef]
- Günthard, H.F.; Calvez, V.; Paredes, R.; Pillay, D.; Shafer, R.W.; Wensing, A.M.; Jacobsen, D.M.; Richman, D.D. Human immunodeficiency virus drug resistance: 2018 recommendations of the international antiviral society-USA panel. Clin. Infect Dis. 2019, 68, 177–187. [Google Scholar] [CrossRef]
- Hemelaar, J.; Gouws, E.; Ghys, P.D.; Osmanov, S. Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 2006, 20, W13–W23. [Google Scholar] [CrossRef]
- Miller, V. International perspectives on antiretroviral resistance. resistance to protease inhibitors. J. Acquir. Immune Defic. Syndr. 2001, 26, S34–S50. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Markowitz, M. Resistance to human immunodeficiency virus type 1 protease inhibitors. Antimicrob. Agents Chemother. 1998, 42, 2775–2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menéndez-Arias, L.; Tözsér, J. HIV-1 protease inhibitors: Effects on HIV-2 replication and resistance. Trends Pharmacol. Sci. 2008, 29, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdi, M.; Szojka, Z.; Mótyán, J.A.; Tőzsér, J. Inhibition profiling of retroviral protease inhibitors using an HIV-2 modular system. Viruses 2015, 7, 6152–6162. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Li, Y.; Schock, H.B.; Hall, D.; Chen, E.; Kuo, L.C. Three-dimensional structure of a mutant HIV-1 protease displaying cross-resistance to all protease inhibitors in clinical trials. J. Biol. Chem. 1995, 270, 21433–21436. [Google Scholar] [CrossRef] [Green Version]
- Geretti, A.M. (Ed.) Antiretroviral Resistance in Clinical Practice; Mediscript: London, UK, 2006. Available online: https://www.ncbi.nlm.nih.gov/books/NBK2239/ (accessed on 20 February 2022).
- Wensing, A.M.J.; Fun, A.; Nijhuis, M. HIV Protease Inhibitor Resistance. In Handbook of Antimicrobial Resistance; Gotte, M., Berghuis, A., Matlashewski, G., Wainberg, M., Sheppard, D., Eds.; Springer: New York, NY, USA, 2017. [Google Scholar]
- Rhee, S.Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Hertogs, K.; Bloor, S.; Kemp, S.D.; van den Eynde, C.; Alcorn, T.M.; Pauwels, R.; van Houtte, M.; Staszewski, S.; Miller, V.; Larder, B.A. Phenotypic and genotypic analysis of clinical HIV-1 isolates reveals extensive protease inhibitor cross-resistance: A survey of over 6000 samples. AIDS 2000, 14, 1203–1210. [Google Scholar] [CrossRef]
- Weber, I.T.; Agniswamy, J. HIV-1 protease: Structural perspectives on drug resistance. Viruses 2009, 1, 1110–1136. [Google Scholar] [CrossRef]
- Menéndez-Arias, L. Molecular basis of human immunodeficiency virus type 1 drug resistance: Overview and recent developments. Antivir. Res. 2013, 98, 93–120. [Google Scholar] [CrossRef]
- Weber, I.T.; Kneller, D.W.; Wong-Sam, A. Highly resistant HIV-1 proteases and strategies for their inhibition. Future Med. Chem. 2015, 7, 1023–1038. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of two coronavirus main proteases: Implications for substrate binding and antiviral drug design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J.; et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Xiong, M.; Su, H.; Zhao, W.; Xie, H.; Shao, Q.; Xu, Y. What coronavirus 3C-like protease tells us: From structure, substrate selectivity, to inhibitor design. Med. Res. Rev. 2021, 41, 1965–1998. [Google Scholar] [CrossRef]
- Kiemer, L.; Lund, O.; Brunak, S.; Blom, N. Coronavirus 3CLpro proteinase cleavage sites: Possible relevance to SARS virus pathology. BMC Bioinform. 2004, 5, 72. [Google Scholar] [CrossRef] [Green Version]
- Koudelka, T.; Boger, J.; Henkel, A.; Schönherr, R.; Krantz, S.; Fuchs, S.; Rodríguez, E.; Redecke, L.; Tholey, A. N-terminomics for the identification of in vitro substrates and cleavage site specificity of the SARS-CoV-2 main protease. Proteomics 2021, 21, e2000246. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jabłońska, J.; Pravda, L.; Vařeková, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Pujadas, G.; Garcia-Vallvé, S. A review of the current landscape of SARS-CoV-2 main protease inhibitors: Have we hit the bullseye yet? Int. J. Mol. Sci. 2021, 23, 259. [Google Scholar] [CrossRef]
- Zephyr, J.; Kurt Yilmaz, N.; Schiffer, C.A. Viral proteases: Structure, mechanism and inhibition. Enzymes 2021, 50, 301–333. [Google Scholar] [PubMed]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Discovery of a novel inhibitor of coronavirus 3CL protease for the potential treatment of COVID-19. bioRxiv 2021. [Google Scholar] [CrossRef]
- Boras, B.; Jones, R.M.; Anson, B.J.; Arenson, D.; Aschenbrenner, L.; Bakowski, M.A.; Beutler, N.; Binder, J.; Chen, E.; Eng, H.; et al. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021, 12, 6055. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Sharma, T.; Ahmad, I.; Abohashrh, M.; Alam, M.M.; Dong, J.J. Is PF-00835231 a Pan-SARS-CoV-2 Mpro inhibitor? A comparative study. Molecules 2021, 26, 1678. [Google Scholar] [CrossRef] [PubMed]
- Vandyck, K.; Deval, J. Considerations for the discovery and development of 3-chymotrypsin-like cysteine protease inhibitors targeting SARS-CoV-2 infection. Curr. Opin. Virol. 2021, 49, 36–40. [Google Scholar] [CrossRef]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Patick, A.K.; Brothers, M.A.; Maldonado, F.; Binford, S.; Maldonado, O.; Fuhrman, S.; Petersen, A.; Smith, G.J., 3rd; Zalman, L.S.; Burns-Naas, L.A.; et al. In vitro antiviral activity and single-dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 2005, 49, 2267–2275. [Google Scholar] [CrossRef] [Green Version]
- Hull, M.W.; Montaner, J.S. Ritonavir-boosted protease inhibitors in HIV therapy. Ann. Med. 2011, 43, 375–388. [Google Scholar] [CrossRef]
- Pfizer, 2021.11.05. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizers-novel-covid-19-oral-antiviral-treatment-candidate (accessed on 15 February 2022).
- Kim, Y.; Lovell, S.; Tiew, K.C.; Mandadapu, S.R.; Alliston, K.R.; Battaile, K.P.; Groutas, W.C.; Chang, K.O. Broad-spectrum antivirals against 3C or 3C-like proteases of picornaviruses, noroviruses, and coronaviruses. J. Virol. 2012, 86, 11754–11762. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Sacco, M.D.; Hurst, B.; Townsend, J.A.; Hu, Y.; Szeto, T.; Zhang, X.; Tarbet, B.; Marty, M.T.; Chen, Y.; et al. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. [Google Scholar] [CrossRef]
- Boceprevir. Drugs RD 2010, 10, 203–210. [CrossRef] [PubMed]
- Ahmad, B.; Batool, M.; Ain, Q.U.; Kim, M.S.; Choi, S. Exploring the binding mechanism of PF-07321332 SARS-CoV-2 protease inhibitor through molecular dynamics and binding free energy simulations. Int. J. Mol. Sci. 2021, 22, 9124. [Google Scholar] [CrossRef] [PubMed]
- Macchiagodena, M.; Pagliai, M.; Procacci, P. Characterization of the non-covalent interaction between the PF-07321332 inhibitor and the SARS-CoV-2 main protease. J. Mol. Graph. Model. 2022, 110, 108042. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2021, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Greasley, S.E.; Noell, S.; Plotnikova, O.; Ferre, R.; Liu, W.; Bolanos, B.; Fennell, K.; Nicki, J.; Craig, T.; Zhu, Y.; et al. Structural basis for Nirmatrelvir in vitro efficacy against the Omicron variant of SARS-CoV-2. bioRxiv 2022. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Chapsal, B.D.; Weber, I.T.; Mitsuya, H. Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc. Chem. Res. 2008, 41, 78–86. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, E.A.; Frey, G.; Namchuk, M.N.; Harrison, S.C.; Hinshaw, S.M.; Windsor, I.W. Recognition of divergent viral substrates by the SARS-CoV-2 main protease. ACS Infect Dis. 2021, 7, 2591–2595. [Google Scholar] [CrossRef]
- Kneller, D.W.; Zhang, Q.; Coates, L.; Louis, J.M.; Kovalevsky, A. Michaelis-like complex of SARS-CoV-2 main protease visualized by room-temperature X-ray crystallography. IUCr J. 2021, 8, 973–979. [Google Scholar] [CrossRef]
- Kneller, D.; Li, H.; Phillips, G.; Weiss, K.; Zhang, Q.; Arnould, M.; Jonsson, C.; Surendranathan, S.; Parvathareddy, J.; Blakeley, M.; et al. Covalent narlaprevir- and boceprevir-derived hybrid inhibitors of SARS-CoV-2 main protease: Room-temperature X-ray and neutron crystallography, binding thermodynamics, and antiviral activity. Res. Sq. 2022, preprint. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Shaqra, A.M.; Zvornicanin, S.; Huang, Q.Y.; Lockbaum, G.J.; Knapp, M.; Tandeske, L.; Barkan, D.T.; Flynn, J.; Bolon, D.N.A.; Moquin, S.; et al. Defining the substrate envelope of SARS-CoV-2 main protease to predict and avoid drug resistance. bioRxiv 2022. [Google Scholar] [CrossRef]
- Cheng, S.C.; Chang, G.G.; Chou, C.Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 2010, 98, 1327–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portelli, S.; Olshansky, M.; Rodrigues, C.H.M.; D’Souza, E.N.; Myung, Y.; Silk, M.; Alavi, A.; Pires, D.E.V.; Ascher, D.B. Author correction: Exploring the structural distribution of genetic variation in SARS-CoV-2 with the COVID-3D online resource. Nat. Genet. 2021, 53, 254, Erratum in Nat. Genet. 2020, 52, 999–1001. [Google Scholar] [CrossRef]
- Torrens-Fontanals, M.; Peralta-García, A.; Talarico, C.; Guixà-González, R.; Giorgino, T.; Selent, J. SCoV2-MD: A database for the dynamics of the SARS-CoV-2 proteome and variant impact predictions. Nucleic Acids Res. 2022, 50, D858–D866. [Google Scholar] [CrossRef]
- Alsulami, A.F.; Thomas, S.E.; Jamasb, A.R.; Beaudoin, C.A.; Moghul, I.; Bannerman, B.; Copoiu, L.; Vedithi, S.C.; Torres, P.; Blundell, T.L. SARS-CoV-2 3D database: Understanding the coronavirus proteome and evaluating possible drug targets. Brief Bioinform. 2021, 22, 769–780. [Google Scholar] [CrossRef]
- Krishnamoorthy, N.; Fakhro, K. Identification of mutation resistance coldspots for targeting the SARS-CoV2 main protease. IUBMB Life 2021, 73, 670–675. [Google Scholar] [CrossRef]
- Sheik Amamuddy, O.; Verkhivker, G.M.; Tastan Bishop, Ö. Impact of early pandemic stage mutations on molecular dynamics of SARS-CoV-2 Mpro. J. Chem. Inf. Model. 2020, 60, 5080–5102. [Google Scholar] [CrossRef]
- Sharma, T.; Abohashrh, M.; Baig, M.H.; Dong, J.J.; Alam, M.M.; Ahmad, I.; Irfan, S. Screening of drug databank against WT and mutant main protease of SARS-CoV-2: Towards finding potential compound for repurposing against COVID-19. Saudi. J. Biol. Sci. 2021, 28, 3152–3159. [Google Scholar] [CrossRef]
- Nakayoshi, T.; Kato, K.; Kurimoto, E.; Oda, A. Virtual alanine scan of the main protease active site in severe acute respiratory syndrome Coronavirus 2. Int. J. Mol. Sci. 2021, 22, 9837. [Google Scholar] [CrossRef]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural plasticity of SARS-CoV-2 3CLMpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef]
- Cross, T.J.; Takahashi, G.R.; Diessner, E.M.; Crosby, M.G.; Farahmand, V.; Zhuang, S.; Butts, C.T.; Martin, R.W. Sequence characterization and molecular modeling of clinically relevant variants of the SARS-CoV-2 main protease. Biochemistry 2020, 59, 3741–3756. [Google Scholar] [CrossRef]
- Ullrich, S.; Ekanayake, K.B.; Otting, G.; Nitsche, C. Main protease mutants of SARS-CoV-2 variants remain susceptible to nirmatrelvir. Bioorg. Med. Chem. Lett. 2022, 62, 128629. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, Y.; Lavrijsen, M.; Lamers, M.M.; de Vries, A.C.; Rottier, R.J.; Bruno, M.J.; Peppelenbosch, M.P.; Haagmans, B.L.; Pan, Q. SARS-CoV-2 Omicron variant is highly sensitive to molnupiravir, nirmatrelvir, and the combination. Cell Res. 2022, 32, 322–324. [Google Scholar] [CrossRef] [PubMed]
- Rai, K.D.; Yurgelonis, I.; McMonagle, P.; Rothan, H.A.; Hao, L.; Gribenko, A.; Titova, E.; Kreiswirth, B.; White, K.M.; Zhu, Y.; et al. Nirmatrelvir, an orally active Mpro inhibitor, is a potent inhibitor of SARS-CoV-2 Variants of Concern. bioRxiv 2022. [Google Scholar] [CrossRef]
- Vangeel, L.; Chiu, W.; De Jonghe, S.; Maes, P.; Slechten, B.; Raymenants, J.; André, E.; Leyssen, P.; Neyts, J.; Jochmans, D. Remdesivir, Molnupiravir and Nirmatrelvir remain active against SARS-CoV-2 Omicron and other variants of concern. Antivir. Res. 2022, 198, 105252. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, M.; Deeks, S.; Boucher, C. Implications of antiretroviral resistance on viral fitness. Curr. Opin. Infect. Dis. 2001, 14, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.W.; Torbett, B.E. Accessory mutations maintain stability in drug-resistant HIV-1 protease. J. Mol. Biol. 2011, 410, 756–760. [Google Scholar] [CrossRef] [Green Version]
- Prabu-Jeyabalan, M.; Nalivaika, E.A.; King, N.M.; Schiffer, C.A. Structural basis for coevolution of a human immunodeficiency virus type 1 nucleocapsid-p1 cleavage site with a V82A drug-resistant mutation in viral protease. J. Virol. 2004, 78, 12446–12454. [Google Scholar] [CrossRef] [Green Version]
- Nijhuis, M.; Schuurman, R.; De Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A.B. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 1999, 13, 2349–2359. [Google Scholar] [CrossRef]
- Su, C.T.; Koh, D.W.; Gan, S.K. Reviewing HIV-1 gag mutations in protease inhibitors resistance: Insights for possible novel gag inhibitor designs. Molecules 2019, 24, 3243. [Google Scholar] [CrossRef] [Green Version]
- Pablos, I.; Machado, Y.; de Jesus, H.C.R.; Mohamud, Y.; Kappelhoff, R.; Lindskog, C.; Vlok, M.; Bell, P.A.; Butler, G.S.; Grin, P.M.; et al. Mechanistic insights into COVID-19 by global analysis of the SARS-CoV-2 3CLpro substrate degradome. Cell Rep. 2021, 37, 109892. [Google Scholar] [CrossRef]
- Goetz, D.H.; Choe, Y.; Hansell, E.; Chen, Y.T.; McDowell, M.; Jonsson, C.B.; Roush, W.R.; McKerrow, J.; Craik, C.S. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry 2007, 46, 8744–8752. [Google Scholar] [CrossRef] [PubMed]
- Chuck, C.P.; Chong, L.T.; Chen, C.; Chow, H.F.; Wan, D.C.; Wong, K.B. Profiling of substrate specificity of SARS-CoV 3CLpro. PLoS ONE 2010, 5, e13197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuck, C.P.; Chow, H.F.; Wan, D.C.; Wong, K.B. Profiling of substrate specificities of 3C-like proteases from group 1, 2a, 2b, and 3 coronaviruses. PLoS ONE 2011, 6, e27228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.M.; Imamichi, H.; Imamichi, T.; Lane, H.C.; Falloon, J.; Vasudevachari, M.B.; Salzman, N.P. Drug resistance during indinavir therapy is caused by mutations in the protease gene and in its Gag substrate cleavage sites. J. Virol. 1997, 71, 6662–6670. [Google Scholar] [CrossRef] [Green Version]
- Doyon, L.; Payant, C.; Brakier-Gingras, L.; Lamarre, D. Novel Gag-Pol frameshift site in human immunodeficiency virus type 1 variants resistant to protease inhibitors. J. Virol. 1998, 72, 6146–6150. [Google Scholar] [CrossRef] [Green Version]
- Wensing, A.M.; van Maarseveen, N.M.; Nijhuis, M. Fifteen years of HIV Protease Inhibitors: Raising the barrier to resistance. Antivir. Res. 2010, 85, 59–74. [Google Scholar] [CrossRef]
- Arts, E.J.; Hazuda, D.J. HIV-1 antiretroviral drug therapy. Cold Spring Harb. Perspect. Med. 2012, 2, a007161. [Google Scholar] [CrossRef]
- Pfizer, 2022.01.18. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-shares-vitro-efficacy-novel-covid-19-oral-treatment (accessed on 15 February 2022).
- Heskin, J.; Pallett, S.J.C.; Mughal, N.; Davies, G.W.; Moore, L.S.P.; Rayment, M.; Jones, R. Caution required with use of ritonavir-boosted PF-07321332 in COVID-19 management. Lancet 2022, 399, 21–22. [Google Scholar] [CrossRef]



| P5 | P4 | P3 | P2 | P1 | P1’ | P2’ | P3’ | P4’ | P5’ | |
|---|---|---|---|---|---|---|---|---|---|---|
| nsp4-nsp5 | S | A | V | L | Q | S | G | F | R | K |
| S496L (3) | A497S (1) | V498I (25) | L499A (1) | - | - | - | F3L (1) | R4K (11) | K5E (1) | |
| V498F (1) | R4G (1) | |||||||||
| nsp5-nsp6 | G | V | T | F | Q | S | A | V | K | R |
| G302C (8) | V303I (4) | T304I (54) | F305L (1) | Q306H (4) | S1N (127) | - | V3M (25) | K4E (36) | R5G (13) | |
| G302S (1) | V303G (1) | T304N (35) | F305C (1) | Q306R (1) | V3L (10) | K4R (21) | ||||
| T304P (1) | V3I (5) | K4Q (6) | ||||||||
| V3A (4) | K4G (1) | |||||||||
| nsp6-nsp7 | V | A | T | V | Q | S | K | M | S | D |
| V286I (22) | A287T (35) | T288I (22) | V289L (20) | Q290H (90) | - | K2R (4) | M3I (1096) | S4A (7) | D5A (7) | |
| V286L (8) | A287V (32) | T288A (3) | V289I (7) | K2E (2) | M3T (12) | S4L (3) | D5E (4) | |||
| A287S (2) | T288S (3) | V289E (1) | M3V (3) | |||||||
| A287D (1) | T288N (2) | |||||||||
| nsp7-nsp8 | R | A | T | L | Q | A | I | A | S | E |
| R79M (5) | A80V (250) | T81I (257) | L82S (2) | Q83H (3) | - | I2V (20) | A3V (26) | S4L (37) | E5D (8) | |
| R79G (4) | A80T (8) | T81A (4) | I2M (6) | A3S (3) | S4T (5) | |||||
| R79S (1) | I2T (3) | A3T (2) | S4A (1) | |||||||
| R79K (1) | I2L (2) | A3D (1) | ||||||||
| nsp8-nsp9 | A | V | K | L | Q | N | N | E | L | S |
| A194V (74) | V195I (35) | - | - | Q198H (31) | - | - | - | - | S5G (11) | |
| V195F (3) | S5N (5) | |||||||||
| V195A (1) | S5R (1) | |||||||||
| nsp9-nsp10 | T | V | R | L | Q | A | G | N | A | T |
| T109I (287) | V110L (1) | R111H (1) | L112T (1) | Q113R (1) | n.a. | n.a. | n.a. | n.a. | n.a. | |
| T109R (1) | V110N (1) | R111C (1) | ||||||||
| nsp10-nsp11 | E | P | M | L | Q | S | A | D | A | Q |
| n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | A2V (44) | D3G (12) | A4V (54) | Q5P (1) | |
| A2T (5) | D3N (6) | A4T (4) | Q5K (1) | |||||||
| A2S (5) | D3E (1) | A4S (1) | Q5L (1) | |||||||
| A2D (2) | A4E (1) | Q5R (1) | ||||||||
| nsp11-nsp12 | H | T | V | L | Q | A | V | G | A | C |
| H928Y (100) | T929I (2) | V930I (14) | L931T (1) | Q932H (41) | - | V2I (3) | G3R (1) | A4V (152) | C5S (1) | |
| H928Q (4) | T929A (1) | V930F (1) | L931Y (1) | A4S (41) | ||||||
| H928L (2) | T929S (1) | V930L (1) | A4T (1) | |||||||
| nsp12-nsp13 | V | A | T | L | Q | A | E | N | V | T |
| V597L (13) | A598S (6763) | T599I (744) | L600Y (1) | Q601H (2) | - | E2A (1) | N3T (6) | V4L (5) | T5I (9) | |
| V597A (11) | A598V (418) | T599N (10) | Q601K (1) | E2G (1) | N3S (2) | V4I (4) | T5A (6) | |||
| V597M (3) | A598T (10) | T599A (1) | E2K (1) | N3H (1) | V4A (1) | |||||
| V597W (1) | A598E (1) | T599L (1) | E2Q (1) | N3I (1) | ||||||
| A598L (1) | E2V (1) | |||||||||
| A598Q (1) | ||||||||||
| nsp13-nsp14 | F | T | R | L | Q | S | L | E | N | V |
| F523C (1) | T524I (114) | R525K (32) | L526F (2) | Q527P (1) | - | - | - | - | - | |
| F523L (1) | T524A (2) | R525I (3) | L526P (1) | |||||||
| T524Q (1) | R525D (1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mótyán, J.A.; Mahdi, M.; Hoffka, G.; Tőzsér, J. Potential Resistance of SARS-CoV-2 Main Protease (Mpro) against Protease Inhibitors: Lessons Learned from HIV-1 Protease. Int. J. Mol. Sci. 2022, 23, 3507. https://doi.org/10.3390/ijms23073507
Mótyán JA, Mahdi M, Hoffka G, Tőzsér J. Potential Resistance of SARS-CoV-2 Main Protease (Mpro) against Protease Inhibitors: Lessons Learned from HIV-1 Protease. International Journal of Molecular Sciences. 2022; 23(7):3507. https://doi.org/10.3390/ijms23073507
Chicago/Turabian StyleMótyán, János András, Mohamed Mahdi, Gyula Hoffka, and József Tőzsér. 2022. "Potential Resistance of SARS-CoV-2 Main Protease (Mpro) against Protease Inhibitors: Lessons Learned from HIV-1 Protease" International Journal of Molecular Sciences 23, no. 7: 3507. https://doi.org/10.3390/ijms23073507